

Abstract
We have previously demonstrated that the proximal tubular cell may contribute to the pathogenesis of renal interstitial fibrosis in diabetes. Transforming growth factor (TGF)-β1 is one of a group of pro-fibrotic cytokines and growth factors, which have been associated with the development of interstitial fibrosis. The aim of the current study was to examine the effect of insulin on the generation of TGF-β1 by proximal tubular cells. HK-2 cells were grown to confluence in the absence of insulin, and serum deprived for 48 hours before all experimental manipulations. Addition of insulin (5 μg/ml) to the culture medium led to a time-dependent increase in TGF-β1 concentration in the cell culture supernatant, and increased incorporation of radiolabeled amino acids into TGF-β1 suggestive of de novo TGF-β1 protein synthesis. Addition of insulin did not alter TGF-β1 mRNA expression as assessed by reverse transcriptase-polymerase chain reaction or Northern analysis. Insulin-induced increase in TGF-β1 concentration was not abrogated by actinomycin D, however, stimulation by insulin, in the presence of cycloheximide led to a dose-dependent decrease in TGF-β1 production. Addition of insulin had no effect on TGF-β1 mRNA stability as assessed by actinomycin D chase, but led to increased binding of a cytoplasmic protein to a putative stem loop structure in the 5′-UTR of TGF-β1 mRNA, previously implicated in the posttranscriptional control of TGF-β1 synthesis. To address the functional significance of insulin-induced alteration in TGF-β1 synthesis, we examined its effect on matrix turnover. Insulin stimulated type IV collagen gene expression and an increase in the concentrations of the type IV collagen laid down in the extracellular matrix. This increase in type IV collagen was abrogated when cells were stimulated by insulin in the presence of an anti-TGF-β1-blocking antibody. In conclusion the data demonstrate that insulin may directly alter the production of TGF-β1 by renal proximal tubular cells by a posttranscriptional mechanism, and that this may have implications for the increase in extracellular matrix that accompanies diabetic nephropathy.
Diabetic nephropathy has now become the largest single disease group among patients with chronic renal failure, and of patients with diabetes up to 40% will develop renal disease.1 Most studies to date have focused on the glomerular abnormalities found in diabetic nephropathy. The importance of pathological changes in the renal cortical interstitium is however now well recognized, suggesting that interstitial fibrosis represents a crucial development leading to progressive renal dysfunction.2,3 With increasing awareness of the importance of these pathological interstitial changes, interest has focused on the role of cells, such as the epithelial cells of the proximal tubule (PTCs) or the interstitial fibroblast, in the initiation of a fibrotic response. Because progressive decline in renal function in diabetes is closely correlated with the degree of renal interstitial fibrosis we have focused on the role of the PTCs in initiating these pathological changes.
Transforming growth factor-β1 (TGF-β1) has emerged as a mediator that is implicated in the pathogenesis of fibrosis in both glomerular and interstitial compartments of the kidney and has been particularly associated with diabetic nephropathy.4 It is now clear that PTCs have the potential to contribute to the pathogenesis of renal fibrosis by the production of profibrotic growth factors, including TGF-β1.5-7 We have previously demonstrated that TGF-β1 production in PTCs may be controlled at the levels of transcription, translation, and secretion of preformed protein as well as at the level of activation of the latent protein.5-8 In the context of diabetic nephropathy we have demonstrated that the synergistic action of elevated concentrations of glucose and cytokines such as platelet-derived growth factor or interleukin-1β stimulate PTC TGF-β1 synthesis.5,6 This induction of TGF-β1 synthesis resulted from translation of glucose-induced TGF-β1 mRNA by either cytokine, and was associated with an alteration in its inherent stability.
Despite the large amount of research into the etiology of renal disease in diabetes mellitus, little is known regarding the relationship between insulin and levels of glycemia in the context of tissue damage. Insulin plays an important role in the overall regulation of protein synthesis. Not only does it cause a global increase in the rate of translation, but it may also bring about marked increases in the translation of specific mRNAs. The aim of this study was therefore to examine the effect of insulin on the synthesis of TGF-β1 by PTCs. In this manuscript we present evidence that insulin specifically stimulates translation of TGF-β1 mRNA, via an interaction with the insulin-like growth factor-1 (IGF-1) receptor. In addition we examine the possible mechanisms involved in mediating these effects of insulin.
Materials and Methods
Cell Culture
All experiments were performed using HK-2 cells, which are human renal proximal tubular epithelial cells immortalized by transduction with human papilloma virus 16 E6/E7 genes.9 Cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F12 (Life Technologies, Paisley, UK) supplemented with 10% fetal calf serum (Biological Industries Ltd., Cumbernauld, UK), glutamine (Life Technologies Ltd., Paisley, UK), transferrin, and sodium selenite (Sigma, Poole, UK). Fresh growth medium was added to cells every 3 to 4 days until confluent. Cells were grown to confluence and growth arrested in serum-free medium for 48 hours. All experiments were subsequently performed under serum-free conditions.
TGF-β1 Protein Quantification
Growth-arrested HK-2 cells were stimulated with insulin (5 μg/ml, Sigma), and supernatant samples collected throughout the subsequent 48 hours. Samples were stored at −70°C before quantification of both TGF-β1 and TGF-β2. In all experiments TGF-β1 protein concentration in the cell culture supernatant was determined by specific enzyme-linked immunosorbent assay (ELISA) (Amersham Pharmacia Biotech Ltd., Little Chalfont, UK). The assay is sensitive to 4 pg/ml, with no cross-reactivity with either TGF-β2 or TGF-β3 isoforms. TGF-β1 was only detected after acidification of the samples indicating that TGF-β1 was produced in its latent form. In the same samples TGF-β2 also quantified by specific ELISA (Quantikine human TGF-β1 ELISA; R&D Systems, Abingdon, UK). The assay is sensitive to 7 pg/ml, with no cross-reactivity with other TGF-β isoforms. TGF-β2 was only detected after acidification of the samples indicating that TGF-β2 was produced in its latent form. In all experiments after removal of the supernatant, cell protein content was determined using a modified Bradford assay,10 and TGF-β (1 + 2) concentrations were corrected for total cell protein concentration.
