

Abstract
Recruitment of lymphocytes is a prominent feature of the inflammatory process in Crohn’s disease (CD). The present study was undertaken to investigate the expression of the novel lymphocyte-specific chemoattractant lymphotactin (Lptn) as a potential regulatory factor for the recruitment of T cells in CD. The expression of Lptn mRNA was quantified in resection specimens of patients with CD in comparison to normal controls without signs of inflammation by real-time quantitative reverse transcriptase-polymerase chain reaction and localized by nonradioactive in situ hybridization. Furthermore, the phenotype of cells expressing Lptn mRNA was characterized. In contrast to normal controls Lptn mRNA was significantly increased in tissue samples affected by CD. Cells expressing Lptn were identified as T cells, mast cells, and unexpectedly dendritic cells. Lptn mRNA was found to be up-regulated on stimulation with phorbol-12-myristate-13-acetate and concanavalin A in T cells isolated from peripheral blood, which could be prevented by dexamethasone, cyclosporine A, and FK506. A similar regulation mechanism could be identified for the Lptn receptor GPR-5 in peripheral T cells. In addition, Lptn mRNA expression could be induced in mature monocyte-derived dendritic cells. The results indicate that local expression of Lptn by activated T cells and to a lesser extent by mast cells and dendritic cells represents a key regulator for lymphocyte trafficking and maintenance of the inflammatory process observed in CD, which might be partly mediated through an autocrine/paracrine pathway of activated T cells.
Crohn’s disease (CD) is a chronic inflammatory disorder of unknown etiology. The histologically observable chronic inflammatory changes in CD include focal lymphocytic infiltrates and occasionally the presence of granulomas with Langhans’-type multinucleated giant cells. 1 The focal mononuclear infiltrates frequently show a perivascular distribution and are mainly localized around small submucosal vessels. An increased recruitment of both memory and naive T cells could be demonstrated in CD, 2 but the mechanisms leading to the recruitment of mononuclear cells within focal infiltrates still remain unclear.
The traffic of inflammatory cells at sites of inflammation includes four distinct phases: circulation, adhesion, diapedesis, and migration. Chemokines are known to play a crucial role in these steps. 3 Chemokines are small secreted proteins stimulating chemotaxis and leading to selective attraction of inflammatory cells. The chemokine superfamily can be subdivided according to the organization of the N-terminal conserved cysteine (C) motif into four groups, which are designated as C-, C-C-, C-X-C-, and C-X3-C-chemokines. 4 The minor differences in the N-terminal structure result in actions on different cell types. 5 C-X-C-chemokines generally attract neutrophils and T cells, C-C-chemokines are chemoattractive for monocytes, eosinophils, basophils, and T cells. 6,7 Whereas the C-X-C- and C-C-subfamilies constitute several members, the C- and C-X3-C-chemokine subfamilies are represented by only one member so far, designated lymphotactin (Lptn) 8 also known as activation-induced T cell-derived and chemokine-related molecule (ATAC) 9 or single C motif-1 (SCM-1) 10 and fractalkine/neurotactin, 11 respectively. Preliminary characterization revealed that in contrast to the members of the C-C- and C-X-C subfamilies, Lptn and fractalkine are powerful attractants for lymphocytes, in particular T cells, but are inactive in inducing the migration of neutrophils and monocytes. 8
In vitro and in vivo Lptn was found to be more chemotactic for CD8+ than CD4+ T cells and, in addition, modestly chemoattractive for natural killer cells. 12-16 In contrast to other chemokine subfamilies Lptn lacks two of the four characteristic cysteine residues, but shows strong amino acid similarities with the C-C-chemokines. 17 Expression of Lptn mRNA was found in certain subsets of T cells, including activated CD8+ and CD4+ T cells, 9 intraepithelial γδ-type T cells, 17 αβ-type thymocytes, 8 mast cells, 18 and natural killer cells. 15 More recently, a unique G-protein-coupled receptor (GPR-5) has been found to function as the natural Lptn ligand. 19,20 Other biological functions of Lptn are still unknown.
So far the expression of Lptn at sites of inflammation has rarely been investigated. The in vitro observed functional properties of Lptn suggest an important function in the recruitment of T cells in the pathogenesis of chronic inflammatory disorders. Because focal lymphocytic infiltrates represent a predominant feature of the chronic inflammatory infiltrate observed in CD, we characterized and determined the phenotype of inflammatory cells expressing this potent chemokine. Our results demonstrate an increased local expression of Lptn in CD, which could be attributed not only to activated T cells, but also to mast cells. In addition, we will show for the first time expression of Lptn mRNA by dendritic cells (DCs) in CD in vivo and by monocyte derived DCs generated in vitro.
Materials and Methods
Tissue Samples
Surgical specimens derived from involved areas of large intestine of 15 patients with active CD were studied. CD was active in all of the patients as confirmed by clinical features, laboratory parameters, as well as macroscopic and histopathological alterations. As controls surgical specimens from six patients with noninflammatory gut disorder were analyzed (four colon carcinomas and two diverticular diseases). For routine morphological analysis, immunohistochemistry, and nonradioactive in situ hybridization appropriate tissue specimens were fixed in formalin and embedded in paraffin. Additional representative samples were snap-frozen in liquid nitrogen, stored at −80°C, and used for RNA isolation.
RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
RNA was isolated from cryopreserved tissue samples and isolated cells using TriReagent according to the manufacturer’s instructions (Sigma, Deisenhofen, Germany) and finally obtained in 30 μl of RNase-free water. RNA content was determined spectrophotometrically. One μg of RNA was used for reverse transcription after a DNase (fast protein liquid chromatography-pure DNaseI; Pharmacia, Freiburg, Germany) digestion step. cDNA synthesis was performed in a total volume of 20 μl of RNase-free water including 1.5 μmol/L of oligo-dT primer, 0.4 mmol/L of each deoxynucleotidetriphosphate (both from MBI Fermentas, St. Leon-Rot, Germany), and 200 U of reverse transcriptase (SuperScriptII; Gibco, Karlsruhe, Germany).
The oligonucleotide primer sequences were determined manually according to the published sequences in GenBank for β-actin (X00351), Lptn (U23772), and GPR-5 (NM005283). PCR conditions were as follows: β-actin (β-actin forward primer: 5′-CTACAATGAGCTGCGTGTGGC-3′; β-actin reverse primer: 5′-CAGGTCCAGACGCAGGATGGC-3′; product size: 270 bp) 3 minutes at 95°C, 35 cycles of 40 seconds at 95°C, 45 seconds at 57°C, 1.5 minutes at 72°C, followed by a final extension step of 7 minutes at 72°C; Lptn (Lptn forward primer: 5′-GTGGAAGGTGTAGGGAGTGAAGTC-3′; Lptn reverse primer: 5′-GTCCATGAGGGTGTAAAGTGAAAT-3′; product size: 354 bp) 3 minutes at 95°C, 35 cycles of 40 seconds at 95°C, 40 seconds at 55°C, 1 minute at 72°C, and final elongation of 7 minutes at 72°C; Lptn receptor GPR-5 (forward primer: 5′-GCTTTCTTCGGGCTGTGATTATTC-3′; reverse primer: 5′-GCGGCAGATGAGCAGGGCGTATT-3′; product size: 310 bp) 3 minutes at 95°C, 35 cycles of 40 seconds at 95°C, 45 seconds at 58°C, 1.30 minutes at 72°C, and final elongation of 10 minutes at 72°C. The PCR products were visualized on 1% agarose gels by staining with ethidium bromide. To exclude amplification of genomic sequences PCR primers were designed to amplify cDNA fragments that span at least one intron of the corresponding gene. cDNA from a human lymph node was used as a positive control template for each primer pair. Negative controls with water instead of cDNA were always included. A PCR reaction was considered to be positive when a single band of correct product length could be detected. Amplification of correct PCR products was controlled by asymmetric restriction enzyme digestion and sequencing with an ABI310 automatic sequencer using the rhodamine-dye terminator kit according to the manufacturer’s instructions (Perkin Elmer, Darmstadt-Weiterstadt, Germany).
