

Abstract
GSTP1 CpG island hypermethylation is the most common somatic genome alteration described for human prostate cancer (PCA); lack of GSTP1 expression is characteristic of human PCA cells in vivo. We report here that loss of GSTP1 function may have been selected during the pathogenesis of human PCA. Using a variety of techniques to detect GSTP1 CpG island DNA hypermethylation in PCA DNA, we found only hypermethylated GSTP1 alleles in each PCA cell in all but two PCA cases studied. In these two cases, CpG island hypermethylation was present at only one of two GSTP1 alleles in PCA DNA. In one of the cases, DNA hypermethylation at one GSTP1 allele and deletion of the other GSTP1 allele were evident. In the other case, an unmethylated GSTP1 allele was detected, accompanied by abundant GSTP1 expression. GSTP1 CpG island DNA hypermethylation was responsible for lack of GSTP1 expression by LNCaP PCA cells: treatment of the cells with 5-azacytidine (5-aza-C), an inhibitor of DNA methyltransferases, reversed the GSTP1 promoter DNA hypermethylation, activated GSTP1 transcription, and restored GSTP1 expression. GSTP1 promoter activity, assessed via transfection of GSTP1 promoter-CAT reporter constructs in LNCaP cells, was inhibited by SssI-catalyzed CpG dinucleotide methylation. Remarkably, although selection for loss of GSTP1 function may be inferred for human PCA, GSTP1 did not act like a tumor suppressor gene, as LNCaP cells expressing GSTP1, either after 5-aza-C treatment or as a consequence of transfection with GSTP1 cDNA, grew well in vitro and in vivo. Perhaps, GSTP1 inactivation may render prostatic cells susceptible to additional genome alterations, caused by electrophilic or oxidant carcinogens, that provide a selective growth advantage.
Somatic genome lesions, including mutations, translocations, amplifications, and deletions, are characteristic of cancer cell DNA. 1-4 Often, these lesions target critical genes involved in cell transformation or in the maintenance of the neoplastic phenotype. At other times, these genome lesions do not seem to target such cancer genes. Somatic changes in deoxycytidine methylation are also frequently found in human cancer cell DNA. 5,6 Many of these DNA methylation changes seem to target critical genes associated with cancer pathogenesis. Other somatic changes in DNA methylation found in cancer cells may not involve critical genes. Ideally, if a cancer cell DNA alteration has targeted a critical gene for cancer development, the DNA lesion has likely provided a selective cell growth or survival advantage at some point during cancer initiation or malignant progression. To infer such selection in vivo for a somatic DNA change found in human cancer cells, the DNA alteration must change the function of a specific gene or its product and must be selectively present in a specific cell population (eg, cancer cells versus normal cells or metastatic cancer cells versus primary site cancer cells).
In a previous study, 7 we reported the detection of somatic changes in deoxycytidine methylation affecting a CpG island encompassing the 5′-regulatory region of the human π-class glutathione S-transferase (GST) gene, GSTP1, in human prostatic carcinomas (PCAs). The specific DNA methylation change, a somatic increase in CpG dinucleotide methylation at a BssHII endonuclease recognition site in the transcriptional promoter near GSTP1, was present in DNA isolated from 20 of 20 PCA specimens. Furthermore, the presence of this DNA alteration correlated with a lack of GSTP1 polypeptide expression in PCA cells in vivo and in vitro, raising the possibility that the DNA methylation change might be associated with gene inactivation. These findings of GSTP1 CpG island DNA methylation and lack of GSTP1 expression in human PCA have now been reported in several subsequent studies from several different laboratories. 8-18 Somatic alterations in CpG dinucleotide methylation, especially alterations targeting CpG dinucleotides clustered into CpG islands at the regulatory region of genes, usually result in changes in gene expression, but not in changes in gene product function. 5,6 To infer selection in vivo for GSTP1 CpG island DNA hypermethylation and loss of GSTP1 function in PCA, GSTP1 CpG island DNA hypermethylation must be associated with gene inactivation and must be selectively present in PCA cells versus normal cells. Furthermore, PCA cells must contain only inactivated GSTP1 genes. GSTP1 is an autosomal gene located at chromosome 11q13. 19-21 To permit selection during prostatic carcinogenesis, prostatic cells must either contain CpG dinucleotide changes affecting both GSTP1 alleles or DNA hypermethylation affecting one GSTP1 allele in association with another gene-inactivating lesion affecting the other GSTP1 allele.
We present here evidence that GSTP1 genes are inactivated in prostatic cells during the pathogenesis of human PCA as a consequence of CpG island DNA hypermethylation, and that cells with inactivated GSTP1 genes may have been selected during human prostatic carcinogenesis. PCA cells in most PCA cases stereotypically fail to express GSTP1 polypeptides. Using a variety of analytic approaches to detect GSTP1 CpG island hypermethylation in PCA cell DNA, we found that all PCA cells in all but one PCA case contained only hypermethylated GSTP1 CpG islands in vivo. In this one PCA case, in which each of the PCA cells carried an unmethylated GSTP1 CpG island allele, all of the cells expressed high levels of GSTP1 polypeptides. In addition, studies of GSTP1 promoter function in LNCaP PCA cells in vitro further supported the notion that CpG island DNA hypermethylation was responsible for GSTP1 transcriptional inactivation. Finally, although PCA cells with GSTP1 CpG island hypermethylation and loss of GSTP1 expression seemed to have been selected during human prostatic carcinogenesis, restoration of GSTP1 expression in fully transformed LNCaP PCA cells, either via 5-aza-C treatment or by transfection with GSTP1 cDNA, failed to reduce LNCaP PCA growth in vitro or tumorigenicity in vivo, suggesting that GSTP1 does not likely function as a tumor suppressor gene in the pathogenesis of PCA.
Materials and Methods
Isolation of Genomic DNA from Normal and Neoplastic Human Cells and Tissues
Genomic DNA was isolated from LNCaP PCA cells, 22 and from PCA tissues, along with normal prostate tissues and normal seminal vesicle tissues, obtained at radical prostatectomy or pelvic lymph node dissection, as previously described. 7,23 The collection of such tissues was conducted as part of a clinical research protocol approved by the Joint Committee on Clinical Investigation at the Johns Hopkins Medical Institutions. Genomic DNA was also isolated from normal and neoplastic tissues, obtained at surgery for carcinomas of the kidney, endometrium, uterine cervix, bladder, and ureter. 24-26 DNA quantity was estimated using a diphenylamine assay. 27
Immunohistochemical Detection of GSTP1, Prostate-Specific Antigen, and Keratin Polypeptides in Human Tissue Sections
Formalin-fixed, paraffin-embedded tissues, were cut into 5-μm sections and stained with anti-GSTP1 antibodies (1:3000 dilution; DAKO, Carpinteria, CA), anti-prostate-specific antigen antibodies (1:25 dilution, DAKO), and anti-prostate-specific acid phosphatase antibodies (1:20,000 dilution, DAKO), using an immunoperoxidase method (ChemMate Universal Detection System; Ventana Medical Systems, Tucson, AZ) with diaminobenzidine as a peroxidase substrate. 7,28 Immunostained tissue sections were counterstained with hematoxylin.
Southern Blot Analyses for GSTP1 CpG Island Hypermethylation and for Other Somatic Genome Alterations
Southern blot analysis of DNA from LNCaP PCA cells, and from normal tissues and PCA tissues, was accomplished as described previously. 7,23 To detect GSTP1 CpG island hypermethylation, purified DNAs were digested first with EcoRI and HindIII, and then with BssHII, an enzyme that will not cut its recognition sequence, GCGCGC, if it contains 5-mC. To detect somatic loss of polymorphic alleles at different chromosomal loci, including 8p, 16q, and 17p, purified DNAs were digested with relevant restriction endonucleases recognizing cutting sites present on only one of two alleles at the various loci. Digested DNAs were electrophoresed on agarose gels, transferred to Zeta-Probe membranes (Bio-Rad, Richmond, CA), hybridized with 32P-labeled GSTP1 cDNA 21 or 32P-labeled genomic probe DNA (probes KS-2, CI-8319, MSR, KSR, and K26 for 8p, HPO-4 for 16q, and YNZ-22 for 17p 23 ), and visualized by autoradiography. Autoradiographs were then subjected to quantitative densitometry using a Scanmaster scanner (Howtek).