To further examine TGF-β1 synthesis, we examined radioactive amino acid incorporation into TGF-β1 in the presence of insulin. Four μCi of 3H-radiolabeled amino acid mixture (20 μCi/ml; Amersham) was added to growth-arrested confluent cells in the presence of insulin (5 μg/ml) for a total of 48 hours. Supernatant samples were subsequently collected for TGF-β1 immunoprecipitation. Before immunoprecipitation supernatant samples (400 μl) were precleared with 25 μl of agarose-protein A beads (Sigma) at 4°C for 1 hour with constant mixing. The beads were removed by centrifugation and the supernatant decanted. Forty μg of polyclonal rabbit anti-human TGF-β1 antibody (Santa Cruz, Autogen Bioclear UK Ltd., Calne, UK) was added to each 400 μl of cleared supernatant and incubated at 4°C with constant mixing for 12 hours. Subsequently 25 μl of agarose beads were added to capture the immune complexes. Separation of the beads was achieved by centrifugation (13,000 × g for 5 minutes) and the supernatant discarded. Twenty-five μl of sodium dodecyl sulfate (SDS) sample buffer was added to each preparation and boiled for 3 minutes at 95°C, which was subsequently run on a 10% SDS-polyacrylamide gel electrophoresis (PAGE). Gels were fixed by incubating in acetic acid/methanol/H20 (1:4:5 by volume) overnight. Finally the gels were soaked in Amplify scintillant (Amersham, Life Science) for 30 minutes and the gels dried before visualization of immunoprecipitated TGF-β1 by autoradiography. Efficiency of immunoprecipitation was confirmed by a second immunoprecipitation of the original supernatant with the primary antibody.
TGF-β1 Gene Expression: Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) and Northern Analysis
After stimulation of confluent growth-arrested HK-2 cells with insulin (5 μg/ml) for up to 48 hours, total cellular RNA was isolated and TGF-β1 mRNA examined by RT, and PCR amplification was performed using specific oligonucleotide primers (Table 1)▶ as previously described.5 PCR was performed for various cycles (28 to 40), to ensure that amplification was in the linear range of the curve. One-tenth of the PCR reaction from both test and control (α-actin) product were mixed and separated by flat bed electrophoresis in 3% w/v NuSieve GTG agarose gels (Flowgen Instruments Ltd., Sittingbourne, UK), stained with ethidium bromide (Sigma) and photographed. The negatives were scanned using a densitometer (model 620 video densitometer; Bio-Rad Laboratories Ltd., Menelhempstead, UK) and the density of the bands compared to those of the housekeeping gene α-actin.
Table 1.
The Sequences of the PCR Amplification Primers
TGF-β1 mRNA expression was also examined by Northern analysis. Fifteen μg of total cellular RNA was fractionated on a formaldehyde-1% agarose gel. RNA was transferred overnight to a positively charged nylon membrane (Roche Diagnostics Ltd, Leises, UK) by capillary action and fixed by UV irradiation. Membranes were hybridized with an internally labeled TGF-β1 DNA probe, that was 32P-radiolabeled by DNA polymerase random priming. Blots were exposed to X-ray film overnight at room temperature. Membranes were stripped by incubating for 2 × 30 minutes in boiling 0.1% (w/v) SDS in RNase-free water, rehybridized with fresh 32P-labeled β-actin oligonucleotide probe, and developed as described above.
Transcription/Translation/mRNA Stability
To further investigate the mechanism of insulin-induced TGF-β1 synthesis, HK-2 cells were stimulated with insulin in the presence of increasing doses of either actinomycin D (0 to 0.5 μg/ml) to inhibit de novo gene transcription or cycloheximide (0 to 5 μg/ml) to inhibit mRNA translation, respectively. Toxicity of actinomycin D and cycloheximide was assessed using cellular ATP measurements. Only doses that did not lead to a reduction in cellular ATP were used. We have previously demonstrated inhibition of gene expression by actinomycin and inhibition of translation by cycloheximide over these dose ranges.5,6 Actinomycin D or cycloheximide was added to growth-arrested HK-2 cells for 1 hour before stimulation with insulin, and supernatant samples were collected throughout the subsequent 48 hours for quantitation of TGF-β1 by ELISA.
The rate of degradation of TGF-β1 mRNA after the addition of actinomycin D was used to assess the effect of insulin on inherent stability of constitutively expressed TGF-β1 mRNA in HK-2 cells.11 Toxicity of actinomycin D was assessed using cellular ATP measurements, and the maximal nontoxic dose was used (0.5 μg/ml).5 Growth-arrested HK-2 cells were incubated with actinomycin D (0.5 μg/ml) for 1 hour before the addition of insulin. In control experiments 5 mmol/L of d-glucose in the absence of insulin was added to actinomycin D-treated cells. The rate of mRNA degradation was determined by isolating total RNA from these cells at various times up to 24 hours after the addition of actinomycin D and performing RT-PCR.
Gel Retardation Analysis
Confluent HK-2 cell monolayers were stimulated with insulin (5 μg/ml) for 48 hours and cytoplasmic proteins were prepared as previously described.12 Briefly after detachment and washing of the cell pellet with phosphate-buffered saline at 4°C, cells were treated with a nondisruptive buffer (10 mmol/L KCl, 10 mmol/L Hepes, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L NaF, 0.5 mmol/L dithiothreitol, 100 μmol/L sodium orthovanadate, 25 mmol/L β-glycerophosphate, 0.2% (v/v) Nonidet P-40, and a cocktail of protease inhibitors) and incubated on ice for a further 10 minutes. Subsequently to remove debris and nuclei, the lysate was centrifuged for 10 minutes at 10,000 × g at 4°C. The supernatant (cytoplasmic extract) was collected and its protein content determined using a modified Bradford assay.10 Aliquots were frozen at −70°C.
A fragment of the TGF-β1 5′-UTR corresponding to bases +1 to +147, was inserted into the HindIII and XbaI sites of the pGEM4 (Promega, Southhampton, UK) and radiolabeled probes for gel retardation analysis generated as previously described.12 The probes (1.5 × cpm) were incubated for 30 minutes at room temperature with the cytoplasmic extract (20 μg) in a total volume of 15 μl containing 10 mmol/L Tris-HCl, pH 7.5, 100 mmol/L NaCl, 5 mmol/L dithiothreitol, 4% glycerol, 1 mmol/L ethylenediaminetetraacetic acid, and 0.1 mg/ml nuclease-free bovine serum albumin. To ensure that proteins did not bind nonspecifically to the target RNA, 1.5 μl of the synthetic polymer poly (dl-dC)-poly (dl-dC) (Pharmacia Biotech) was added to the final mixture. Complexes were resolved from the free probe by electrophoresis through 6% nondenaturing polyacrylamide gel and visualized by autoradiography.
Role of Insulin versus IGF Receptors
To determine whether the effects of insulin were mediated via binding to its own receptor or via the IGF receptor, three approaches were used. 1) Cells were stimulated with insulin over a range of insulin concentrations that would lead to interaction primarily with the insulin receptor (0 to 500 ng/ml). 2) At the higher dose (5 μg/ml), which led to alterations in TGF-β1 generation, the role of the IGF-1 receptor in mediating these effects was determined by stimulating cells with insulin in the presence of a monoclonal anti-human IGF-1R-blocking antibody (R&D Systems). 3) Finally, cells were stimulated with recombinant IGF-1 (R&D Systems), to determine whether the effect of insulin could be mimicked by IGF-1 over a dose range that would lead to it interacting primarily with the IGF-1 receptor. In all experiments supernatant samples were collected at time points up to 48 hours, and stored at −70°C before quantification of TGF-β1.