Cloning of PCR Products
For generation of the Lptn and GPR-5 riboprobes for quantitative real-time RT-PCR or in situ hybridization PCR products were cloned into the blunt-end SmaI restriction site of the transcription vector pBluescript II KS plasmid. Briefly, blunt-end PCR products were generated by using Vent polymerase (Pharmacia, Heidelberg, Germany) according to the PCR-cycling conditions as mentioned above. After microspin column purification (Pharmacia) the blunt-end PCR products were ligated into a SmaI restricted pBluescript II KS plasmid using T4-DNA ligase at 16°C overnight and transformed into the CaCl2-competent Escherichia coli strain DH5α. After propagation under standard culture conditions the plasmids were isolated using the alkaline lysis procedure. Cloning of Lptn- and GPR-5-specific PCR fragments was confirmed by cycle-sequencing with the Rhodamine-dye terminator kit using an ABI 310 Sequencer according to the manufacturer’s instructions (Perkin-Elmer). Riboprobes were generated by in vitro transcription using T3- or T7-polymerase (Boehringer, Mannheim, Germany). The amount of cRNA transcripts was monitored by 1% agarose gel electrophoresis and determined spectrophotometrically.
Real-Time Quantitative RT-PCR Analysis
Quantification of Lptn and GPR-5 mRNA expression in a RT-PCR set up was performed on an iCycler iQ real-time detection system (Bio-Rad, Hercules, CA). For RT-PCR a one-step RT-PCR kit (Qiagen, Hilden, Germany) was used. The 15-μl reaction from this kit was supplemented with 150 ng of total cellular RNA, 0.6 μmol/L each of the gene-specific primers, 4 U of RNase inhibitor (Promega, Mannheim, Germany) and 0.5 μl of SYBRgreen I dye (Molecular Probes, MoBiTec, Göttingen, Germany) and diluted in 1× RT-PCR buffer (Qiagen) to 12.5×. The dilution was checked spectrophotometrically at 260 nm. The final concentration of SYBRgreen I dye was 0.4×. The reaction mix was loaded into rows of eight 200-μl PCR reaction tubes (Sarstedt, Nümbrecht, Germany) and covered with optical sealing tape (Bio-Rad). The thermal cycling proceeded as follows: 50°C for 30 minutes for reverse transcription, followed by 15 minutes 95°C for denaturation of reverse transcriptase, and activation of HotStar Taq polymerase. A maximum of 50 PCR cycles was run in the following profile: initial denaturation at 95°C, annealing at 55°C, and extension at 72°C for 30 seconds, each. Acquisition of fluorescence signals was monitored on the iCycler and terminated when all reactions reached an amplification plateau and template-free control stayed at basement levels. A PCR standard curve was generated using the same reaction mix including in vitro generated Lptn and GPR-5 cRNA (dilution range from 1 to 1000 attomoles) instead of total cellular RNA. Data analysis was done with iCycler iQ real-time detection system software (Bio-Rad). Results derived from the PCR standard curve for Lptn and GPR-5 mRNA expression are given in attomoles per μg total cellular RNA.
To verify that only specific PCR products evoked fluorescence signals a melting curve of PCR products was obtained in a cycler program of denaturation (95°C), renaturation (50°C), and a temperature ramp from 60°C to 95°C with 0.5°C increments for 10 seconds. Melting peaks were related to PCR product size on 1% agarose gels.
Nonradioactive in Situ Hybridization
For in situ hybridization of Lptn- and GPR-5-specific mRNA transcripts paraffin sections were mounted on slides coated with 3-aminopropyl-triethoxysilane (Sigma) under RNase-free conditions. The slides were allowed to dry at 37°C overnight. Digoxigenin-11-dUTP-labeled antisense and sense probes were generated by in vitro transcription using T3 or T7 polymerase (Boehringer). The amount of transcripts was monitored by 1% agarose gel electrophoresis and labeling efficiency was controlled by dot-blot analysis of serial probe dilutions.
In situ hybridization was performed modified according to the method described by Breitschopf and colleagues. 21 In brief, tissue sections were deparaffinized, rehydrated in serial dilutions of ethanol, and postfixed in 4% Tris-buffered saline (TBS)- buffered paraformaldehyde. Samples were permeabilized using proteinase K (10 μg/ml) for 30 minutes at 37°C. Digestion was stopped by washing the samples in a phosphate-buffered saline solution (pH 7.4) and the slides were dehydrated in serial dilutions of ethanol. Digoxigenin-labeled riboprobes were diluted in hybridization buffer (Amersham, Braunschweig, Germany). After application of sense or antisense cRNA probes the samples were covered with sterile coverslips and placed on a hot plate at 85°C for 5 minutes for probe and target denaturation. Hybridization was performed overnight at 58°C in a sealed humid chamber containing 50% formamide. Nonspecific binding or unbound probes were removed by the following posthybridization washes: 1× standard saline citrate/0.1% sodium dodecyl sulfate at room temperature (2 × 5 minutes) and 0.2× standard saline citrate/0.1% sodium dodecyl sulfate at 58°C (2 × 10 minutes) followed by RNase digestion (20 μg/ml, Gibco) for 30 minutes at 37°C. Finally, sections were washed in TBS containing 0.1% Tween-20 (TBST, Boehringer). Hybridization signals were detected using a sheep polyclonal antibody F(ab)2-fragment against digoxigenin conjugated with alkaline phosphatase (1:500, Boehringer). Alkaline phosphatase activity was visualized using 5-bromo-4-chloro-3-indolyl phosphate as substrate and nitro blue tetrazolium as coupler (Boehringer). For each tissue section sense riboprobes served as controls.
Detection of GPR-5-specific mRNA required signal amplification. Briefly, hybridization signals were detected using a sheep polyclonal antibody F(ab)2-fragment against digoxigenin conjugated with horseradish peroxidase (1:300, Boehringer) and applying the tyramide signal amplification method as described below in the next section using diaminobenzidine as chromogen for visualization.