A CpG Dinucleotide Methylation-Sensitive Endonuclease/Polymerase Chain Reaction (PCR) Assay for the Simultaneous Discrimination of Maternal and Paternal GSTP1 Alleles and Detection of GSTP1 CpG Island Hypermethylation
Purified DNAs were digested extensively with HpaII, with MspI, or left undigested, and then subjected to PCR amplification using primers encompassing a polymorphic [ATAAA]n repeat sequence and two HpaII/MspI sites in the 5′ region of GSTP1 (GenBank positions −535 to −509, 5′-AGCCTGGGCCACAGCGTGAGACTACGT-3′, and −246 to −266, 5′-GGAGTAAACAGACAGCAGGAAGAGGAC-3′) using reaction conditions described previously. 13 As a control, the DNAs were also subjected to PCR amplification with primers encompassing the polymorphic [ATAAA]n repeat sequence but not the two HpaII/MspI sites (GenBank positions −535 to −509, 5′-AGCCTGGGCCACAGCGTGAGACTACGT-3′, and −364 to −337, 5′-TCCCGGAGCTTGCACACCCGCTTCACA-3′). PCR products were visualized, after end-labeling the downstream primer with [γ-32P]ATP using T4 polynucleotide kinase, by electrophoresis on 6% polyacrylamide DNA sequencing gels containing 8 mol/L urea run at 60 W for 2.5 hours, gel mounting, and drying on filter paper (Whatman), and exposure to X-OMAT film (Eastman-Kodak, Rochester, NY).
A Bisulfite Genomic-Sequencing Approach for the Detection of Somatic GSTP1 CpG Island DNA Hypermethylation
To map CpG dinucleotide changes throughout the GSTP1 CpG island, bisulfite genomic sequencing, which permits discrimination of 5-mC from C, 29 was undertaken. Purified DNAs (200 ng) were digested with EcoRI, admixed with salmon sperm DNA (2.5 μg), and then treated with sodium bisulfite as described previously. 30 Bisulfite-treated DNA was then subjected to two rounds of PCR to amplify GSTP1 CpG island alleles, using primers that recognize antisense strand GSTP1 sequences after conversion of C to T (first PCR reaction primers: GenBank positions −636 to −613, 5′-ACA/GCAACCTATAATTCCACCTACTC-3′, and +117 to +94, 5′-GTT/CGGGAGTTGGGGTTTGATGTTG-3′; second PCR reaction primers: GenBank positions −535 to −512, 5′-AACCTAAACCACAACA/GTAAAACAT-3′, and +89 to +66, 5′-TTGGTTTTATGTTGGGAGTTTTGA-3′). The first PCR reaction contained 100 ng bisulfite-treated DNA, 1 μmol/L primers, 250 μmol/L deoxyribonucleotide triphosphates, and 2.5 Units Platinum Taq polymerase (Life Technologies, Inc., Rockville, MD) in OptiPrime buffer no. 7 (Stratagene, La Jolla, CA). The reaction mixture was heated to 94°C for 2 minutes, then subjected to PCR with incubation at 94°C for 1 minute, 58°C for 2 minutes, and 72°C for 3 minutes for five cycles, followed by incubation at 94°C for 30 seconds, 63°C for 2 minutes, and 72°C for 1.5 minutes for 25 cycles before a final extension at 72°C for 6 minutes. The second nested PCR reaction mixture, which contained 15 ng of DNA, 1 μmol/L of primers, 250 μmol/L of deoxyribonucleotide triphosphates, and 2.5 U of Taq polymerase in OptiPrime buffer no. 8 (Stratagene), was heated to 94°C for 2 minutes, then subjected to PCR with incubation at 94°C for 1 minute, 57°C for 2 minutes, and 72°C for 3 minutes for five cycles, followed by incubation at 94°C for 30 seconds, 62°C for 2 minutes, and 72°C for 1.5 minutes for 25 cycles before a final extension at 72°C for 6 minutes. To permit DNA sequencing of individual GSTP1 CpG island alleles, PCR products were first purified by separation on 1% agarose gels (Life Technologies), isolated from the agarose (using a QIAquick gel extraction kit; Qiagen, Valencia, CA), and recovered by ethanol precipitation, and then cloned by ligation into pCR 2.1pTOPO cloning vectors (using a TOPO kit; Invitrogen, Carlsbad, CA) followed by introduction into TOP 10 One-Shot competent bacteria. Plasmid DNAs isolated from independent drug-resistant bacterial clones (a minimum of 10 clones for each PCR reaction product) were subjected to DNA sequence analysis using a cycle-sequencing approach with M13-sequencing primers dye-labeled terminators (Abi Prism Dye Terminator Cycle Sequencing Ready Reaction kit; Perkin Elmer, Emeryville, CA), and an ABI automated sequencer.
Propagation of LNCaP Human PCA Cells in Vitro and in Vivo, Assessment of Effects of GSTP1 CpG Island Methylation on GSTP1 Regulation in LNCaP Human PCA Cells, and Isolation of LNCaP Variants Expressing GSTP1 Polypeptides
LNCaP PCA cells, which contain hypermethylated GSTP1 CpG island alleles and fail to express GSTP1, 7 and PC-3 PCA cells, which contain unmethylated GSTP1 CpG island alleles and express abundant GSTP1, 7,31 were propagated in vitro in RPMI 1640 (Mediatech) supplemented with 10% fetal calf serum (Life Technologies). GSTP1 transcription by isolated nuclei from LNCaP and from PC-3 was assessed via nuclear run-on transcription assay accomplished as previously described, 32 using GSTP1 genomic DNA, hAR cDNA and TOP1 cDNA as hybridization targets for radiolabeled nuclear RNA. To reverse GSTP1 CpG island DNA hypermethylation in LNCaP PCA cells, the cells were treated with 5 μmol/L 5-aza-C in complete growth medium. GSTP1 expression was monitored via Northern blot analysis, using radiolabeled GSTP1 cDNA probes (with TOP1 and H4 cDNA probes as controls), and immunoblot analysis, using anti-GSTP1 antibodies (with anti-lamin B antibodies as controls), in a manner previously described. 7 The LNCaP-5azaC subline, isolated by treatment of LNCaP cells with 5-aza-C for more than 30 generations, was maintained by propagation in vitro in growth medium containing 5-aza-C.
To ascertain the effect of CpG island DNA hypermethylation on GSTP1 promoter function in LNCaP PCA cells, GSTP1 transcriptional regulatory sequences (GenBank positions −408 to +36) were isolated, treated with SssI (New England BioLabs, Beverly, MA), a bacterial CpG methylase, or left untreated, and then ligated to a linearized pCAT-Basic vector (Promega, Madison, WI), without propagation in bacteria, before transfection into LNCaP PCA cells using Lipofectamine Plus reagent (Life Technologies). GSTP1 promoter activity in LNCaP PCA cells was also evaluated using a series of unmethylated GSTP1 promoter/CAT reporter constructs as previously described for MCF-7 breast cancer cells. 33 CAT reporter expression was assessed 48 hours after transfection using an enzyme activity assay (Flash Cat nonradioactive assay kit, Stratagene). The plasmids pCAT-Control (Promega) and pCMV-β-gal (Stratagene) served as controls for transient transfection analyses.
LNCaP-GSTP1 subclones were generated via transfection of pCMV-GSTP1neo, prepared by ligating GSTP1 cDNA 21 into pCMV-neo, selection of G418 (Life Technologies)-resistant subclones, and verification of GSTP1 expression by immunoblot analysis using anti-GSTP1 antibodies. Growth rates of LNCaP cells, LNCaP-5-aza-C cells, and LNCaP-GSTP1 subclones were determined by estimation of cell number throughout time during propagation in vitro in complete growth medium (in the absence of 5-aza-C or G418). Tumorigenicity for LNCaP cells and each of the LNCaP variants was assessed by inoculation of 10 6 cells in 0.1 ml of saline solution admixed with 75% Matrigel into the subcutaneous region of the flanks of athymic mice. 34 Tumor size was determined by caliper measurement. At 8 weeks after inoculation, tumors were excised and subjected to immunohistochemical staining with anti-GSTP1 antibodies as described above.