Signaling Pathways
Involvement of the MAP/ERK kinase, PI 3 kinase, and p70 S6 kinase in insulin-induced TGF-β1 stimulation, was examined by stimulation of growth-arrested cells with insulin (5 μg/ml) in the presence of either PD 98059 (2 μmol/L), wortmannin (10 nmol/L), or rapamycin (30 nmol/L), which are specific inhibitors of each of these kinases. That the inhibitors did not influence cell viability was confirmed, as cellular ATP was not affected by the addition of each of the inhibitors. All inhibitors were obtained from Calbiochem (Nottingham, UK) and the dose was derived from their known IC50, and their previously published effects in proximal tubular cells.13 In all experiments the inhibitor was added for 20 minutes before the addition of insulin in the presence of the inhibitor. Supernatant samples were collected at time points up to 48 hours and stored at −70°C before quantification of TGF-β1.
Functional Significance of Increased TGF-β1 Synthesis
Analysis of alteration in collagen IV concentration in the extracellular matrix was examined by Western analysis. Briefly cells were stimulated with insulin (5 μg/ml) for 48 hours under serum-free conditions in the presence or absence of a TGF-β-blocking antibody (Genzyme Diagnostics, Cambridge MA). After detachment of the cell monolayer by the addition of 0.02% ethylenediaminetetraacetic acid, the extracellular matrix laid down by the cells was extracted by the addition of 4 mol/L guanidine hydrochloride-2% Triton X-100 in 25 mmol/L of Tris/HCl in the presence of the protease inhibitors benzamidine, n-ethylmaliamide, phenylmethyl sulfonyl fluoride, and e-amino-n-caproic acid. Matrix samples were subsequently prepared in SDS sample buffer and boiled for 3 minutes at 95°C. Equal amounts of total protein were loaded onto 3 to 12% SDS-PAGE gradient gels and electrophoresis was performed under reducing conditions according to the procedure of Laemmli.14 After electrophoresis the separated proteins were transferred to a nitrocellulose membrane (Amersham, Little Chalfont, UK). The membrane was blocked with Tris-buffered saline containing 5% nonfat powdered milk for 1 hour and then incubated with the primary antibody (goat polyclonal anti-collagen type IV; ICN Pharmaceuticals, Inc., Baisingstoke, UK) in Tris-buffered saline containing 1% bovine serum albumin and 0.05% Tween 20 (Tris-buffered saline-Tween) for 1 hour at room temperature. The blots were subsequently washed in Tris-buffered saline-Tween and then incubated with an appropriate horseradish peroxidase-conjugated secondary antibody (Sigma) in Tris-buffered saline-Tween. Proteins were visualized using enhanced chemiluminescence (Amersham) according to the manufacturer’s instructions. By Western analysis, the higher molecular weight band represents intact collagen whereas the lower molecular weight bands demonstrate its partial degradation.
Gelatinase activity in cell culture supernatant was determined by zymography. Samples were run at 4°C on 7.5% nonreducing SDS-polyacrylamide gels containing gelatin at a final concentration of 1 mg/ml at 4°C. The gels were then washed in 2.5% Triton at room temperature for 1 hour, before incubation overnight at 37°C in 50 mmol/L of Tris-HCl, pH 7.6, containing 10 mmol/L of CaCl2 and 0.05% Brij. The presence of gelatinase activity was demonstrated by zones of lysis in the Coomassie Blue-stained gel.15
Statistics
Statistical analysis was performed using the unpaired Student’s t-test, with a value of P < 0.05 considered to represent a significant difference. The data are presented as means ± SD of n experiments. For each individual experiment the mean of duplicate determinations was calculated.
Results
Stimulation of TGF-β1 by Insulin
Addition of insulin to growth-arrested HK-2 cells led to an increase in TGF-β1 concentration in the cell culture medium (Figure 1)▶ . This effect was significant 24 hours after the addition of insulin (655.2 ± 12.8 pg/ng cell protein versus 445.9 ± 10.6 pg/ng cell protein, insulin versus control, mean ± SD, n = 8, P = 0.003). Despite an increase in TGF-β1 generated after the addition of insulin, no alteration in the expression of TGF-β1 mRNA was seen, as assessed by either RT-PCR or Northern analysis (Figure 2)▶ . Northern analysis however confirmed constitutive expression of a single 2.5-kb TGF-β1 transcript. Autoradiography of immunoprecipitated TGF-β1 after stimulation by insulin in the presence of radiolabeled amino acids demonstrated increased incorporation of radiolabel into TGF-β1 thus confirming stimulation of de novo TGF-β1 synthesis by insulin (Figure 3)▶ .
Figure 1.

Insulin stimulates an increase in TGF-β1 generation. Confluent monolayers of HK-2 cells were growth arrested and insulin (5 μg/ml) added under serum-free conditions (filled bars). In control experiments after growth arrest fresh medium containing 5 mmol/L of d-glucose without additional insulin was added (shaded bars). Supernatant samples were collected throughout the following 48 hours for determination of TGF-β1 by ELISA. Cell protein concentration of the remaining monolayer was determined and results expressed as TGF-β1 per ng of total cell protein. Results represent mean ± SD of eight individual experiments.
Figure 2.

Stimulation of TGF-β1 is not associated with alteration in the expression of its mRNA. Serum-deprived HK-2 cells were either stimulated with insulin (5 μg/ml), in the absence of serum, or in control experiments 5 mmol/L d-glucose was added in the absence of insulin. RNA was isolated at time points up to 48 hours. RT-PCR was performed and PCR products separated by electrophoresis on a 3% agarose gel (A). Scanning densitometry confirmed the lack of induction of TGF-β1 after addition of insulin. One representative experiment of four individual experiments is shown. After isolation of RNA, TGF-β1 mRNA was also examined by Northern analysis (B).
Figure 3.

Incorporation of radiolabeled amino acids into TGF-β1. Growth-arrested cells were stimulated under serum-free conditions with insulin (5 μg/ml) in the presence of 4 μCi of 3H-radiolabeled amino acid mixture. Supernatant samples were subsequently collected after 48 hours and TGF-β1 immunoprecipitated as described in Materials and Methods. After immunoprecipitation, radiolabeled TGF-β1 was visualized by autoradiography.
Previously we have demonstrated that 25 mmol/L of d-glucose may prime PTCs for TGF-β1 synthesis after the addition of a second stimulus. The effect of 25 mmol/L of d-glucose on insulin-induced TGF-β1 synthesis was investigated by pretreatment of growth-arrested HK-2 cells with either 25 mmol/L of d-glucose or 5 mmol/L of d-glucose for 48 hours before stimulation with insulin. In contrast to our previous studies, pretreatment with 25 mmol/L of d-glucose did not alter the amount of TGF-β1 generated in response to insulin [48 hours after addition of insulin −655.2 ± 12.8 pg/ng cell protein versus 765 ± 10.9 pg/ng cell protein, 5 mmol/L d-glucose (n = 8) versus 25 mmol/L d-glucose (n = 4), mean ± SD, P = 0.065].