To determine the specified cell types expressing Lptn mRNA hybridized specimens were incubated with the monoclonal antibodies (mAbs) Ki-M1P recognizing monocytes/macrophages 22 (1:1000; kindly provided by Prof. Parwaresch, Kiel, Germany) or CD3 for T cells (undiluted; Beckmann Coulter, Heidelberg, Germany) followed by a rabbit anti-mouse Ig fluorescein isothiocyanate (1:50; DAKO, Hamburg, Germany) for evaluation by immunofluorescence. In case of the antigens CD4 (1:50) and CD8 (1:50) for T-cell subsets (both Novocastra, Dossenheim, Germany), CD20 for B-cells (1:50), CD57 for natural killer cells (1:50), tryptase for mast cells (1:300; all DAKO), and S100 protein recognizing DCs (undiluted; Beckman Coulter) immunohistochemistry was performed on serial sections. To visualize bound primary antibodies, the following detector components were applied as recommended by the manufacturer: a biotin-streptavidin-amplified system containing biotinylated secondary antibody (multilinker 1:20), alkaline-phosphatase-conjugated streptavidin (label 1:20), fast red (chromogen), and naphthol phosphate (substrate; Biogenex, Hamburg, Germany).
Immunohistochemistry and Double Immunofluorescence for Lptn
Paraffin-embedded sections were immunostained applying the tyramide signal amplification system method according to the manufacturer’s instructions (Dupont-NEN, Boston, MA). For the detection of Lptn (1:600, rabbit anti-human Lptn; TEBU, Frankfurt, Germany) sections were pretreated by repeated boiling in citrate buffer (pH 6.1) in a microwave (700 W, 5 × 3 minutes). After cooling to room temperature nonspecific binding was blocked with 20% bovine serum albumin (Sigma) in 0.05 mol/L Tris/HCl-buffered NaCl solution (TBS, pH 7.4) followed by incubation with the primary antibody overnight at 4°C in a humidified chamber. After rinsing in TBS the slides were incubated for 30 minutes with a biotinylated goat anti-rabbit Ig polyclonal antibody (1:500, DAKO) diluted in Tris-HCl buffered NaCl blocking buffer (TNB, pH 7.5) (Dupont-NEN). After washing three times in TBS slides were incubated with streptavidin-horseradish peroxidase complex (diluted 1:100 in TNB) for an additional 30 minutes. Three washes with TBS were followed by application of biotin-tyramide diluted 1:100 in Amplification Diluent (Dupont-NEN) for 10 minutes. Reactive sites were detected with streptavidin-horseradish peroxidase (1:100 in TBS, 30 minutes at room temperature). Peroxidase activity was visualized by using aminoethyl carbazole as chromogen. In control reactions irrelevant primary antibody was applied or primary antibodies were omitted.
To characterize in situ the co-expression of Lptn and other antigens such as CD3 (DAKO), CD4, CD8 (both Novocastra), CD31, CD45RA (both DAKO), CD103 (HML-1; Santa Cruz, Heidelberg, Germany), tryptase, and the proliferation-associated antigen Ki67 (Dianova, Hamburg, Germany) dual-color digital confocal laser microscopy on paraffin-embedded sections was performed. Pretreatment of the deparaffinized slides was performed as mentioned above. The Lptn mAb was applied first, incubated overnight in a humidified chamber at 4°C, and detected by using the biotin-tyramide signal amplification system (Dupont-NEN): biotinylated secondary antibodies (goat anti-rabbit antibody conjugated with biotin, DAKO) were diluted in TNB (1:500) and applied for 30 minutes at room temperature. After washing three times in TBS slides were incubated in streptavidin-horseradish peroxidase complex (diluted 1:100 in TNB) for an additional 30 minutes. Three washes with TBS were followed by application of biotin-tyramide diluted 1:100 in amplification diluent (Dupont-NEN) for 5 minutes. Reactive sites were detected with streptavidin-fluorescein isothiocyanate (1:50 in TBS, 30 minutes at room temperature). Subsequently, the polyclonal antibody CD3 and the mAbs CD4, CD8, CD31, CD45RA, CD103, Ki67, and tryptase were applied overnight at 4°C. The mAbs were detected by indirect immunofluorescence using rhodamine-Red-X-conjugated F(ab)-fragments of rabbit anti-mouse Ig (1:50, Dianova). CD3 was detected by application of rhodamine-Red-X-conjugated goat anti-rabbit Ig (1:50, Dianova). After each incubation the sections were thoroughly washed in TBS and embedded in fluoromount fluorescent mounting media before examination to prevent fading (DAKO). Irrelevant antibodies or omission of first and second step antibodies served as negative controls. Analysis of fluorescence signals was performed on a Leica TCS NT laser scanning microscope (Heidelberg, Germany).
Separation of Monocytes and T Cells from Blood
Monocytes and T lymphocytes were obtained by Ficoll (Biochrom, Berlin, Germany) gradient centrifugation of peripheral mononuclear cells separated from the buffy coats of blood samples from healthy volunteers. For positive selection of interphase cells the mAbs anti-CD14 and anti-CD3 directly coupled to magnetic beads were used for enrichment of monocytes and T lymphocytes, respectively (all Miltenyi Biotec, Bergisch-Gladbach, Germany). All selection steps were repeated once. Separated cells were >95% pure as determined by flow cytometry (data not shown).
T-Cell Stimulation Experiments
For cell stimulation experiments T cells were cultured at a density of 10 6 T cells/ml RPMI 1640 culture medium supplemented with 10% fetal calf serum, 2 μmol/L l-glutamine, and gentamicin (50 μg/ml), further referred as complete culture medium, in the presence of 5% CO2 at 37°C. Cells were stimulated for 12 hours with phorbol-12-myristate-13-acetate (PMA, 50 ng/ml) or concanavalin A (ConA, 200 μg/ml). Dexamethasone (1 μmol/L), cyclosporine A (500 ng/ml), and FK506 (100 ng/ml) were added immediately before stimulation. To investigate a possible regulatory effect of Lptn itself on its expression by activated T cells, a blocking anti-human Lptn antibody (TEBU, Frankfurt, Germany) was supplemented to the cultures at a neutralizing concentration of 10 μg/ml of complete culture medium. The expression of Lptn and GPR-5 was determined by real-time quantitative RT-PCR analysis as described above.
Dendritic Cell Differentiation
For the generation of mature DCs, CD14-positive selected monocytes were cultured as described previously. 23 Briefly, the monocytes were plated in 6-well tissue-culture plates at a density of 2 × 10 6 cells/well in 4 ml of complete culture medium. Granulocyte-macrophage colony-stimulating factor and interleukin (IL)-4 were added at final concentrations of 800 and 1000 U/ml, respectively. Cultures were fed on days 2 and 4 by removing 1 ml of the medium and adding back 1.5 ml of fresh medium with cytokines (1600 U/ml granulocyte-macrophage colony-stimulating factor and 1000 U/ml IL-4). Cells were harvested on day 6 and transferred into new wells and cultured for 1 more day with fresh granulocyte-macrophage colony-stimulating factor and IL-4 supplemented with 25% monocyte-conditioned media, which was prepared by culturing 10 7 peripheral blood mononuclear cells on immunoglobulin-coated bacteriological plates for 1 hour at 37°C in 8 ml of complete culture medium. The DCs harvested at day 7 proved to be fully mature as determined by their strong expression of the CD83 antigen, the expression of which ranged from 55 to 85% of the population as shown by fluorescence-activated cell sorting analysis (data not shown). Total mRNA of DCs at different stages of maturation was analyzed for expression of Lptn by RT-PCR as described above.