Results
Southern Blot Analyses Reveal that Most PCA Cells Contain Only Hypermethylated GSTP1 CpG Island Sequences in Vivo
Most PCA tissues are composed of admixtures of normal and neoplastic cells. Normal cells, including fibroblasts, vascular endothelial cells, and inflammatory cells, may comprise up to 30 to 50% or more of the cells in different prostate tumor specimens. Not surprisingly, analyses of DNA isolated from such tumors for the presence of somatic genome alterations are frequently confounded by the presence of normal cell DNA among the tumor DNA in the various samples. In our initial study, we used Southern blot analysis to assess GSTP1 CpG island hypermethylation in DNA from 20 matched normal tissue and PCA specimens. 7 Hypermethylated GSTP1 CpG island sequences were detected as GSTP1 sequences that failed to cut with the 5-mC-sensitive restriction endonuclease BssHII, an enzyme that cuts at the sequence GCGCGC in DNA only when the sequence does not contain 5-mCpG. Using this approach, we found a varied abundance of abnormal hypermethylated GSTP1 promoter alleles amid normal unmethylated GSTP1 promoter alleles in the PCA DNA samples. 7 To determine whether the normal unmethylated GSTP1 promoter sequences in the PCA DNA specimens were present in PCA cells or were present only in normal cells located in the tumor specimens, we compared the abundance of unmethylated and methylated GSTP1 alleles against the abundance of retained and lost polymorphic sequences on chromosomes 8p, 16q, and 17p for each matched normal tissue and PCA DNA specimen (Figure 1) ▶ . In the majority of cases studied (eight of nine), an equivalent level of retained polymorphic DNA sequences at chromosomal loci exhibiting allelic loss and retained unmethylated GSTP1 alleles were present in PCA DNA specimens (Figure 1B) ▶ . These retained normal alleles were likely contributed by normal cells admixed with tumor cells in the PCA specimens. For one case (case no. 96), a significantly greater level of retained unmethylated GSTP1 alleles than retained polymorphic DNA sequences at an allelic loss locus was evident in the PCA DNA specimen (Figure 1B) ▶ . The simplest explanation for the discrepancy in the level of retained normal alleles present in this case was that some or all of the PCA cells contained unmethylated GSTP1 promoter alleles or that some or all of the PCA cells contained less extensively methylated GSTP1 promoter alleles. To evaluate this possibility, strategies for assessing allele-specific GSTP1 hypermethylation and for determining the extent of hypermethylation throughout the GSTP1 CpG island region were used.
Figure 1.
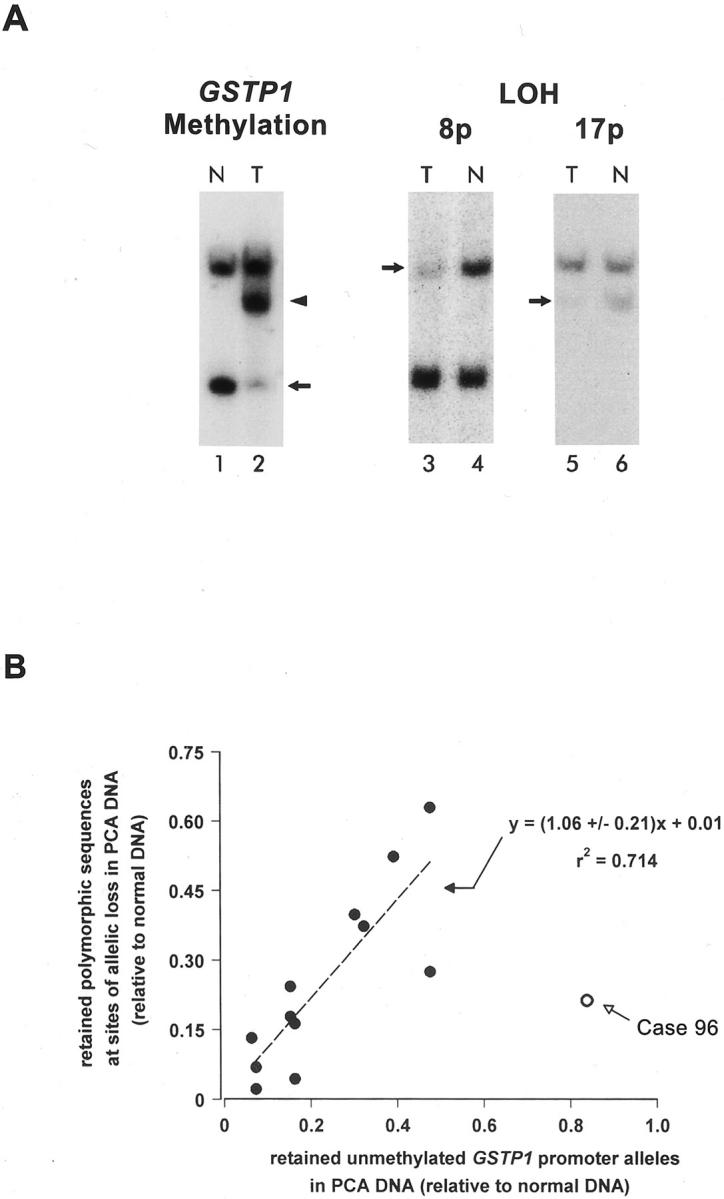
Equivalence of GSTP1 CpG island hypermethylation and chromosome deletions in DNA from prostate cancer (PCA) cases. Southern blot analysis (see Materials and Methods) was used to determine the abundance of normal unmethylated GSTP1 alleles 7 and of retained polymorphic sequences at sites of allelic loss on chromosomes 8p, 16q, and 17p 23 for DNA from PCA (T; lanes 2, 3, and 5) and from matched normal tissues (N; lanes 1, 4, and 6). A: Representative Southern blots for one PCA case are displayed. To discriminate GSTP1 CpG island hypermethylation (lanes 1 and 2), DNAs were digested first with EcoRI and HindIII, and then with BssHII, an enzyme that will not cut its recognition sequence, GCGCGC, if it contains 5-mC. An arrow denotes the position of normal unmethylated GSTP1 alleles; the position of hypermethylated GSTP1 alleles is indicated by an arrowhead. Loss of polymorphic alleles (LOH) at chromosomal loci on 8p (lanes 3 and 4) and 17p (lanes 5 and 6) were discriminated by digestion with relevant restriction endonucleases recognizing sites present on only one of two alleles at each locus. Arrows denote normal retained polymorphic sequences at sites of allelic loss. B: The quantities of retained unmethylated GSTP1 alleles for nine PCA cases were plotted as a function of the quantities of retained polymorphic DNA sequences at chromosomal loci exhibiting allelic loss. PCA DNA from case 96 exhibited a significantly greater level of retained unmethylated GSTP1 alleles than retained polymorphic DNA sequences at an allelic loss locus.