The effect of insulin on the generation of TGF-β1 was specific in that addition of insulin did not stimulate TGF-β2 generation (1097.5 ± 235 pg/ng cell protein versus 839.9 ± 313.4 pg/ng cell protein, control versus insulin stimulated, mean ± SD, n = 8, P = 0.08). The lack of induction of TGF-β2 in response to insulin was apparent for both 5 mmol/L and 25 mmol/L of d-glucose-pretreated cells (data not shown).
TGF-β1 Synthesis Is Dependent on mRNA Translation
Inhibition of de novo gene transcription by the addition of actinomycin D before addition of insulin in HK-2 cells had no effect on insulin-induced TGF-β1 generation (Figure 4A)▶ . In contrast inhibition of mRNA translation and protein synthesis by addition of cycloheximide (Figure 4B)▶ led to a dose-dependent decrease in TGF-β1 generation after the addition of insulin ]764.2 ± 316.5 pg/ng cell protein versus 338.2 ± 96.8 pg/ng cell protein control versus cycloheximide (5 μg/ml), mean ± SD, n = 8, P = 0.02].
Figure 4.

Insulin stimulation of TGF-β1 generation is dependent on translation but not on de novo gene transcription. Confluent monolayers of HK-2 cells were growth arrested and insulin (5 μg/ml) added under serum-free conditions in the presence of increasing doses of either actinomycin (filled bars) or cycloheximide (shaded bars). Supernatant samples were collected 48 hours after the addition of insulin and TGF-β1 quantified by ELISA. Data represents mean TGF-β1 concentration corrected for total cell protein ± SD of eight individual experiments (*, P < 0.05).
Insulin Did Not Influence TGF-β1 mRNA Stability
We have previously demonstrated in glucose-primed cells that stimuli that induce TGF-β1 translation may do so by mechanisms that influence the stability of glucose-induced TGF-β1 mRNA. In contrast in the current study, results of actinomycin D chase experiments demonstrate that addition of insulin did not affect the stability of constitutively expressed TGF-β1 mRNA (Figure 5)▶ .
Figure 5.

Insulin does not alter TGF-β1 mRNA stability. HK-2 cells were growth arrested before inhibition of transcription by the addition of actinomycin-D (500 ng/ml). Total cellular RNA was isolated throughout the following 12 hours either from untreated cells (constitutive TGF-β1 mRNA stability) or after addition of insulin (5 μg/ml) (A). Alteration in the stability of d-glucose-induced TGF-β1 mRNA was determined by RT-PCR (B). Scanning densitometry confirmed that there was no difference in the ratio of TGF-β1 mRNA to the housekeeping gene between the two experimental conditions (one representative experiment of four is shown).
Insulin Induced Increased Binding of a Cytoplasmic Protein to TGF-β1 5′-UTR
Proteins that bind to the 5′-UTR of a mRNA can also regulate translation. Furthermore binding of a cytoplasmic factor to a putative stem loop structure in the 5′ UTR of TGF-β1 has been demonstrated to regulate TGF-β1 translation. Protein binding to this region (bases +1 to +147) was therefore investigated by gel retardation analysis. When a radiolabeled probe transcribed from this fragment was incubated with HK-2 cytoplasmic extract derived from cells stimulated with insulin, the mobility of the probe was reduced, indicating that a factor in the extract formed a complex with the probe (Figure 6)▶ . This second band was not seen when the probe was incubated with cytoplasmic extract derived from HK-2 cells to which 5 mmol/L of d-glucose was added in the absence of insulin. Specificity of probe binding was confirmed by competition with unlabeled probe added to the binding reaction. Under these conditions formation of the complex was abolished.
Figure 6.

Binding of a cytoplasmic factor to TGF-β1 5′-UTR in the presence of insulin. Radiolabeled probe generated against sequences in the 5′-UTR of TGF-β1 mRNA spanning +1 to +147 was incubated with cytoplasmic extract of HK-2 cells treated with either 5 mmol/L d-glucose in the absence of insulin (lanes 1 and 2) or insulin (5 μg/ml) (lanes 3 and 4) for 48 hours. The mixture was resolved on a nondenaturing polyacrylamide gel. The arrow indicates complex formation between the probe and factor(s) in the extract. Competition was with an excess of unlabeled RNA probe (lane 5).
Insulin versus the IGF-1 Receptor as the Mediator of TGF-β1 Translation
Addition of insulin at doses <1 μg/ml had no effect on the generation of TGF-β1 (Figure 7A)▶ . The effect of insulin at the higher doses (1 to 5 μg/ml) was however inhibited in a dose-dependent manner by the addition of a monoclonal antibody blocking ligand binding to the IGF-1 receptor (Figure 7B)▶ . Addition of recombinant IGF-1 mimicked the effect of addition of the high dose of insulin leading to a dose-dependent increase in TGF-β1 generation. This was also inhibited by the addition of a monoclonal antibody to the IGF-1 receptor (Figure 8)▶ .
Figure 7.
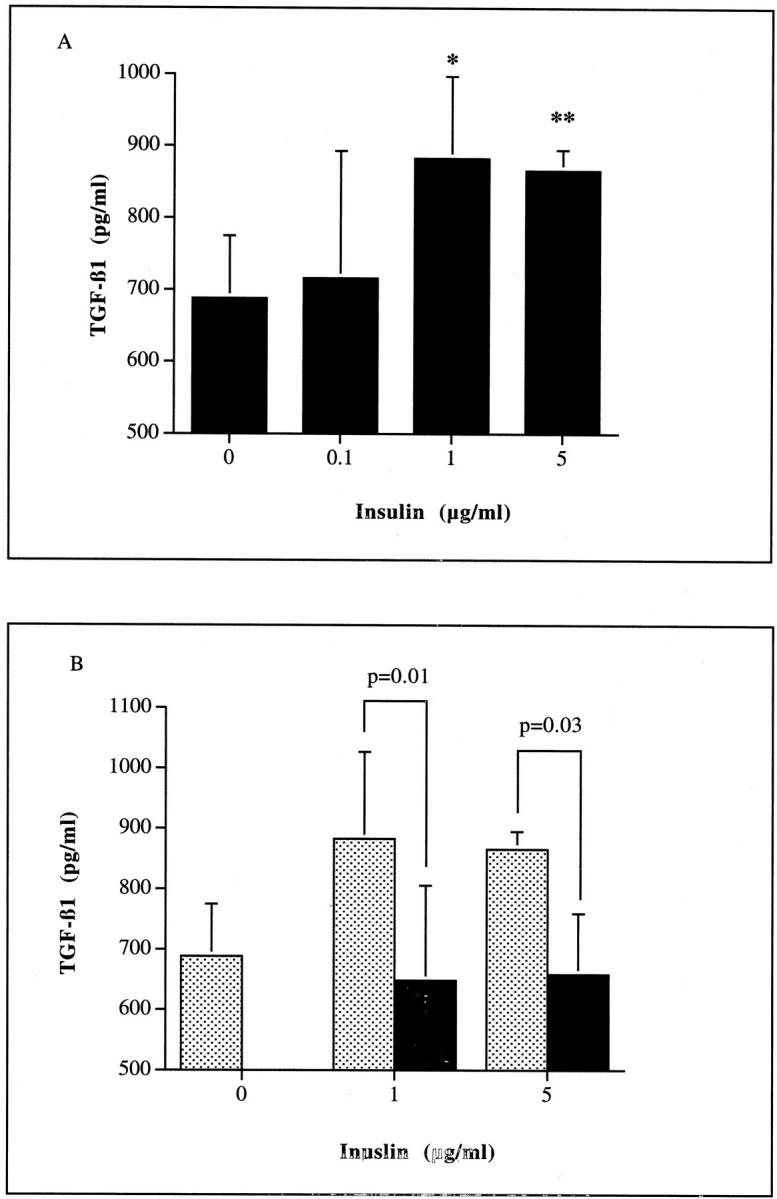
Stimulation of TGF-β1 requires the addition of high doses of insulin (A), and was abrogated by the addition of a blocking antibody to the IGF-1 receptor (B). Confluent monolayers of HK-2 cells were growth arrested and insulin (0 to 5 μg/ml) added under serum-free conditions (A). In addition, insulin (0, 1.0, or 5 μg/ml) was added to confluent HK-2 cells, under serum-free conditions in the presence (filled bars) or absence (shaded bars) of 50 ng/ml of a monoclonal antibody to IGF1R (B). Supernatant samples were collected after 48 hours for determination of TGF-β1 by ELISA. For both experiments, cell protein concentration of the remaining monolayer was determined and results expressed as TGF-β1 per ng of total cell protein. Results represent mean ± SD of four individual experiments (*, P < 0.05; **, P < 0.005).