Statistical Analysis
Statistical data analysis was performed using the InStat statistical program (GraphPad-Software, San Diego, CA). Statistical differences were evaluated by unpaired Student’s t-test. P values were determined by a two-sided calculation. Results were considered to be statistically significant if P values were <0.05.
Results
Quantitative RT-PCR of Lptn mRNA in CD
Lptn mRNA expression was analyzed in tissue samples affected by active CD (n = 6) and from normal controls without signs of inflammation (n = 6) by RT-PCR with oligonucleotide primers specific for the human Lptn cDNA. As internal control RT-PCR for the housekeeping gene β-actin was performed. Lptn-specific products could be amplified from mRNA probes isolated from all tissue samples investigated.
For quantification of Lptn mRNA expression in tissue samples a method of real-time quantitative RT-PCR was established using iCycler technology. Lptn mRNA levels of equal amounts of total RNA from the tissue samples were calculated by reference to a standard curve, which was obtained by one-step RT-PCR containing different amounts of in vitro generated Lptn cRNA (Lptn standards), showing a dilution range from 1 to 1000 attomoles (Figure 1, A and B) ▶ . Quantification results were given as attomoles per μg total cellular RNA of each investigated tissue sample. In the total RNA isolated from tissue with active CD a significant increased expression of Lptn mRNA could be determined in comparison to RNA samples obtained from bowel tissue without signs of inflammation (P < 0.001). In RNA samples from CD Lptn mRNA was found to be approximately eightfold more expressed than in controls (Figure 1C) ▶ .
Figure 1.

Quantitative real-time RT-PCR analysis of Lptn mRNA. Total RNA was analyzed for Lptn mRNA expression by quantitative RT-PCR on RNA from tissue samples affected by CD (n = 6) and not inflammatory affected bowel (n = 6). The amount of Lptn mRNA expression per μg total mRNA was derived from a standard curve using increasing amounts of in vitro generated Lptn cRNA. Threshold cycle values of Lptn-specific amplification products were deducted from changes in SYBR Green fluorescence (RFU) during each PCR cycle for Lptn standards and Lptn samples from CD (A, left) and control (A, right). The threshold (dashed line) was set according to RFU variations in early PCR cycles. The calibration curve was derived from threshold cycle values (shown in A) for each known amounts of Lptn cRNA standard (1, 10, 100, and 1000 attomoles) (B). Lptn mRNA expression was found to be significantly elevated in CD when compared to normal controls (C, P < 0.001). Data are shown from one of three independent experiments.
Distribution and Cellular Characterization of Lptn-Expressing Cells
As Lptn mRNA expression was shown to be increased in active CD the results of quantitative RT-PCR were confirmed by nonradioactive in situ hybridization using digoxigenin-labeled cRNA probes and by immunohistochemistry with a Lptn-specific antibody.
In control tissue specimens only few Lptn-positive cells could be observed. Cells expressing Lptn mRNA and protein were almost restricted to the lamina propria (Figure 2, a and b) ▶ . In situ hybridization experiments and immunostainings on tissue specimens from CD revealed numerous Lptn-positive cells within the inflammatory infiltrate of the entire bowel wall. In the mucosa numerous positive cells could be observed within the lymphocytic infiltrates of the lamina propria and additionally within the epithelium (Figure 2, c and d) ▶ . Furthermore, numerous Lptn-positive cells were observed adjacent to blood vessels. The perivascular pattern was predominantly found in the deeper layers of the bowel wall comprising the submucosa, the muscularis propria, and the subserosal tissue (Figure 2e) ▶ . Granulomas, occasionally observed in CD, did not show any expression of Lptn mRNA or protein by epithelioid cells or giant cells, which was in contrast to the strong intracytoplasmic Lptn expression by surrounding lymphoid cells (Figure 2f) ▶ . Furthermore, a clear relationship was observed between the increased numbers of Lptn-expressing cells and the extent of lymphocytic infiltration in CD versus controls without significant signs of inflammation.
Figure 2.
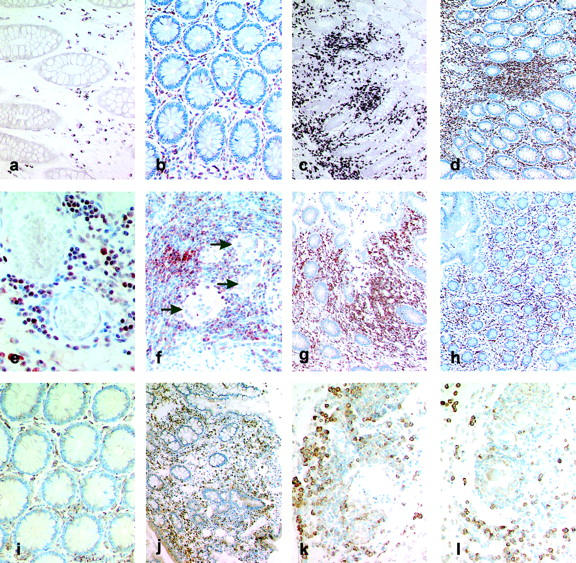
Localization of Lptn and GPR-5 expression in CD. a–h: Immunohistochemistry and mRNA in situ hybridization for Lptn. In control tissue samples of the large bowel Lptn mRNA (a) and protein (b) expression was found to be restricted to a few lymphocytes within the lamina propria. In tissue specimens affected by CD increased numbers of cells expressing Lptn mRNA (c) and protein (d) were found predominantly within lymphocytic aggregates of the lamina propria and submucosa. Within the subserosa Lptn-positive cells were predominantly found in a perivascular pattern (e, immunohistochemistry). Giant cells (arrows) or epithelioid cells of granulomas did not show any Lptn protein expression in contrast to surrounding T cells (f). Biopsy specimens obtained from a patient with active CD (severity activity index) demonstrated an increased number of Lptn-positive cells (g, immunohistochemistry), which were found to be significantly reduced on therapy with systemic steroids (severity activity index, 35) (h, immunohistochemistry). i–l: In situ hybridization for GPR-5 mRNA. In noninflammatory-affected bowel the expression of GPR-5 mRNA was restricted to cells within the lamina propria (i). In CD an increased number of GPR-5 mRNA-positive cells were found within the lamina propria (j), whereas cells with the strongest intracytoplasmic expression for GPR-5 mRNA were found adjacent to submucosal vessels (k) and granulomas (l). Original magnifications: ×100 (c, d, g, h, j); ×200 (a, b, f, i, k, l); ×400 (e).
To investigate the modulation of Lptn expression under therapy additional immunostainings were performed on biopsy specimens from a patient taken at the time of active CD and after therapy with systemic steroids (prednisolone 10 mg/day). The biopsy specimen taken at the time point of active CD (severity activity index of Goebell, 285; Figure 2g ▶ ) showed a significant increased number of Lptn-positive cells within the lamina propria, which were found to be significantly reduced in remission on therapy with steroids 8 months later (severity activity index, 35; Figure 2h ▶ ). However, these data have to be confirmed by further cases.