Somatic GSTP1 CpG Island DNA Hypermethylation Changes Affect Both Maternal and Paternal GSTP1 Alleles in Most PCA Cases
GSTP1 CpG island hypermethylation might contribute to the neoplastic transformation of PCA cells or might appear in PCA cells as a consequence of the process of prostatic carcinogenesis. To infer selection of inactivating GSTP1 promoter hypermethylation during the pathogenesis of prostate cancer, GSTP1 DNA hypermethylation must affect both GSTP1 alleles in prostatic cells, or if present at one GSTP1 allele, must be accompanied by other somatic genome lesions affecting the other GSTP1 allele. To determine whether GSTP1 promoter DNA hypermethylation was present at one or both GSTP1 alleles, a PCR strategy was used to distinguish DNA hypermethylation at maternal and paternal GSTP1 alleles (Figure 2) ▶ . After treatment of DNA from matched normal and neoplastic prostate tissues with the restriction endonuclease HpaII, which cuts at the sequence CCGG but not at the sequence C5-mCGG, or with MspI, which cuts both sequences CCGG and C5-meCGG, the digested DNA specimens were subjected to PCR amplification using oligonucleotide primers targeting a polymorphic [ATAAA]n repeat sequence near two HpaII/MspI sites at the GSTP1 regulatory region (Figure 2) ▶ . The amplification of polymorphic GSTP1 promoter sequences after HpaII digestion, but not after MspI digestion, indicated the presence of CpG dinucleotide methylation at the HpaII/MspI sites in the DNA analyzed. Using this approach, GSTP1 CpG island DNA hypermethylation was detected in the majority of PCA DNA specimens (40 of 42 or 95%) and not in normal prostate DNA specimens (Table 1) ▶ . Furthermore, no GSTP1 CpG island DNA hypermethylation was detected in any of the GSTP1 alleles present in either normal or neoplastic tissues from kidney, bladder, ureter, uterus, or uterine cervix (Table 1) ▶ . Of informative PCA cases containing DNA heterozygous for polymorphic GSTP1 [ATAAA]n repeat sequences, 28 of 33 (85%) exhibited DNA hypermethylation affecting both GSTP1 alleles, 1 of 33 (3%) exhibited allelic loss, 2 of 33 (6%) exhibited DNA hypermethylation affecting one of two GSTP1 alleles (cases no. 96 and no. 419, see Figure 3 ▶ ), and 2 of 33 (6%) failed to exhibit DNA hypermethylation at either GSTP1 allele.
Figure 2.

Discrimination of DNA hypermethylation at maternal and paternal GSTP1 alleles using a PCR strategy. DNA from matched normal (normal) and neoplastic (tumor) prostate tissues was left untreated (U; lanes 1, 4, 7, and 10), or was treated with HpaII (H; lanes 2, 5, 8, and 11), which cuts CCGG but not C5-mCGG, or treated with MspI (M; lanes 3, 6, 9, and 12), which cuts CCGG and C5-mCGG, before being subjected to PCR amplification using oligonucleotide primers targeting a polymorphic [ATAAA]n repeat sequence near the GSTP1 regulatory region. For primer set B, the amplification of polymorphic GSTP1 promoter sequences after HpaII digestion, but not after MspI digestion, indicated the presence of CpG dinucleotide methylation at the HpaII/MspI sites in the DNA analyzed.
Table 1.
Detection of GSTP1 CpG Island Hypermethylation in Cancer DNA Using an Assay Capable of Discriminating CpG Hypermethylation Affecting Maternal and Paternal GSTP1 Alleles13
| Cancer organ site* | Number of cancer cases with GSTP1 CpG island hypermethylation† |
|---|---|
| Prostate‡ | 40/42 |
| Noninformative (homozygous for GSTP1 [ATAAA]n repeats) | 11/11 |
| Informative (heterozygous for GSTP1 [ATAAA]n repeats) | 29/31 (27 cases with 2 hypermethylated GSTP1 alleles, 2 cases with 1 hypermethylated GSTP1 allele, and 2 cases with 0 hypermethylated GSTP1 alleles) |
| Kidney | 1/10 |
| Endometrium | 0/10 |
| Uterine cervix | 0/10 |
| Bladder/ureter | 0/5 |
*For each case, DNA was isolated from cancer tissues, and from normal tissues, as described in the Materials and Methods.
†None of the DNA isolated from normal tissues displayed any GSTP1 CpG island DNA hypermethylation.
‡Control normal DNA for prostate cancer cases included DNA from normal prostate tissue adjacent to cancer, DNA from seminal vesicles without cancer involvement, and DNA from white blood cells.
Figure 3.
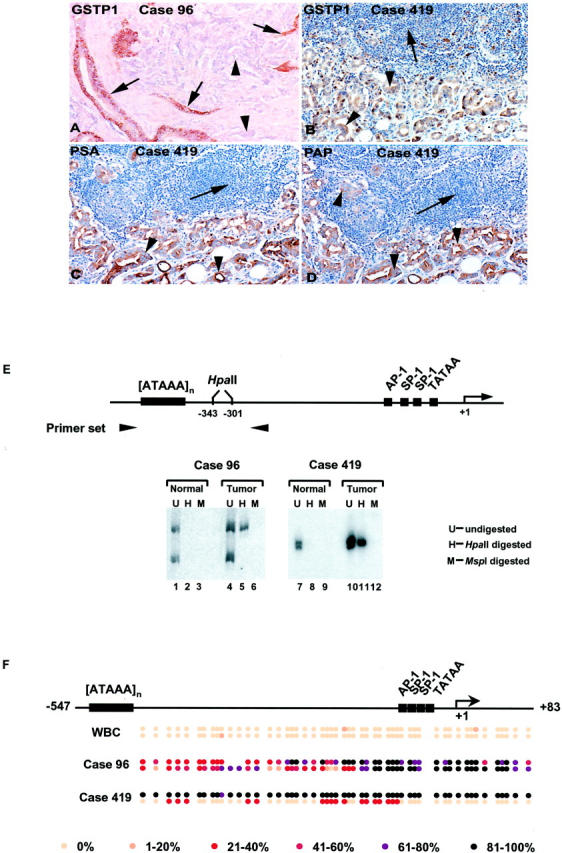
Analysis of GSTP1 expression and of GSTP1 CpG island methylation for prostate cancer (PCA) case 96 and case 419. Both case 96 and case 419 showed GSTP1 hypermethylation affecting only one of two GSTP1 alleles in PCA DNA by the 5-mCpG-sensitive restriction endonuclease/PCR assay (see Figure 2 ▶ and Table 1 ▶ ). Immunohistochemical staining with anti-GSTP1 antibodies revealed an absence of GSTP1 expression in PCA cells (arrowheads) versus normal cells (arrows) in case 96 (A), but an abundance of GSTP1 expression in PCA cells (arrowheads) in case 419 (B). PCA cells in case 419 nonetheless appeared to express prostate-specific antigen (C) and prostate-specific acid phosphatase (D) as evidenced by immunohistochemical staining with appropriate antibodies. E: DNA from case 96 and from case 419 was subjected to analysis using the 5-mC-sensitive restriction endonuclease-PCR assay described for Figure 2 ▶ . DNA from matched normal (normal) and neoplastic (tumor) prostate tissues was left untreated (U; lanes 1, 4, 7, and 10), or was treated with HpaII (H; lanes 2, 5, 8, and 11), which cuts CCGG but not C5-mCGG, or treated with MspI (M; lanes 3, 6, 9, and 12), which cuts CCGG and C5-mCGG, before being subjected to PCR amplification using oligonucleotide primers targeting a polymorphic [ATAAA]n repeat sequence near the GSTP1 regulatory region. DNA from both of the PCA cases was also subjected to bisulfite genomic sequencing analysis (F), using an assay capable of distinguishing CpG dinucleotide methylation patterns at both maternal and paternal GSTP1 alleles (see Materials and Methods). For each case, a minimum of eight PCR clones was sequenced; the fraction of PCR clones with 5-mC at each CpG site is indicated for each polymorphic [ATAAA]n repeat allele using the gray scale provided. For case 96, although the extent of CpG dinucleotide methylation throughout each GSTP1 CpG island allele was different, both GSTP1 alleles displayed CpG dinucleotide hypermethylation, particularly near known cis regulatory elements. For case 419, GSTP1 DNA hypermethylation appeared to be present on only one of two GSTP1 CpG island alleles.