Figure 8.

Insulin stimulation of TGF-β1 was mimicked by the addition of recombinant IGF-1. HK-2 cells were stimulated with increasing doses of IGF-1 (0 to 250 ng/ml) under serum-free conditions in the presence (filled bars) or absence (shaded bars) of 50 ng/ml of the monoclonal antibody to IGF1R (B). Supernatant samples were collected for determination of TGF-β1 after 48 hours. Data represents mean ± SD (n = 8; *, P < 0.05 versus unstimulated control and IGF stimulated in the presence of antibody).
The Effect of Insulin Was Dependent on PI 3-Kinase Activity
Induction of TGF-β1 by insulin was inhibited by the addition of the PI 3-kinase inhibitor wortmannin (Figure 9)▶ . Significant inhibition of insulin (5 μg/ml) induced TGF-β1 production was seen with 10 nmol/L of wortmannin after 48 hours [482.4 ± 131.1 pg/ng cell protein versus 655.2 ± 12.8 pg/ng cell protein, insulin plus wortmannin (n = 5) versus insulin (n = 8), mean ± SD, P = 0.03]. Similarly significant inhibition of insulin-induced TGF-β1 generation was also seen after inhibition of p70 S6 kinase by the addition of 30 nmol/L of rapamycin [487.4 ± 57.7 pg/ng cell protein versus 655.2 ± 12.8 pg/ng cell protein, insulin plus rapamycin (n = 8) versus insulin (n = 8), mean ± SD, P = 0.0003]. Inhibition of TGF-β1 protein synthesis by either wortmannin or rapamycin was also associated with inhibition of binding of insulin-induced cytoplasmic protein to the probe spanning the sequences base +1 to +147 of the 5′-UTR of TGF-β1 (Figure 10)▶ . In contrast inhibition of MAP/ERK kinase by the addition of PD 98059 (2 μmol/L) had no effect on insulin-induced TGF-β1 generation [711.5 ± 27.6 pg/ng cell protein versus 655.2 ± 12.8 pg/ng cell protein, insulin plus PD 98059 (n = 4) versus insulin (n = 8), mean ± SD, P = 0.4).
Figure 9.

Inhibition of insulin-stimulated TGF-β1 production by wortmannin or rapamycin. Confluent monolayers of HK-2 cells were growth arrested and insulin (5 μg/ml) added under serum-free conditions either in the absence of inhibitor (▪ bars, n = 8), or in the presence of 10 nmol/L of wortmannin ( ▨ , n = 5), 30 nmol/L of rapamycin ( ▧ , n = 8), or 2 μmol/L of PD 98059 (□, n = 4). Control experiments were performed by adding 5 mmol/L of d-glucose in the absence of insulin ( , n = 8). Supernatant samples were collected at time points up to 48 hours for determination of TGF-β1 by ELISA. Cell protein concentration of the remaining monolayer was determined and results expressed as TGF-β1 per ng of total cell protein. Results represent mean ± SD; *, P < 0.05 versus the unstimulated control (5 mmol/L d-glucose alone).
Figure 10.

Inhibition of binding of cytoplasmic factor to TGF-β1 5′-UTR by wortmannin or rapamycin. Radiolabeled probe generated against sequences in the 5′-UTR of TGF-β1 mRNA spanning +1 to +147 was incubated with cytoplasmic extract of HK-2 cells treated with insulin alone (5 μg/ml) or insulin in the presence of either wortmannin (10 nmol/L) or rapamycin (30 nmol/L) for 48 hours. The mixture was resolved on a nondenaturing polyacrylamide gel. Competition was with an excess of unlabeled RNA probe.
Insulin Modulation of Extracellular Matrix Generation
To determine the functional importance of altered TGF-β1 synthesis in response to insulin, we examined the effect of insulin on collagen generation and its dependence on TGF-β1. Stimulation of cells with insulin led to increased expression of type IV collagen mRNA (α1) and increased deposition in the extracellular matrix laid down by the cells (Figure 11)▶ . Addition of insulin also led to an increase in gelatinase activity, seen predominantly as increased gelatinolytic activity at a molecular weight corresponding to 95-kd gelatinase B/MMP9. Stimulation of cells with insulin in the presence of the TGF-β-blocking antibody abrogated the increased gelatinase activity (Figure 12)▶ . Because of this inhibition of gelatinase activity, total type IV collagen concentration in the cell culture supernatant was not affected by this TGF-β-blocking antibody (Figure 12)▶ .
Figure 11.