To characterize the phenotype of inflammatory cells expressing Lptn mRNA or protein combined nonradioactive in situ hybridization combined with immunofluorescence or double-immunofluorescence investigations were performed. The great majority of Lptn-positive cells could be identified as CD3+ T cells (Figure 3a ▶ and Figure 4a ▶ ) comprising both the CD4+ and CD8+ subtype (Figure 3, c and d) ▶ . Other cells expressing Lptn were identified as mostly perivascular located CD117/c-Kit-positive mast cells (Figure 3e) ▶ and in addition occasionally S100 protein-positive DCs (Figure 3f) ▶ . The latter were mostly observed within the upper part of the lamina propria or in the vicinity of subserosal vessels. By extensive further investigations no expression of Lptn mRNA or protein was found by other cell types such as natural killer cells (detected by mAb CD57), monocytes/macrophages, or granuloma histiocytes (detected by mAb Ki-M1P; Figure 3b ▶ ), and B-lymphocytes (detected by mAb CD20). Furthermore, no expression was found by epithelial cells of the mucosa or granulocytes as determined by morphology.
Figure 3.
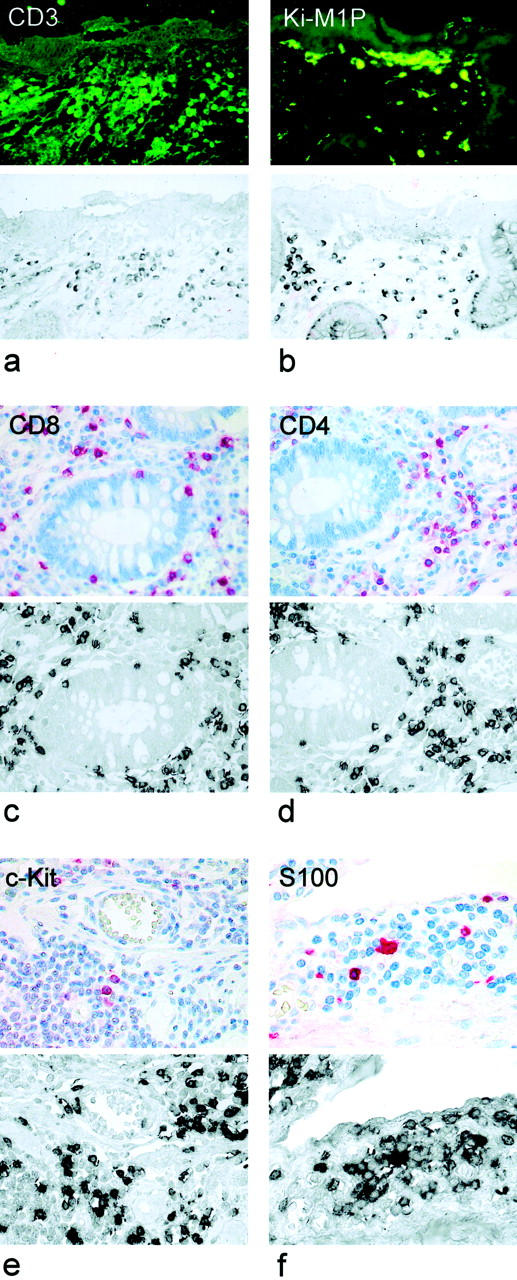
Cellular expression of Lptn in CD. Lptn mRNA was detected by nonradioactive in situ hybridization combined with indirect immunofluorescence or by immunostaining of serial sections. The majority of Lptn mRNA-positive cells were found to be T cells showing co-expression for CD3 (a), whereas no expression was found by Ki-M1P-positive monocytes or macrophages (b). CD4- and CD8-positive T cells express Lptn mRNA (c and d). In addition, Lptn mRNA could be identified in perivascular c-Kit-positive mast cells (e) and S100-positive DCs (f). Original magnifications: ×400 (a–e); ×600 (f).
Figure 4.

Lptn expression by immigrating and proliferating T cells in CD. Two-color immunofluorescence was used to determine the expression of Lptn (fluorescein isothiocyanate, green fluorescence) by double labeling with different antibodies (rhodamine-red X, red fluorescence) for CD3 (a), the CD103 (b), CD31 (c), CD45RA (d), and Ki67 (e). Lptn-positive cells proved to express the pan T-cell antigen CD3 (a) and the gut homing molecule CD103 (b). Perivascular-located Lptn-positive cells were often positive for CD31 (c) and CD45RA (d), characteristic for immigrating naïve T cells. Regular expression for CD31 was found on blood vessels (arrow, c). Additionally, Lptn expression could be observed by Ki67-positive cells (e) representing both CD4+ (f, arrows) and CD8+ (g, arrows) T cells. Nonspecific staining was excluded by negative controls (h). Original magnifications, ×600.
By double-label immunofluorescence we found the Lptn-positive cells to express the gut-homing molecule CD103 (Figure 4b) ▶ . Furthermore, numerous predominantly perivascular-located cells expressed the antigens CD31 (Figure 4c) ▶ and CD45RA (Figure 4d) ▶ thus representing immigrating naïve inflammatory cells. Additionally, Lptn-positive cells proved to express the proliferation-associated antigen Ki67 (Figure 4e) ▶ thus comprising predominantly proliferating CD4+ or CD8+ T cells within the inflammatory infiltrate (Figure 4, f and g) ▶ . In parallel, performed negative controls did not show any specific staining pattern (Figure 4h) ▶ .
Expression of GPR-5 mRNA in CD
The expression of GPR-5 mRNA was investigated by nonradioactive in situ hybridization. In controls GPR-5 mRNA expression was found to be restricted to cells of the lamina propria (Figure 2i) ▶ . In CD numerous GPR-5 mRNA-positive cells could be observed within the inflammatory infiltrate of the entire bowel wall. Within the mucosa increased numbers of positive cells were found within the lymphocytic infiltrates of the lamina propria (Figure 2j) ▶ . Occasionally, expression of GPR-5 mRNA could be observed by intraepithelial lymphocytes. Furthermore, abundant GPR-5 mRNA could be observed in cells adjacent to blood vessels (Figure 2k) ▶ and in lymphocytes surrounding granulomas within the deeper layers of the inflammatory affected bowel. No expression of GPR-5 mRNA could be observed by epithelioid cells, giant cells (Figure 2l) ▶ , and epithelial cells.
The comparison of biopsy specimens before and after conservative therapy with steroids demonstrated comparable results to those obtained for Lptn: in nonactive CD a significant reduced number of GPR-5 mRNA-positive cells could be found compared to the increased number of GPR-5 mRNA-positive cells observed within the inflammatory affected mucosa from active CD (data not shown).