Bisulfite Genomic Sequencing Analyses Reveal that DNA from One PCA Case, Containing PCA Cells that Express High Levels of GSTP1 Polypeptides, Displays CpG Island Hypermethylation Affecting One GSTP1 Allele but Not the Other
For the four cases that did not appear to contain somatic GSTP1 CpG island DNA hypermethylation at both maternal and paternal GSTP1 alleles using the allele-specific GSTP1 PCR DNA methylation assay described, the failure to detect CpG island hypermethylation could have been a result of a true absence of somatic GSTP1 CpG island hypermethylation in PCA cells. Alternatively, GSTP1 CpG island hypermethylation may have been present in PCA cell DNA, but not at the specific CpG dinucleotides sampled in the assay used (an assay false-negative). To resolve this issue, genomic DNA from each of these four cases was subjected to analysis using a bisulfite genomic-sequencing approach capable of ascertaining the extent of CpG island DNA hypermethylation at maternal versus paternal GSTP1 alleles. One of the prostate cancer cases (case no. 96) that showed GSTP1 hypermethylation affecting only one of two GSTP1 alleles in PCA DNA by the 5-mCpG-sensitive restriction endonuclease/PCR assay (Figure 3E ▶ and Table 1 ▶ ) also showed less GSTP1 promoter methylation, relative to loss of polymorphic DNA sequences at an allelic loss locus, by Southern blot analysis (Figure 1B) ▶ . When DNA from this PCA case was subjected to bisulfite genomic-sequencing analysis (Figure 3F) ▶ , GSTP1 hypermethylation was evident at both GSTP1 alleles, although the extent of CpG dinucleotide methylation throughout each GSTP1 CpG island allele was different, with the most dense area of CpG dinucleotide methylation clustered near the known cis promoter regulatory elements. 19,20,33,35-41 Immunohistochemical-staining analysis of PCA tissues from this case revealed an absence of GSTP1 expression in all PCA cells, consistent with inactivation of both GSTP1 alleles (Figure 3A) ▶ . Similarly, DNA from both of the PCA cases that appeared not to contain GSTP1 hypermethylation at either GSTP1 allele when assessed using the allele-specific GSTP1 PCR DNA methylation assay did contain GSTP1 DNA hypermethylation affecting both GSTP1 alleles when assessed using bisulfite genomic sequencing (not shown). Neither of these cases expressed immunoreactive GSTP1 in PCA cells when PCA tissues were stained with anti-GSTP1 antibodies (not shown). The remaining PCA case that showed GSTP1 hypermethylation at only one of two GSTP1 CpG island alleles (case no. 419, see Figure 3E ▶ ) when assessed using the allele-specific GSTP1 PCR DNA methylation assay appeared also to contain GSTP1 DNA hypermethylation at only one of two GSTP1 CpG island alleles when assessed using bisulfite genomic sequencing (Figure 3F) ▶ . Immunohistochemical staining of PCA tissues from this PCA case revealed abundant GSTP1 expression (Figure 3B) ▶ , as well as expression of prostate-specific antigen (Figure 3C) ▶ and prostate-specific acid phosphatase (Figure 3D) ▶ consistent with uninhibited transcription of the unmethylated GSTP1 promoter alleles present in PCA cells in this PCA case. Of interest, the PC-3 and DU145 PCA cell lines also contain both unmethylated and hypermethylated GSTP1 CpG island alleles, and each cell line also exhibits high-level GSTP1 mRNA and GSTP1 polypeptide expression. 7 Also, although GSTP1-expressing PCA cells are extremely rare in PCAs at the time of initial presentation, GSTP1-expressing PCA cells have been detected in locally recurrent or persistent PCAs after radiation therapy in as many as 62% cases, 42 suggesting that reactivation of GSTP1 expression may well occur under certain circumstances in vivo as well as in vitro. For case no. 419, whether the expressed GSTP1 allele carries a somatic mutation that affects GSTP1 function has not been determined.
GSTP1 CpG Island Hypermethylation Prevents GSTP1 Expression in LNCaP PCA Cells
We previously reported that LNCaP PCA cells contain only hypermethylated GSTP1 CpG island alleles and fail to express either GSTP1 mRNA or GSTP1 polypeptides. 7 To determine whether diminished GSTP1 transcription might be responsible for the lack of GSTP1 mRNA expression in LNCaP cells, nuclear run-on transcription analysis was undertaken. Significantly reduced GSTP1 transcription in LNCaP PCA cells was evident in comparison with PC-3 PCA cells (Figure 4A) ▶ , known to contain unmethylated GSTP1 CpG island alleles and to express high levels of GSTP1 mRNA and GSTP1 polypeptides. 7 Treatment with inhibitors of DNA methyltransferases has been reported to result in reversal of GSTP1 CpG island hypermethylation and restoration of GSTP1 expression in MCF-7 breast cancer cells 43 and in Hep3B liver cancer cells. 30 To ascertain whether the GSTP1 CpG island hypermethylation might contribute to the reduced GSTP1 transcription in LNCaP PCA cells, we subjected LNCaP PCA cells propagated in vitro to treatment with the DNA methyltransferase inhibitor 5-aza-C. Exposure of LNCaP PCA cells to 5-aza-C resulted in a reversal of GSTP1 DNA hypermethylation evident by Southern blot analysis (Figure 4B) ▶ and a restoration of GSTP1 mRNA and GSTP1 polypeptide expression seen using Northern blot and immunoblot analyses, respectively (Figure 4, C and D) ▶ . Increased GSTP1 expression by 5-aza-C-treated LNCaP cells did not seem to be merely the result of 5-aza-C induction of GSTP1 transcription. LNCaP cells containing unmethylated GSTP1 promoter alleles after 5-aza-C treatment expressed similar amounts GSTP1 mRNA and GSTP1 polypeptides in the presence or absence of 5-aza-C (Figure 4, C and D) ▶ .
Figure 4.

Contribution of GSTP1 CpG island hypermethylation to lack of GSTP1 expression by LNCaP prostate cancer (PCA) cells. A: Nuclear run-on transcription analyses of GSTP1, hAR, and TOP1, using nuclei from LNCaP PCA cells, which fail to express GSTP1 mRNA, and PC-3 PCA cells, which express high levels GSTP1 mRNA, were undertaken. The amount of 32P-UTP-labeling of GSTP1 and hAR transcripts, relative to 32P-UTP-labeling of TOP1 transcripts, is displayed. B–D: LNCaP PCA cells propagated in vitro were treated with the DNA methyltransferase inhibitor 5-aza-C. By Southern blot analysis (B), 5-aza-C treatment resulted in the appearance of unmethylated GSTP1 CpG island alleles in LNCaP DNA, as evidenced by the appearance of unmethylated BssHII recognition sites in the GSTP1 promoter region. By Northern blot analysis (C) and by immunoblot analyses (D), 5-aza-C treatment triggered a restoration of GSTP1 expression in LNCaP PCA cells, detected whether or not 5-aza-C was present in the growth medium.
To directly determine the effect of CpG island DNA hypermethylation on GSTP1 promoter function, we conducted transient expression assays using hypermethylated and unmethylated GSTP1 promoter/CAT reporter DNA constructs, prepared by ligating SssI CpG-methylase-treated and untreated GSTP1 promoter sequences to unmethylated CAT reporter sequences, transfected into LNCaP cells (Figure 5) ▶ . In initial experiments using unmethylated GSTP1 promoter/CAT reporter constructs transfected into LNCaP cells, transcriptional enhancing sequences were evident at −408 to −291 and at −73 to −65 5′ of the transcription start site (Figure 5A) ▶ . The region −73 to −65 has also been found to augment GSTP1 promoter function in human MCF-7 breast cancer (BCA) cells in previous studies. 33,35,38,39 No evidence for a cis-acting transcriptional silencer, as has been reported at −105 to −86 5′ of the transcription start site for MCF-7 cells, 39 was seen (Figure 5A) ▶ . However, when hypermethylated GSTP1 promoter/CAT reporter constructs were transfected into LNCaP cells, a reduction in CAT reporter activity, in comparison to unmethylated GSTP1 promoter/CAT reporter-transfected LNCaP cells, was found (Figure 5A) ▶ , consistent with an inhibitory effect of GSTP1 CpG island hypermethylation on GSTP1 transcription in PCA cells. Of note, although 5-aza-C treatment of unmethylated SV2 promoter/CAT reporter-transfected LNCaP cells resulted in a substantial induction of CAT reporter expression, 5-aza-C treatment of unmethylated GSTP1 promoter/CAT reporter-transfected LNCaP cells triggered only minimal increases in GSTP1 promoter activity (Figure 5B) ▶ , confirming that 5-aza-C treatment of LNCaP cells was unlikely to have elevated GSTP1 mRNA and GSTP1 polypeptide expression (Figure 4) ▶ via GSTP1 promoter trans-activation.
Figure 5.