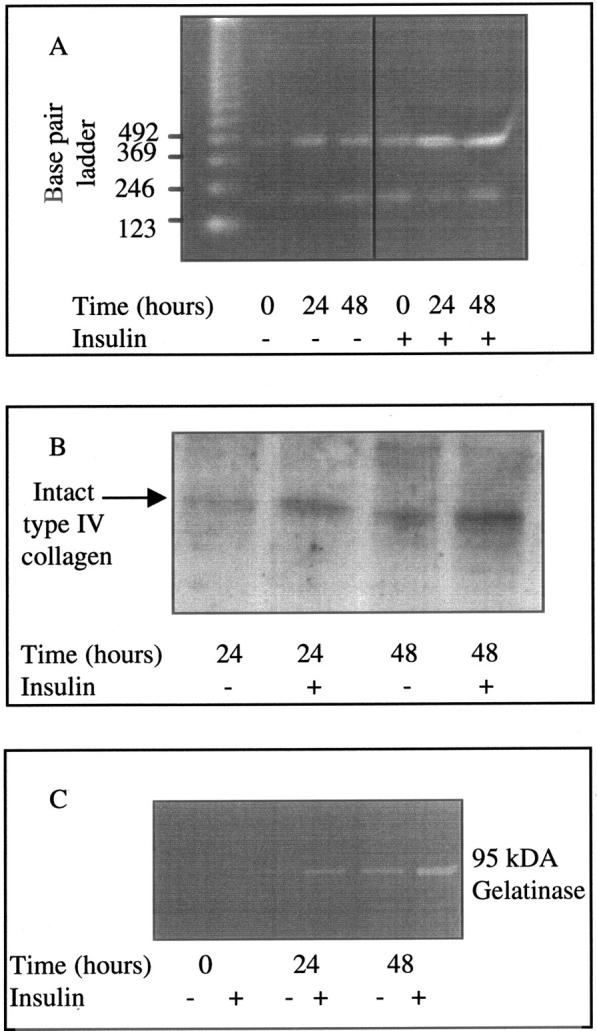
Expression of type IV (αI) collagen mRNA (A), type IV collagen protein (B), and gelatinolytic activity (C) after stimulation with insulin. Cells were grown to confluence and growth arrested before stimulation with insulin (5 μg/ml) under serum-free conditions. Total cellular RNA was isolated at time points up to 48 hours after stimulation and type IV collagen mRNA expression examined by RT-PCR as described in Materials and Methods. For type IV collagen, PCR amplification was performed for 28 cycles. PCR amplification for β-actin was performed for 28 cycles. Ethidium bromide-stained PCR products were separated on a 3% agarose gel (A). One representative experiment of four individual experiments performed is shown. For examination of matrix collagen deposition after detachment of the cell monolayer the extracellular matrix was prepared as described in Materials and Methods. Type IV collagen was subsequently examined by Western analysis after separation on SDS-PAGE gradient gels and electrophoresis performed under reducing conditions (B). One representative experiment of four individual experiments is shown. Gelatinolytic activity in cell culture supernatant samples 48 hours after insulin stimulation, was determined by substrate zymography (C) as described in Materials and Methods. One representative experiment of four individual experiments performed is shown.
Figure 12.

Effect of TGF-β blocking antibody on insulin-mediated alterations in the extracellular matrix. Confluent monolayers of growth-arrested cells were stimulated with insulin (5 μg/ml) in the presence of a TGF-β-blocking antibody (30 ng/ml). Supernatant samples were collected 48 hours after cell stimulation. Gelatinolytic activity (A) and type IV collagen (B) were examined by zymography and Western analysis, respectively, as described in Materials and Methods. One representative experiment of three is shown.
Discussion
It is now clear that the rate of progression of renal disease is closely related to the degree of cortical interstitial fibrosis. TGF-β1 is an important cytokine that has been implicated in the pathogenesis of numerous fibrotic diseases, and its generation by the renal proximal tubular cell suggests that this cell type may also influence pathological events in the renal interstitium. Our previous studies have demonstrated that elevated concentrations of glucose modulate TGF-β1 production by PTCs suggesting that this cell may contribute to the pathogenesis of diabetic interstitial fibrosis.
Although it is clear that hyperglycemia is an important factor in the initiation of renal disease in diabetes because only 40% of diabetic patients develop nephropathy, other factors must play an important part in both initiating and perpetuating renal injury. Despite the large body of research into the pathogenesis of diabetic nephropathy little is know regarding the effect of insulin. Hyperinsulinemia is a feature of type 2/non-insulin-dependent diabetes, and furthermore insulin therapy in type 1/insulin-dependent diabetes is also associated with periods of hyperinsulinemia. Insulin plays an important role in the overall regulation of protein synthesis and may specifically influence mRNA translation. Not only does it cause a global increase in the rate of translation, but, superimposed on this it also brings about marked increases in the translation of specific mRNAs.16 The data presented in the current article demonstrate that insulin stimulated the de novo synthesis of TGF-β1. Furthermore we have shown that this increase in TGF-β1 synthesis was not associated with induction of TGF-β1 mRNA, and did not require de novo gene transcription because it was unaffected by actinomycin; rather, stimulation of TGF-β1 synthesis in PTCs after addition of insulin involved stimulation of translation of TGF-β1 mRNA. The potential functional importance of increased TGF-β1 synthesis was illustrated by the associated alteration in extracellular matrix turnover that was antagonized by inhibition of TGF-β activity.
The data also demonstrate that the effect of insulin was specific because the addition of insulin did not stimulate the production of TGF-β2. The mRNA coding for TGF-β1 contains an unusually long 5′-UTR that is highly rich in GC content.17 In other systems these sequences tend to be highly structured and are thought to inhibit translation. In contrast to TGF-β1, the long 5′-UTR of TGF-β2 does not have such a high GC content,18 and is therefore not subject to the same posttranscriptional influences.
Three transcripts have been reported for TGF-β1 (2.5, 1.9, and 1.4 kb) that differ in the length of their GC-rich 5′-UTR.19-21 In other systems 5′-UTR with a rich GC content tend to be highly structured and are thought to inhibit translation by impeding the progress of the 40S-scanning ribosomal subunit toward the initiation codon. It is thought that translation of the truncated TGF-β1 transcripts may therefore be more efficient owing to the deletion of inhibitory elements in the 5′ leader sequence. We show that PTCs only express the poorly translated 2.5-kb transcript, and furthermore its expression was not affected by the addition of insulin. Elements spanning bases +1 to +147 5′-UTR of TGF-β1 mRNA have previously been shown to be involved in the inhibition of TGF-β1 translation although the inhibitory effect of this fragment is cell-type-dependent.22 It has been suggested that a cytosolic factor may regulate TGF-β1 mRNA translation by binding to these elements.12 Most recently is has been postulated that the poor translational efficiency of the 2.5-kd TGF-β1 mRNA in vivo may be because of a combination of poor initiation sequence context, and inhibitory interactions of limiting trans-acting factors with cis-inhibitory elements in an otherwise stimulatory 5′-UTR.23 In contrast we have identified a possible stimulatory trans-acting factor binding a portion of the 5′-UTR mRNA spanning sequences +1 to +147 previously identified as an inhibitory element.
We have previously demonstrated that TGF-β1 production in PTCs may be controlled at the levels of transcription, translation, and secretion of preformed protein, as well as at the level of activation of the latent protein. These studies suggest that alteration of TGF-β1 mRNA stability may be involved in facilitation of glucose-induced mRNA translation when cells were pretreated with glucose before the addition of a second stimulus such as the proinflammatory cytokine interleukin-1β that facilitates mRNA translation.6 Increased TGF-β1 mRNA is not however universally associated with facilitation of its translation. In fact under certain circumstances stabilization of TGF-β1 is associated with an inhibitory influence on its translation.12 In the current article we have demonstrated that insulin-induced TGF-β1 mRNA translation was not associated with alteration in its stability. This suggests that numerous independent and stimulus-specific mechanisms influence TGF-β1 mRNA translation.