Regulation of Lptn mRNA Expression by T Cells in Vitro
Quantitative real-time RT-PCR was performed on unstimulated or Con A-stimulated or PMA-stimulated CD3-positive selected T cells from peripheral human blood. Freshly isolated T cells demonstrated a very weak expression of Lptn mRNA expression, which was strongly enhanced by addition of Con A or PMA, whereby PMA was more powerful by inducing Lptn mRNA expression than ConA. The addition of a neutralizing Lptn antibody at saturating concentrations during the culture period did not show any regulatory effect on the expression of Lptn mRNA by T cells either without or with stimulation by ConA or PMA (data not shown).
Dexamethasone, FK506, and cyclosporine A are powerful immunosuppressive drugs, which are successfully used for therapy in CD. Their therapeutic effect is partly mediated by inhibiting transcriptional activation of different cytokine genes such as of the IL-2 gene. 24 Addition of FK506 and cyclosporine A prevented the observed up-regulation of Lptn mRNA by PMA, whereas dexamethasone led to a decrease of Lptn mRNA expression below the amount of Lptn mRNA expression of untreated T cells (Figure 5A) ▶ .
Figure 5.

Expression of Lptn and of the Lptn receptor GPR-5 by in vitro-stimulated T cells. By quantitative real-time RT-PCR both Lptn (A) and the Lptn receptor GPR-5 (B) were found to be up-regulated on stimulation with ConA and PMA in CD3+ T cells isolated from peripheral blood. Addition of the immunosuppressive-acting drugs cyclosporine A (CyA), FK506, and dexamethasone (Dexa) prevented the up-regulation of Lptn and Lptn receptor GPR-5 by PMA in a similar manner.
In further experiments the expression of the Lptn receptor GPR-5 was analyzed by real-time RT-PCR. Unstimulated T cells demonstrated weak amplification products for GPR-5 mRNA, but under stimulation with ConA and PMA a dramatic increase in GPR-5 mRNA expression was observed. As shown for Lptn simultaneous application of the immunosuppressive drugs dexamethasone, FK506, and cyclosporine prevented PMA mediated up-regulation (Figure 5B) ▶ .
Lptn mRNA Expression by Monocyte-Derived Dendritic Cells in Vitro
Because of the unexpected expression of Lptn mRNA by small numbers of S100-positive DCs, monocyte-derived DCs were generated in vitro. 23 Lptn mRNA expression was investigated at different stages of DC differentiation and maturation. RT-PCR analysis of freshly isolated monocytes and day 3-harvested immature DCs showed no expression of Lptn mRNA. First Lptn-specific amplification products became detectable on day 5 of in vitro differentiation, the expression of which increased until day 7, when full maturation with conditioned media was induced (Figure 6) ▶ . The results demonstrate a maturation-dependent expression of Lptn mRNA in DCs.
Figure 6.

Lptn mRNA expression by DCs. Monocyte-derived DCs were generated from highly purified CD14+ monocytes by culture with granulocyte-macrophage colony-stimulating hormone and IL-4. CD14+ monocytes and early day (d) 3 immature DCs showed no Lptn-specific amplification products by RT-PCR, which was in contrast to mRNA isolated from DCs harvested on days 5 and 7, representing late-stage immature (day 5) and mature DCs (day 7), respectively. Quality of cDNA was checked by amplification for β-actin (bottom lane). As positive control served total mRNA from a human lymph node. For negative control water was used as template.
Discussion
The inflammatory infiltrate in CD is characterized by an increased number of T cells, which belong either to the freshly immigrated CD45RA+ subtype representing naive T cells or to T cells expressing CD45RO representing the memory subtype. 2 It is hypothesized that cell renewal by immigration of naive T cells into the inflamed bowel wall plays an important mechanism in the perpetuation of the inflammatory process observed in CD. 2 The precise mechanisms, however, that are responsible for the recruitment of both naive and memory T cells in CD remain unclear. To investigate the potential role of Lptn, a potent chemokine that predominantly attracts T cells, the expression of Lptn mRNA and protein in CD was investigated; cell types expressing Lptn in CD were further characterized.
Our results show a statistically significant increase of Lptn mRNA expression in CD versus normal controls (P < 0.001) as determined by quantitative real-time RT-PCR analysis (Figure 1) ▶ . The amount of Lptn mRNA per μg total RNA was found to be approximately eight times higher in CD than in controls. These results could be confirmed by in situ hybridization experiments for Lptn mRNA. In CD increased numbers of Lptn mRNA and protein-expressing cells were found within the entire inflammatory affected bowel wall (Figure 2) ▶ . No differences could be observed by comparing the staining pattern of Lptn mRNA by in situ hybridization or protein by immunohistochemistry. A comparable distribution could be observed for the Lptn receptor GPR-5 in CD and controls as determined by in situ hybridization (Figure 2) ▶ . Cells demonstrating the most abundant GPR-5 mRNA expression were predominantly located adjacent to blood vessels, a finding that is in accordance to the in vitro observed chemoattractive function of Lptn.
By applying simultaneous immunostaining of serial sections we found most of these Lptn-positive cells to express the T-cell antigen CD3, which comprised by subset analysis both CD4- and CD8-positive T cells (Figures 3 and 4) ▶ ▶ . Furthermore, we found the Lptn-positive cells to express the integrin CD103, which is known to play a key role for homing of inflammatory cells such as T cells into the gut. In addition, numerous Lptn-positive cells demonstrated expression of the antigens CD31 and CD45RA (Figure 4) ▶ , which have been shown to be expressed by naive T cells in CD. 2 Because CD31/CD45RA and Lptn co-expressing T cells were found predominantly in a perivascular pattern, these cells represent newly immigrated inflammatory cells.
By additional investigations we found a substantial number of Lptn-positive cells to express the proliferation-associated antigen Ki67 in CD. The proliferating T cells were found to represent both the CD4+ and CD8+ subset (Figure 4) ▶ . Thus, active proliferation of Lptn-positive cells is at least in part responsible for the observed increase of Lptn-expressing cells in CD.
More recently, different abilities of Lptn have been demonstrated on proliferation and stimulation of CD4+ and CD8+ T cells. Lptn could be shown to inhibit CD4+ T-cell proliferation in vitro through a decreased production of Th1 (IL-2, interferon-γ) but not Th2 (IL-4, IL-13) cytokines. In contrast, Lptn exerted a potent stimulatory effect on CD8+ T-cell proliferation and IL-2 secretion. CD4+ T-cell proliferation was completely restored by addition of IL-2. 25 Because the inflammatory process in CD represents a Th1-mediated immune response reflected by an increased expression of interferon-γ, tumor necrosis factor-α, and IL-2, increased expression of IL-2 might compensate the inhibitory effect of Lptn on CD4+ T-cell proliferation in CD as observed by us.
The observed expression of Lptn mRNA or protein by CD4- and CD8-positive T cells is in accordance with previous reports demonstrating that both activated T-cell subsets represent the most important source of Lptn. 14 In addition, Lptn production has been detected in αβ-type thymocytes 8,9 and murine intraepithelial γδ-type T cells. 17 The latter finding seems to be of special importance, because Lptn was found to be the most abundant chemokine produced by intraepithelial γδ T cells, peripheral blood γδ T cells did not express Lptn at all in mice. 17 This latter finding is in accordance with our finding of intraepithelial lymphocytes demonstrating expression of Lptn in CD but not in controls.