Effects of CpG island DNA hypermethylation on GSTP1 promoter function in LNCaP prostate cancer (PCA) cells. A: Unmethylated GSTP1 promoter/CAT reporter constructs were used for GSTP1 promoter mapping, 33 revealing transcriptional enhancing sequences at −408 to −291 and at −73 to −65 5′ of the transcription start site after transfection into LNCaP PCA cells. When methylated GSTP1 promoter sequences (black dots), prepared by treatment with SssI methylase, were ligated to CAT reporter sequences and transfected into LNCaP PCA cells, a reduction in CAT reporter activity, in comparison to unmethylated GSTP1 promoter/CAT reporter-transfected LNCaP cells, was evident. B: The trans-activation effects of 5-aza-C exposure (black bars) on the activity of unmethylated CMV, SV2, and GSTP1 promoters in LNCaP PCA cells were assessed. 5-Aza-C treatment of unmethylated GSTP1 promoter/CAT reporter-transfected LNCaP cells triggered only minimal increases in GSTP1 promoter activity.
Restoration of GSTP1 Expression in LNCaP Cells Fails to Abrogate LNCaP Proliferation in Vitro or Tumorigenicity in Vivo
Somatic GSTP1 inactivation seems to be selected during human prostatic carcinogenesis. Adler and colleagues 44 have reported that π-class GSTs inhibit Jun N-terminal kinase (JNK) activity. If expression of GSTP1 in PCA cells inhibited PCA growth by interfering with growth-promoting signal transduction pathways, loss of GSTP1 function might provide a selective growth advantage for PCA cells. To determine whether restoration of GSTP1 expression affected PCA growth, GSTP1 expression was restored in LNCaP cells, either by 5-aza-C treatment or by transfection with pCMV-GSTP1. When the proliferation of LNCaP cells, LNCaP-5-aza-C cells, LNCaP-neo cells, and three independent LNCaP-GSTP1 subclones, in tissue culture flasks in vitro was assessed, no consistent inhibition of cell growth was evident (Table 2) ▶ . In addition, when each of the cell lines was admixed with Matrigel and injected subcutaneously into immunodeficient mice, no consistent differences in tumorigenicity was seen (Table 2) ▶ .
Table 2.
Forced GSTP1 Expression in LNCaP Cells Fails to Reduce Proliferation in Vitro or Tumorigenicity in Vivo
| Cell line | GSTP1 expression* | Doubling time in vitro (days) | Tumorigenicity in vivo (fraction of mice with tumors at 8 weeks)† |
|---|---|---|---|
| LNCaP | − | 1.11± 0.07 | 9 /15 |
| LNCaP-5-aza-C | + | Not determined | 8 /10 |
| LNCaP-neo | − | 0.09± 0.14 | 15 /15 |
| LNCaP-GSTP1-1 | + | 1.04± 0.04 | 15 /15 |
| LNCaP-GSTP1-3 | + | 0.88± 0.04 | 15 /15 |
| LNCaP-GSTP1-5 | + | 1.06± 0.10 | 10 /15 |
*GSTP1 expression assessed by immunoblot analysis with anti-GSTP1 antibodies.
†Cells (106) admixed with Matrigel were inoculated subcutaneously into athymic mice. At 8 weeks after inoculation, animals were sacrificed and the appearance of tumors >4 mm3 was scored.
Discussion
Hypermethylation CpG island sequences encompassing the transcriptional promoter of GSTP1 has been reported to be the most common somatic genome alteration in human PCA. 7-12 Furthermore, loss of GSTP1 function seems to occur very early in prostatic carcinogenesis, as loss of GSTP1 expression and GSTP1 CpG island DNA hypermethylation have been detected in the majority of prostatic intraepithelial neoplasia lesions. 13 The data presented in this study, which focused on localized PCA removed at prostatectomy, revealed that somatic GSTP1 defects, whether CpG island hypermethylation or gene deletions, were present in all of the PCA cases studied. For the PCA cases in which PCA cells failed to express GSTP1 in vivo, defective GSTP1 alleles, and only defective GSTP1 alleles, were present in all of the cancer cells. For LNCaP PCA cells propagated in vitro, which contained only defective GSTP1 alleles and also failed to express GSTP1, reversal of abnormal GSTP1 CpG island DNA hypermethylation resulted in restoration of GSTP1 expression. The GSTP1 CpG island DNA hypermethylation also likely prevented GSTP1 expression by PCA cells in vivo. In the single case studied in which PCA cells expressed abundant GSTP1 polypeptides, although one of the GSTP1 alleles carried CpG island DNA hypermethylation, the other allele was free of any somatic GSTP1 defects. To be subject to selection in cancer cells, somatic genome alterations, including CpG island DNA hypermethylation, must be maintained through cell division and must affect gene and/or gene product function. CpG dinucleotide methylation patterns can be maintained through mitosis by the action of DNA methyltransferases at the site of DNA replication. 45-47 Taken together, all of the data collected for this manuscript strongly suggest that selection for GSTP1 inactivation during the pathogenesis of human PCA can be inferred for most PCA cases.
The mechanisms by which critical genes, such as GSTP1, acquire somatic CpG island DNA hypermethylation during cancer pathogenesis have not been established. Nonetheless, abnormal actions of DNA methyltransferases likely play some sort of role. Forced expression of DNA methyltransferases in immortalized mammalian cells has been shown to result both in de novo hypermethylation and in transformation in vitro. 48-50 Transformation by c-fos seems to require DNA methyltransferase expression. 51 Mice carrying defective Apc alleles and disrupted Dnmt1 alleles exhibit fewer intestinal polyps. 52 Often, silenced genes manifest a repressed chromatin conformation along with carrying increased CpG island hypermethylation. In fact, recent data have suggested that DNA methyltransferases and 5-mC-binding proteins may interact directly with chromatin remodeling enzymes, such as histone deacetylases, to repress gene expression. 53-62 In contrast, transcriptionally active genes seem relatively resistant to de novo CpG island DNA methylation. 63,64 Whether a possible coordination of DNA methyltransferase activity and transcriptional inactivity may lead to specific gene silencing during the development of human cancers has not been determined. Nonetheless, an inducible gene such as GSTP1 might be especially vulnerable to inactivation, while in a nonexpressed state, via this type of mechanism. Genes encoding GSTs are characteristically expressed at very low levels in many tissues until induced, via an increase in transcriptional promoter activity, on exposure to oxidants and electrophiles. 65-67 Perhaps, in the absence of inducer exposure, low level GSTP1 transcription might render the GSTP1 CpG island vulnerable to de novo DNA hypermethylation.
How might the phenotype of lack of GSTP1 expression be subject to selection during prostatic carcinogenesis? In one selection model, GSTP1 might act like a tumor suppressor gene, which when inactivated leads to tumor growth. Favoring this type of model, Adler and colleagues 44 have reported that π-class GSTs can interfere with N-terminal c-Jun kinase signaling. Against this model, our studies of LNCaP PCA cell growth and tumorigenicity discerned no role for GSTP1 expression in abrogation of LNCaP PCA cell proliferation in vitro or in vivo. In another selection model, GSTP1 might act like a caretaker gene, which when inactivated leads to additional somatic genome alterations that promote tumor growth. 4 GSTP1, like other GSTs, can catalyze the detoxification of oxidants and electrophiles that threaten genome damage. 66 As an example, mice carrying disrupted Gstp alleles display enhanced skin tumorigenesis on exposure to 7,12-dimethylbenz anthracene. 68 In addition, recent data indicate that GSTP1 may provide prostate cells protection against DNA adduct formation associated with ingestion of dietary heterocyclic aromatic amine carcinogens, such as 2-amino-1-methyl-6-phenylimidazo[4,5-β]pyridine (PhIP), present in many foods in the stereotypical North American diet, particularly well-done or charred meats. 69 However, in these studies, when LNCaP cells were genetically modified to express GSTP1, the resultant cells appeared protected not only against DNA adduct formation on exposure to N-OH-PhIP, an activated PhIP metabolite, but also against N-OH-PhIP cytotoxicity. 69 Loss of GSTP1 function thus rendered LNCaP cells vulnerable to both genome damaging and cell killing effects of N-OH-PhIP. For lack of GSTP1 expression to be selected in the face of PhIP exposure, PhIP-mediated genome damage must target another gene involved in prostate cell growth regulation. In this way, loss of GSTP1 caretaker function might indirectly lead to selection during prostatic carcinogenesis. The data presented in this article permit only the inference that selection for GSTP1 inactivation during the pathogenesis of human PCA has likely occurred. To prove selection, model studies demonstrating a selective growth or survival advantage for loss of GSTP1 function in prostate cells will be required.