Insulin interacts, albeit with different affinities with both the insulin and the IGF-1 receptor. The receptors for insulin and IGF-1 are also structurally related and highly homologous, and not surprisingly the signaling mechanisms of these receptors are also broadly similar.24,25 The insulin receptor demonstrates high-affinity binding to insulin and lower affinity binding to IGF-1, whereas the IGF-1 receptor binds IGF-1 (and IGF-2) with a higher affinity than it does insulin.25 Thus at low concentrations insulin will interact primarily with the insulin receptor, but at higher concentrations, such as those used in our experiments, the insulin may interact with either receptor. We have demonstrated that the effects of insulin at our hyperinsulinemic concentrations are likely to be mediated via binding to the IGF-1 receptor, in that we could mimic the effect of insulin by the addition of recombinant IGF-1 and antagonize the effects of insulin by blockade of the IGF-1 receptor. Although the effects of insulin were mimicked by addition of recombinant IGF-1, the significance of IGF-1 as a stimulus for renal TGF-β1 synthesis in the context of progressive diabetic nephropathy is unclear. Studies of circulating IGF-1 levels in patients with diabetes are conflicting, in part because of problems with assay methodologies. Studies with the greatest number of patients however have demonstrated decreased IGF-1 levels (50 to 90% of normal) in patients with both type 1 and type 2 diabetes.26 That IGF-1 is not involved in the progression of nephropathy is also supported by data from the streptozotocin-induced diabetes that demonstrates that the rapid increase in renal growth seen in this mode, is preceded by a rise in renal tissue expression of IGF-1 related to alteration in renal IGF receptors rather that an alteration in local IGF-1 production. This increase in renal IGF-1 however returns to normal 4 days after the induction of diabetes.27 Although we cannot rule out a role for IGF-1 in modulation of TGF-β1 synthesis in diabetic nephropathy, it is likely that insulin/hyperinsulinemia via IGF-1 receptor signaling modulates renal TGF-β1 in diabetes.
The renal proximal tubular cell is a bipolar epithelial cell. Its plasma membrane is differentiated into a basolateral portion that is in contact with the peritubular circulation and a brush border portion that is bathed in glomerular ultrafiltrate. Previously we have demonstrated that induction of TGF-β1 synthesis after sequential stimulation with glucose and cytokines was a polar phenomenon. Induction of TGF-β1 only occurred after application of glucose to the basal aspect of the cell, and similarly stimulation of TGF-β1 synthesis was only seen after addition of cytokines also to the basolateral cell surface.8 It is known that the IGF-1 receptor has an asymmetric distribution, being present primarily on the basolateral cell surface.28 The actions of IGF-1 on TGF-β1 synthesis in the proximal tubule, like those of glucose and cytokines, are therefore likely to be mediated via interaction of circulating peptide with specific receptors on the basolateral membrane.
Many similarities exist in the postreceptor signaling pathways between the insulin and the IGF receptor. The effects of these ligands are initiated by their binding to the extracellular domain of the receptors and subsequent activation of intracellular tyrosine kinase activity. This leads to autophosphorylation of the receptor and activation of Ras and phosphatidylinositol-3-kinase and hence to the activation of a number of serine/threonine protein kinases activating the mitogen-activated kinase (MAP/ERK kinase) cascade, protein kinase B (PKB) and the activation of the 70-kd ribosomal S6 protein kinase (p70 S6 kinase). Previous studies with insulin have demonstrated that alteration in signaling via each of these pathways may influence protein translation by their regulation of eukaryotic initiation factors (eIFs), and elongation factors (eEFs).16,29,30 Our data demonstrate that the effect of insulin on TGF-β1 synthesis was mediated via the PI-3 kinase-p70 S6 kinase cascade because it was inhibited by both rapamycin and wortmannin, but not inhibited by inhibition of MAPK/Erk kinase. Studies to date have demonstrated that the acute effects of insulin and growth factors on protein synthesis are achieved by regulating the function of multiple components of the initiation and elongation complexes involved in mRNA translation. The eukaryotic initiation factor 2b (eIF2b) is phosphorylated in response to insulin by a pathway involving PI 3-kinase.31 An insulin-induced reduction in phosphorylation of eukaryotic elongation factor-2 (eEF2) is associated with an increase in translation, and both rapamycin and wortmannin block this effect.32 These inhibitors also inhibit insulin-stimulated phosphorylation of the eukaryotic initiation factor 4E (eIF-4E)-binding protein (4EBP-1).33,34 These known effects of insulin and the pattern of inhibition of insulin-stimulated TGF-β1 synthesis that we have demonstrated suggests that these initiation and elongation factors may be involved in facilitation of translation of TGF-β1 and may represent useful avenues for future studies to identify the cytoplasmic factors that affect this process.
It is well recognized that the control of TGF-β1 bioactivity is complex, and that independent regulatory mechanisms may influence transcription, translation, secretion, activation, and postreceptor signaling events. The data in the current article further emphasized the complexity of control of TGF-β1 synthesis. Together with previously published studies it is now clear that numerous different stimulus-specific mechanisms influence TGF-β1 translation. We have also demonstrated that in diabetes numerous mechanisms exist that may alter the generation of TGF-β1 by the proximal tubular epithelial cell. Furthermore the data demonstrate that insulin stimulates TGF-β1-mediated alterations in extracellular matrix turnover suggesting that insulin and hyperinsulinemia may therefore influence the pathological changes in the renal interstitium associated with this condition.
Footnotes
Address reprint requests to Dr. A. O. Phillips, Institute of Nephrology, University of Wales College of Medicine, Heath Park, Cardiff CF14 4XN, Wales, UK. E-mail: phillipsao@cf.ac.uk.
Supported by the British Diabetic Association (to K. M.), the National Kidney Research Fund (to R. E.) and by an Advanced Training Fellowship from the Wellcome Trust (to A. P.).