The control of Lptn gene expression is not well understood. Although it is clear that expression is essentially activation-dependent, there are only a few studies that have examined this in detail. For murine T cells it could be shown that Th1 clones and CD8+ T cells express Lptn maximally on engagement of the T cell receptor and that co-stimulation through CD28 inhibited expression. 26 In humans this effect seems to be restricted to the CD4+ subpopulation, whereas CD8+ T cells are resistant. In our study Con A and PMA induced up-regulation of Lptn mRNA expression in T cells isolated from peripheral blood, showing that signals either provided by T-cell receptor engagement (Con A) or directly by activation of protein kinase C (PMA) induce the expression of Lptn as observed in CD (Figure 5) ▶ . Unexpectedly, we found an increased expression of Lptn in unstimulated T cells after 24 hours in culture, which may be because of the separation method used in this study by applying CD3-positive selection. CD3 itself has been shown to activate T cells by T-cell receptor ligation. 25 Furthermore it has been shown that T-cell stimulation by CD3 caused a significant up-regulation of Lptn. 7 Thus, the T cells investigated by us were at least partly activated by CD3 stimulation leading to an increased basal Lptn mRNA expression. To investigate possible effects of Lptn by regulating its expression via an autocrine feedback loop, additional experiments applying a neutralizing polyclonal antibody at saturating concentrations during the culture period were performed. In these specific blocking experiments no significant modulation on the expression of Lptn mRNA in T cells could be observed. Therefore, an autocrine regulation mechanism controlling the expression of Lptn seems not to be likely. This conclusion is further supported by our results obtained in tissue specimens in CD demonstrating that almost the entire T-cell population expressed Lptn.
Beside glucocorticosteroids the immunosuppressive drugs FK 506 and cyclosporine A are used for the therapy of CD. We found all these three agents to prevent completely the PMA mediated up-regulation of Lptn mRNA by T cells (Figure 5) ▶ . This finding is in agreement with the observation by Wang and colleagues, 3 who demonstrated Lptn expression by activated lymphoid cells in the acute phase of rejection in a rat model of renal graft rejection, whereas no expression of Lptn mRNA was observed in chronic rejection; expression of Lptn was found to depend on the activation of lymphocytes and could be down-regulated by cyclosporine A and FK506. A similar regulation mechanism on stimulation and inhibition by immunosuppressive drugs as for Lptn could be shown in peripheral T cells for its receptor GPR-5 (Figure 5) ▶ . Thus, the expression of Lptn and its receptor seems to be tightly linked to activation of T cells, which points to a possible autocrine or paracrine regulation mechanism of the recruitment of activated T cells in autoimmune disease such as CD. This finding is further supported by the observed decrease in the number of cells expressing Lptn and its receptor GPR-5 in biopsy specimens of CD after sufficient therapy by systemic steroids as shown in this study (Figure 2) ▶ .
Other cells expressing Lptn mRNA were identified as mast cells or DCs (Figure 3) ▶ . Expression of Lptn mRNA could not be observed by other cell types including granulocytes, natural killer cells, B-lymphocytes, monocytes/macrophages, or fibroblasts as also shown by other authors. 27
The capacity of mast cells to produce Lptn provides a possible link between mast cell activation and recruitment of T cells. Particularly, because mast cells are predominantly found in the vicinity of blood vessels, Lptn expressed by these cells may function in the recruitment of T cells from the blood stream. Furthermore, mast cells have been shown to express a wide range of chemokines, some of which possess chemotactic activities for T cells such as RANTES, IL-8, IL-16, MCP-1, macrophage inflammatory protein-1α, and macrophage inflammatory protein-1β. 28 Recently, the expression of Lptn by mast cells as well as by the mast cell line HMC-1 and the basophil cell line kU812 has been examined in vitro. 18 It could be demonstrated that expression of Lptn was dependent on aggregation of the FcεRI and that its expression was inhibited by cyclosporine A and dexamethasone as described for T cells in this study.
Interestingly, we observed by careful examination expression of Lptn mRNA by DCs in CD (Figure 3) ▶ . To confirm this result further experiments were performed with in vitro-generated monocyte-derived DCs. No amplification products for Lptn mRNA could be found in CD14+ monocytes and immature DCs on day 3. Lptn mRNA expression appeared after 5 to 7 days of culturing, when DC maturation has occurred (Figure 6) ▶ . Thus, Lptn mRNA expression seems to be restricted to the mature stages of DCs. This finding is in accordance with our observations that functionally immature DCs such as the Langerhans cells of the epidermis do not show any Lptn mRNA expression, whereas interdigitating DCs within the lymph node paracortex, which represent fully mature T cell accessory DCs, demonstrate expression of Lptn mRNA (data not shown). Furthermore, we found expression of Lptn within some germinal centers probably representing germinal center DCs, an observation that has also been suggested previously. 27
More recently, the obvious capability of Lptn to induce recruitment of T cells in immune response 29 has successfully been used in a model of tumor immune therapy in mice. Mouse bone marrow-derived DCs were genetically modified with the Lptn gene by adenovirus vector-mediated transfection. Immunization with tumor RNA-pulsed Lptn-modified DCs improved the therapeutic efficacy of tumor rejection by enhanced chemotaxis of DCs to CD4+ and CD8+ T cells resulting in a higher efficiency of antigen peptide delivery for induction of T-cell-mediated tumor rejection. 30,31 These results provide evidence that induction of expression and release of Lptn by mature DCs would not only represent a powerful tool for novel therapy approaches using DCs for tumor vaccination, but also point to the possible Lptn-mediated chemotactic properties of mature DCs in chronic inflammatory diseases such as CD.
The results indicate that increased expression of Lptn by activated T cells, mast cells, and DCs in CD seems to provide a powerful chemotactic signal for circulating T lymphocytes; persistent expression of Lptn at sites of inflammation seems to be involved in the continuous recruitment of peripheral T cells from the blood stream expressing the Lptn receptor. Down-regulation mRNA expression of Lptn and its receptor in T cells by dexamethasone, FK506, and cyclosporine A represents a possible mechanism preventing activation of T cells and being responsible for the beneficial effects of these immunomodulatory-acting drugs in CD. It seems worthwhile to look for Lptn expression in other autoimmune diseases that are characterized by increased recruitment of activated T cells such as sarcoidosis, rheumatoid arthritis, or atopic dermatitis.
Acknowledgments
We thank Mrs. Maren Reffelmann for excellent technical help.
Footnotes
Address reprint requests to Dr. med. Peter Middel, Department of Pathology, Georg-August-University of Göttingen, Robert-Koch-Str. 40, 37075 Göttingen. E-mail: pmiddel@med.uni-goettingen.de.