In our study, using a combination of assays, GSTP1 CpG island hypermethylation was detected in DNA from every prostate cancer case surveyed. As such, sensitive and specific detection of GSTP1 CpG island hypermethylation might offer an opportunity for molecular detection, diagnosis, and staging of human PCA. Thus far, two basic PCR strategies have emerged. The first features the use of 5-mCpG-sensitive restriction endonucleases before PCR amplification of GSTP1 CpG island sequences. One version of this PCR strategy seems capable of detecting PCA DNA in 91% of PCA cases at a limiting sensitivity of 2 pg. This assay has been reported to detect as little as 2 ng PCA DNA when the PCA DNA is admixed with 1 μg of white blood cell DNA. 9 The second PCR strategy for detecting hypermethylated GSTP1 CpG island sequences involves the use of the bisulfite reaction followed by PCR, which results in the conversion of C, but not of 5-mC, to T. Primers specific for converted target sequences derived from 5-mCpG-containing versus CpG-containing GSTP1 alleles are then used to selectively amplify products from hypermethylated versus unmethylated GSTP1 CpG islands (methylation-specific PCR or MSP). 30,70,71 In a recent report, a version of this PCR strategy, able to discriminate as few as 200 LNCaP PCA cells, detected PCA DNA in 94% of PCA tissues, 72% of plasma or serum specimens, 50% of ejaculates, and 36% of urine specimens from men with known PCA. 8 As more data become available regarding consensus GSTP1 CpG island DNA methylation patterns characteristic of PCA, both of these PCR strategies can be refined to discriminate a greater fraction of PCA cases, perhaps permitting GSTP1 CpG island DNA hypermethylation to serve as a potentially useful molecular biomarker for PCA detection, diagnosis, and staging.
Acknowledgments
We thank Kathleen R. Cho and Lora H. Ellenson for providing genomic DNA from endometrial and uterine cervix cancer specimens.
Footnotes
Address reprint requests to William G. Nelson, M.D., Ph.D., Bunting-Blaustein Cancer Research Building, Room 151, 1650 Orleans St., Baltimore, MD 21231-1000. E-mail: bnelson@jhmi.edu.
Supported by National Institutes of Health/National Cancer Institute grants CA58236 and CA70196.
Wen-Hsiang Lee, William B. Isaacs, and William G. Nelson have a patent (U.S. patent 5552,277) entitled “Genetic Diagnosis of Prostate Cancer.”
References
- 1.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C: Genetic instability and Darwinian selection in tumours. Trends Cell Biol 1999, 9:M57-M60 [PubMed] [Google Scholar]
- 2.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B: Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386:761-763 [DOI] [PubMed] [Google Scholar]
- 5.Baylin SB, Herman JG: DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000, 16:168-174 [DOI] [PubMed] [Google Scholar]
- 6.Robertson KD, Jones PA: DNA methylation: past, present and future directions. Carcinogenesis 2000, 21:461-467 [DOI] [PubMed] [Google Scholar]
- 7.Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG: Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA 1994, 91:11733-11737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goessl C, Krause H, Muller M, Heicappell R, Schrader M, Sachsinger J, Miller K: Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res 2000, 60:5941-5945 [PubMed] [Google Scholar]
- 9.Lee WH, Isaacs WB, Bova GS, Nelson WG: CG island methylation changes near the GSTP1 gene in prostatic carcinoma cells detected using the polymerase chain reaction: a new prostate cancer biomarker. Cancer Epidemiol Biomarkers Prev 1997, 6:443-450 [PubMed] [Google Scholar]
- 10.Millar DS, Ow KK, Paul CL, Russell PJ, Molloy PL, Clark SJ: Detailed methylation analysis of the glutathione S-transferase pi (GSTP1) gene in prostate cancer. Oncogene 1999, 18:1313-1324 [DOI] [PubMed] [Google Scholar]
- 11.Santourlidis S, Florl A, Ackermann R, Wirtz HC, Schulz WA: High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 1999, 39:166-174 [DOI] [PubMed] [Google Scholar]
- 12.Suh CI, Shanafelt T, May DJ, Shroyer KR, Bobak JB, Crawford ED, Miller GJ, Markham N, Glode LM: Comparison of telomerase activity and GSTP1 promoter methylation in ejaculate as potential screening tests for prostate cancer. Mol Cell Probes 2000, 14:211-217 [DOI] [PubMed] [Google Scholar]
- 13.Brooks JD, Weinstein M, Lin X, Sun Y, Pin SS, Bova GS, Epstein JI, Isaacs WB, Nelson WG: CG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev 1998, 7:531-536 [PubMed] [Google Scholar]
- 14.Cookson MS, Reuter VE, Linkov I, Fair WR: Glutathione S-transferase PI (GST-pi) class expression by immunohistochemistry in benign and malignant prostate tissue. J Urol 1997, 157:673-676 [PubMed] [Google Scholar]
- 15.Montironi R, Mazzucchelli R, Stramazzotti D, Pomante R, Thompson D, Bartels PH: Expression of pi-class glutathione S-transferase: two populations of high grade prostatic intraepithelial neoplasia with different relations to carcinoma. Mol Pathol 2000, 53:122-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montironi R, Mazzucchelli R, Pomante R, Thompson D, Duval da Silva V, Vaught L, Bartels PH: Immunohistochemical expression of pi class glutathione S-transferase in the basal cell layer of benign prostate tissue following chronic treatment with finasteride. J Clin Pathol 1999, 52:350-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray GI, Taylor VE, McKay JA, Weaver RJ, Ewen SW, Melvin WT, Burke MD: The immunohistochemical localization of drug-metabolizing enzymes in prostate cancer. J Pathol 1995, 177:147-152 [DOI] [PubMed] [Google Scholar]
- 18.Moskaluk CA, Duray PH, Cowan KH, Linehan M, Merino MJ: Immunohistochemical expression of pi-class glutathione S-transferase is down-regulated in adenocarcinoma of the prostate. Cancer 1997, 79:1595-1599 [DOI] [PubMed] [Google Scholar]
- 19.Cowell IG, Dixon KH, Pemble SE, Ketterer B, Taylor JB: The structure of the human glutathione S-transferase pi gene. Biochem J 1988, 255:79-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrow CS, Cowan KH, Goldsmith ME: Structure of the human genomic glutathione S-transferase-pi gene. Gene 1989, 75:3-11 [DOI] [PubMed] [Google Scholar]
- 21.Moscow JA, Fairchild CR, Madden MJ, Ransom DT, Wieand HS, O’Brien EE, Poplack DG, Cossman J, Myers CE, Cowan KH: Expression of anionic glutathione-S-transferase and P-glycoprotein genes in human tissues and tumors. Cancer Res 1989, 49:1422-1428 [PubMed] [Google Scholar]
- 22.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP: LNCaP model of human prostatic carcinoma. Cancer Res 1983, 43:1809-1818 [PubMed] [Google Scholar]
- 23.Carter BS, Ewing CM, Ward WS, Treiger BF, Aalders TW, Schalken JA, Epstein JI, Isaacs WB: Allelic loss of chromosomes 16q and 10q in human prostate cancer. Proc Natl Acad Sci USA 1990, 87:8751-8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kessis TD, Slebos RJ, Han SM, Shah K, Bosch XF, Munoz N, Hedrick L, Cho KR: p53 gene mutations and MDM2 amplification are uncommon in primary carcinomas of the uterine cervix. Am J Pathol 1993, 143:1398-1405 [PMC free article] [PubMed] [Google Scholar]
- 25.Ronnett BM, Burks RT, Cho KR, Hedrick L: DCC genetic alterations and expression in endometrial carcinoma. Mod Pathol 1997, 10:38-46 [PubMed] [Google Scholar]
- 26.Brooks JD, Bova GS, Marshall FF, Isaacs WB: Tumor suppressor gene allelic loss in human renal cancers. J Urol 1993, 150:1278-1283 [DOI] [PubMed] [Google Scholar]
- 27.Burton K: Determination of DNA concentration with diphenylamine. Methods Enzymol 1968, 12B:163-166 [Google Scholar]
- 28.De Marzo AM, Marchi VL, Epstein JI, Nelson WG: Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol 1999, 155:1985-1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark SJ, Harrison J, Paul CL, Frommer M: High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994, 22:2990-2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tchou JC, Lin X, Freije D, Isaacs WB, Brooks JD, Rashid A, De Marzo AM, Kanai Y, Hirohashi S, Nelson WG: GSTP1 CpG island DNA hypermethylation in hepatocellular carcinomas. Int J Oncol 2000, 16:663-676 [DOI] [PubMed] [Google Scholar]