References
- 1.United States Renal Data System’s. 1990 Annual Data Report: causes of ESRD. Am J Kidney Dis 1990, 15:S22–S27 [PubMed]
- 2.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Muller GA: The pathogenesis of chronic renal failure in diabetic nephropathy. Path Res Pract 1991, 187:251-259 [DOI] [PubMed] [Google Scholar]
- 3.Lane PH, Steffes MW, Fioretto P, Mauer SM: Renal interstitial expansion in insulin-dependant diabetes mellitus. Kidney Int 1993, 43:661-667 [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA: Expression of transforming growth factor-β is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 1993, 90:1814-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips AO, Steadman R, Topley N, Williams JD: Elevated d-glucose concentrations modulate TGF-β1 synthesis by human cultured renal proximal tubular cells: the permissive role of platelet derived growth factor. Am J Pathol 1995, 147:362-374 [PMC free article] [PubMed] [Google Scholar]
- 6.Phillips AO, Topley N, Steadman R, Morrisey K, Williams J: Induction of TGF-β1 synthesis in d-glucose primed human proximal tubular cells: differential stimulation by the macrophage derived pro-inflammatory cytokines IL-1β and TNFα. Kidney Int 1996, 50:1546-1554 [DOI] [PubMed] [Google Scholar]
- 7.Phillips AO, Topley N, Morrisey K, Williams JD, Steadman R: Basic fibroblast growth factor stimulates the release of pre-formed TGF-β1 from human proximal tubular cells in the absence of de-novo gene transcription or mRNA translation. Lab Invest 1997, 76:591-600 [PubMed] [Google Scholar]
- 8.Phillips AO, Morrisey R, Steadman R, Williams JD: Polarity of stimulation and secretion of TGF-β1 by proximal tubular cells. Am J Pathol 1997, 150:101-111 [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA, Torok-Storb B: HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 1994, 45:48-57 [DOI] [PubMed] [Google Scholar]
- 10.Redinbaugh MG, Campell WH: Adaptation of the dye-binding assay to microtitre plates. Ann Biochem 1985, 147:144-147 [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Lin G, Baarsch MJ, Scamurra RW, Murtaugh MP: Interleukin-4 suppresses inflammatory cytokine gene transcription in porcine macrophages. J Leukoc Biol 1994, 56:507-513 [DOI] [PubMed] [Google Scholar]
- 12.Romeo DS, Park K, Roberts AB, Sporn MB, Kim SJ: An element of the transforming growth factor-β1 5′-untranslated region represses translation and specifically binds a cytosolic factor. Mol Endocrinol 1993, 7:759-766 [DOI] [PubMed] [Google Scholar]
- 13.Dixon R, Brunskill NJ: Activation of mitogenic pathways by albumin in kidney proximal tubule epithelial cells: implications for the pathophysiology of proteinuric states. J Am Soc Nephrol 1999, 10:1487-1497 [DOI] [PubMed] [Google Scholar]
- 14.Laemmli UK: Cleavage of structural proteins during the assembly the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 15.Phillips AO, Morrisey K, Martin J, Williams JD, Steadman R: Exposure of human renal proximal tubular cells to glucose leads to accumulation of type IV collagen and fibronectin by decreased degradation. Kidney Int 1997, 52:973-984 [DOI] [PubMed] [Google Scholar]
- 16.Proud CG, Denton RM: Molecular mechanisms for the control of translation by insulin. Biochem J 1997, 328:329-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, Roberts AB, Sporn MB, Goeddel DV: Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316:701-705 [DOI] [PubMed] [Google Scholar]
- 18.Noma T, Glick AB, Geiser AG, O’Reilly MA, Roberts AB, Sporn MB: Molecular cloning and structure of the human transforming growth factor β2 gene promoter. Growth Factors 1991, 4:247-255 [DOI] [PubMed] [Google Scholar]
- 19.Qian SW, Kondaiah P, Casscells W, Roberts AB, Sporn MB: A second messenger RNA species of transforming growth factor β1 in infarcted rat heart. Cell Regul 1991, 2:241-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostrowski LE, Gray TE, Randell SH, Nettesheim P: Characterization of a third transforming growth factor β1 transcript in rat tracheal epithelial cells. Cell Growth Differ 1993, 4:985-991 [PubMed] [Google Scholar]
- 21.Romeo D, Allison RSH, Kondaiah P, Wakefield LM: Recharacterisation of the start sites for the major human transforming growth factor β1 mRNA. Gene 1997, 189:289-295 [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, Park K, Koellers D, Kim KY, Wakefield LM: Post-transcriptional regulation of the human transforming growth factor beta1 gene. J Biol Chem 1992, 267:13702-13707 [PubMed] [Google Scholar]
- 23.Allison RSH, Mumy ML, Wakefield LM: Translational control elements in the major human transforming growth factor β1 mRNA. Growth Factors 1998, 16:89-100 [DOI] [PubMed] [Google Scholar]
- 24.Werner H, LeRoith D: Insulin-like growth factor 1 receptor: structure, signal transduction and function. Diabetes Rev 1995, 3:28-37 [Google Scholar]
- 25.Adamo M, Roberts CT, LeRoith D: How distinct are the insulin and insulin-like growth factor 1 signalling systems? Biofactors 1992, 3:151-157 [PubMed] [Google Scholar]
- 26.Bach LA, Rechler MM: Insulin-like growth factors and diabetes. Diabetes Metab Rev 1992, 8:221-257 [DOI] [PubMed] [Google Scholar]
- 27.Flyvbjerg A: Role of growth hormone, insulin-like growth factors (IGFs) and IGF-binding proteins in the renal complications of diabetes. Kidney Int 1997, 52:S12-S19 [PubMed] [Google Scholar]
- 28.Hammerman MR, Rogers S: Distribution of IGF receptors in the plasma membrane of proximal tubular cells. Am J Physiol 1987, 253:F841-F847 [DOI] [PubMed] [Google Scholar]
- 29.Shepherd PR, Withers DJ, Siddle K: Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 1998, 333:471-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moule SK, Denton RM: Multiple signalling pathways involved in the metabolic effects of insulin. Am J Cardiol 1997, 80:41A-49A [DOI] [PubMed] [Google Scholar]
- 31.Welsh GI, Stokes CM, Wang X, Sakaue H, Ogawa W, Kasuga M, Proud CG: Activation of translation initiation factor eIF2B by insulin requires phosphatidyl inositol 3-kinase. FEBS Lett 1997, 410:418-422 [DOI] [PubMed] [Google Scholar]
- 32.Redpath NT, Foulstone EJ, Proud CG: Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J 1966, 15:2291-2297 [PMC free article] [PubMed] [Google Scholar]
- 33.Diggle TA, Moule SK, Avison MB, Flynn A, Foulstone EJ, Proud CG, Denton RM: Both rapamycin-sensitive and -insensitive pathways are involved in the phosphorylation of the initiation factor-4E-biinding protein in response to insulin in rat epididymal fat-cells. Biochem J 1996, 316:447-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flynn A, Vries RGJ, Proud CG: Signalling pathways which regulate eIF4E. Biochem Soc Trans 1997, 25:192. [DOI] [PubMed] [Google Scholar]
- 35.Brinker JM, Gudas LJ, Loidl HR, Wang SY, Rosenbloom J, Kefalides NA, Myers JC: Restricted homology between human alpha-1 type IV and other procollagen chains. Proc Nat Acad Sci USA 1985, 82:3649-3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neubauer A, Neubauer B, Liu E: Polymerase chain reaction based assay to detect allelic loss in human DNA: loss of beta interferon gene in chronic myelogeneous leukemia. Nucleic Acids Res 1990, 18:993-998 [DOI] [PMC free article] [PubMed] [Google Scholar]