- 31.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW: Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 1979, 17:16-23 [PubMed] [Google Scholar]
- 32.Celano P, Berchtold CM, Giardiello FM, Casero RA, Jr: Modulation of growth gene expression by selective alteration of polyamines in human colon carcinoma cells. Biochem Biophys Res Commun 1989, 165:384-390 [DOI] [PubMed] [Google Scholar]
- 33.Moffat GJ, McLaren AW, Wolf CR: Involvement of Jun and Fos proteins in regulating transcriptional activation of the human pi class glutathione S-transferase gene in multidrug-resistant MCF7 breast cancer cells. J Biol Chem 1994, 269:16397-16402 [PubMed] [Google Scholar]
- 34.Passaniti A, Isaacs JT, Haney JA, Adler SW, Cudjik TJ, Long PV, Kleinman HK: Stimulation of human prostatic carcinoma tumor growth in athymic mice and control of migration in culture by extracellular matrix. Int J Cancer 1992, 51:318-324 [DOI] [PubMed] [Google Scholar]
- 35.Jhaveri MS, Morrow CS: Contribution of proximal promoter elements to the regulation of basal and differential glutathione S-transferase P1 gene expression in human breast cancer cells. Biochim Biophys Acta 1998, 1396:179-190 [DOI] [PubMed] [Google Scholar]
- 36.Morrow CS, Goldsmith ME, Cowan KH: Regulation of human glutathione S-transferase pi gene transcription: influence of 5′-flanking sequences and trans-activating factors which recognize AP-1-binding sites. Gene 1990, 88:215-225 [DOI] [PubMed] [Google Scholar]
- 37.Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, Wolf CR, Yamamoto M: The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans 2000, 28:33-41 [DOI] [PubMed] [Google Scholar]
- 38.Moffat GJ, McLaren AW, Wolf CR: Sp1-mediated transcriptional activation of the human Pi class glutathione S-transferase promoter. J Biol Chem 1996, 271:1054-1060 [DOI] [PubMed] [Google Scholar]
- 39.Moffat GJ, McLaren AW, Wolf CR: Functional characterization of the transcription silencer element located within the human Pi class glutathione S-transferase promoter. J Biol Chem 1996, 271:20740-20747 [DOI] [PubMed] [Google Scholar]
- 40.Xia C, Hu J, Ketterer B, Taylor JB: The organization of the human GSTP1–1 gene promoter and its response to retinoic acid and cellular redox status. Biochem J 1996, 313:155-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia CL, Cowell IG, Dixon KH, Pemble SE, Ketterer B, Taylor JB: Glutathione transferase pi its minimal promoter and downstream cis-acting element. Biochem Biophys Res Commun 1991, 176:233-240 [DOI] [PubMed] [Google Scholar]
- 42.Cheng L, Sebo TJ, Cheville JC, Pisansky TM, Slezak J, Bergstralh EJ, Pacelli A, Neumann RM, Zincke H, Bostwick DG: p53 protein overexpression is associated with increased cell proliferation in patients with locally recurrent prostate carcinoma after radiation therapy. Cancer 1999, 85:1293-1299 [PubMed] [Google Scholar]
- 43.Jhaveri MS, Morrow CS: Methylation-mediated regulation of the glutathione S-transferase P1 gene in human breast cancer cells. Gene 1998, 210:1-7 [DOI] [PubMed] [Google Scholar]
- 44.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z: Regulation of JNK signaling by GSTp. EMBO J 1999, 18:1321-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Oakeley EJ, Sun L, Jost JP: Multiple domains are involved in the targeting of the mouse DNA methyltransferase to the DNA replication foci. Nucleic Acids Res 1998, 26:1038-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araujo FD, Knox JD, Szyf M, Price GB, Zannis-Hadjopoulos M: Concurrent replication and methylation at mammalian origins of replication. Mol Cell Biol 1998, 18:3475-3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leonhardt H, Page AW, Weier HU, Bestor TH: A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71:865-873 [DOI] [PubMed] [Google Scholar]
- 48.Wu J, Issa JP, Herman J, Bassett DE, Jr, Nelkin BD, Baylin SB: Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc Natl Acad Sci USA 1993, 90:8891-8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J, Herman JG, Wilson G, Lee RY, Yen RW, Mabry M, de Bustros A, Nelkin BD, Baylin SB: Expression of prokaryotic HhaI DNA methyltransferase is transforming and lethal to NIH 3T3 cells. Cancer Res 1996, 56:616-622 [PubMed] [Google Scholar]
- 50.Vertino PM, Yen RW, Gao J, Baylin SB: De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol Cell Biol 1996, 16:4555-4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakin AV, Curran T: Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science 1999, 283:387-390 [DOI] [PubMed] [Google Scholar]
- 52.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, Weinberg RA, Jaenisch R: Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995, 81:197-205 [DOI] [PubMed] [Google Scholar]
- 53.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP: DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 2000, 25:338-342 [DOI] [PubMed] [Google Scholar]
- 54.Rountree MR, Bachman KE, Baylin SB: DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet 2000, 25:269-277 [DOI] [PubMed] [Google Scholar]
- 55.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T: DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 2000, 24:88-91 [DOI] [PubMed] [Google Scholar]
- 56.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB: Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999, 21:103-107 [DOI] [PubMed] [Google Scholar]
- 57.Ng HH, Jeppesen P, Bird A: Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol 2000, 20:1394-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D, Bird A: MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet 1999, 23:58-61 [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D: Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 1999, 13:1924-1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A: Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393:386-389 [DOI] [PubMed] [Google Scholar]
- 61.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP: Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 1999, 23:62-66 [DOI] [PubMed] [Google Scholar]
- 62.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP: Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998, 19:187-191 [DOI] [PubMed] [Google Scholar]
- 63.Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, Temper V, Razin A, Cedar H: Sp1 elements protect a CpG island from de novo methylation. Nature 1994, 371:435-438 [DOI] [PubMed] [Google Scholar]
- 64.Macleod D, Charlton J, Mullins J, Bird AP: Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev 1994, 8:2282-2292 [DOI] [PubMed] [Google Scholar]
- 65.Henderson CJ, McLaren AW, Moffat GJ, Bacon EJ, Wolf CR: Pi-class glutathione S-transferase: regulation and function. Chem Biol Interact 1998, 111–112:69-82 [DOI] [PubMed] [Google Scholar]
- 66.Hayes JD, Pulford DJ: The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol 1995, 30:445-600 [DOI] [PubMed] [Google Scholar]
- 67.Daniel V: Glutathione S-transferases: gene structure and regulation of expression. Crit Rev Biochem Mol Biol 1993, 28:173-207 [DOI] [PubMed] [Google Scholar]
- 68.Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, Wolf CR: Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci USA 1998, 95:5275-5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nelson CP, Kidd LCR, Sauvegeot J, Isaacs WB, De Marzo AM, Groopman JD, Nelson WG, Kensler TW: Protection against 2-hydroxyamino-1-methyl-6-phenylaimidazo[4,5-b]pyridine cytotoxicity and DNA adduct formation in human prostate by glutathione S-transferase P1. Cancer Res 2001, 61:103-109 [PubMed] [Google Scholar]
- 70.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG: Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 1998, 58:4515-4518 [PubMed] [Google Scholar]