References
- 1.Whithead R: Pathology of Crohn’s disease. Kirsner J Shorter R eds. Inflammatory Bowel Disease. 1980, :pp 296-307 Lea & Febiger, Philadelphia [Google Scholar]
- 2.Burgio V, Fais S, Boirivant M, Perrone A, Pallone F: Peripheral monocyte and naive T-cell recruitment and activation in Crohn’s disease. Gastroenterology 1995, 109:1029-1038 [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Nonomura N, Takahara S, Li B, Azuma H, Ichimaru N, Kokado Y, Matsumiya K, Miki T, Suzuki S, Okuyama A: Lymphotactin: a key regulator of lymphocyte trafficking during acute graft rejection. Immunology 1998, 95:56-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rollins B: Chemokines. Blood 1997, 90:909-929 [PubMed] [Google Scholar]
- 5.Oppenheim J, Zachariae C, Mukaida N, Matsushima K: Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol 1991, 9:617-648 [DOI] [PubMed] [Google Scholar]
- 6.Bagglioni M, Dewald B, Moser B: Interleukin-8 and related chemotactic cytokines CXC- and CC-chemokines. Adv Immunol 1994, 55:97-179 [PubMed] [Google Scholar]
- 7.Olive D, Cerdan C: CD28 co-stimulation results in down-regulation of lymphotactin expression in human CD4+ but not CD8+ T cells via an IL-2-dependent mechanism. Eur J Immunol 1999, 29:2443-2453 [DOI] [PubMed] [Google Scholar]
- 8.Kelner G, Kennedy J, Bacon K, Kleyensteuber S, Largaespada D, Jenkins N, Copeland NG, Bazan J, Moore K, Schall T, Zlotnik A: Lymphotactin: a cytokine that represents a new class of chemokine. Science 1994, 266:1395-1399 [DOI] [PubMed] [Google Scholar]
- 9.Müller S, Dorner B, Karthäuser U, Mayes H, D’Apuzzo M, Senger G, Kroczek R: Cloning of ATAC, an activation-induced chemokine-related molecule exclusively expressed in CD8+ T lymphocytes. Eur J Immunol 1995, 25:1744-1748 [DOI] [PubMed] [Google Scholar]
- 10.Yoshida T, Imai T, Kakizuki M, Yoshie O: Molecular cloning of a novel C or γ chemokine, SCM-1. FEBS Lett 1995, 360:155-159 [DOI] [PubMed] [Google Scholar]
- 11.Bazan J, Bacon K, Hardiman G, Wang W, Soo K, Rossi D, Greaves D, Zlotnik A, Schall T: A new class of membrane-bound chemokine with CX3C motif. Nature 1997, 385:640-644 [DOI] [PubMed] [Google Scholar]
- 12.Kennedy J, Kelner G, Kleyensteuber S, Schall T, Weiss M, Yssel H, Schneider P, Cocks B, Bacon K, Zlotnik A: Molecular cloning and functional characterization of human lymphotactin. J Immunol 1995, 155:203-209 [PubMed] [Google Scholar]
- 13.Bianchi G, Sozzani S, Zlotnik A, Montovani A, Allavena P: Migratory response of human natural killer cells to lymphotactin. Eur J Immunol 1996, 26:3238-3241 [DOI] [PubMed] [Google Scholar]
- 14.Dorner B, Müller S, Entschladen F, Schröder J, Franke P, Kraft R, Friedl P, Clark-Lewis I, Kroczek R: Purification, structural analysis, and function of natural ATAC, a cytokine secreted by CD8+ T cells. J Biol Chem 1997, 272:8817-8823 [DOI] [PubMed] [Google Scholar]
- 15.Hedrick J, Saylor S, Figueroa D, Mizoue L, Xu Y, Menon S, Abrams J, Handel T, Zlotnik A: Lymphotactin is produced by NK cells and attracts both NK cells and T cells in vivo. J Immunol 1997, 158:1533-1540 [PubMed] [Google Scholar]
- 16.Maghazachi A, Skalhegg B, Rolstad B, Al-Aoukaty A: Interferon-inducible protein-10 and lymphotactin induce the chemotaxis and mobilization of intracellular calcium in natural killer cells through pertussis toxin-sensitive and -insensitive heterotrimeric G-proteins. FASEB J 1997, 11:765-774 [DOI] [PubMed] [Google Scholar]
- 17.Boismenu R, Feng L, Xia Y, Chang J, Havran W: Chemokine expression by intraepithelial γδ T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J Immunol 1996, 157:985-992 [PubMed] [Google Scholar]
- 18.Rumsaeng V, Vliagoftis H, Oh C, Metcalfe D: Lymphotactin gene expression in mast cells following Fc (ε) receptor I aggregation: modulation by TGF-b, IL-4, dexamethasone, and cyclosporine A. J Immunol 1997, 158:1353-1360 [PubMed] [Google Scholar]
- 19.Yoshida T, Imai T, Kakizaki M, Nishimura S, Takayi S, Yoshie O: Identification of single-motif-1/lymphotactin receptor XCR1. J Biol Chem 1998, 273:16551-16554 [DOI] [PubMed] [Google Scholar]
- 20.Yoshida T, Izawa D, Nakayama T, Nakahara K, Kakizaki M, Imai T, Suzuki R, Miyasaka M, Yoshie O: Molecular cloning of mXCR1, the murine SCM-1/lymphotactin receptor. FEBS Lett 1999, 458:37-40 [DOI] [PubMed] [Google Scholar]
- 21.Breitschopf H, Suchanek G, Gould R, Colman D, Lassmann H: In situ hybridization with digoxigenin-labeled probes: sensitive and reliable detection method applied to myelinating rat brain. Acta Neuropathol 1992, 84:581-587 [DOI] [PubMed] [Google Scholar]
- 22.Radzun H, K Hansmann M, Heidebrecht H, Bödewadt-Radzun S, Wacker H, Kreipe H, Lumbeck H, Hernandez C, Kuhn C, Parwaresch M: Detection of a monocyte/macrophage differentiation antigen in routinely processed paraffin-embedded tissues by monoclonal antibody Ki-M1P. Lab Invest 1991, 65:306-315 [PubMed] [Google Scholar]
- 23.Bender A, Sapp M, Schuler G, Steinmann R, Bhardwai N: Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods 1996, 196:121-135 [DOI] [PubMed] [Google Scholar]
- 24.Adorini L, Guery J, Rodriguez-Tarduchy G, Trembleau S: Selective immunosuppression. Trend Pharmacol Sci 1993, 14:285-289 [DOI] [PubMed] [Google Scholar]
- 25.Cerdan C, Serfling E, Olive D: The C-class chemokine, lymphotactin, impairs the induction of Th1-type lymphokines in human CD4+ T cells. Blood 2000, 96:420-428 [PubMed] [Google Scholar]
- 26.Hautamaa D, Merica R, Chen Z, Jenkins M: Gene structure, post-translational modification and inhibition of expression by CD28 costimulation. Cytokine 1997, 9:375-382 [DOI] [PubMed] [Google Scholar]
- 27.Hedrick JA, Zlotnik A: Molecule of the month: lymphotactin. Clin Immunol Immunopathol 1998, 87:218-222 [DOI] [PubMed] [Google Scholar]
- 28.Mekori Y, Metcalfe D: Mast cell T cell interactions. J Allergy Clin Immunol 1999, 104:517-523 [DOI] [PubMed] [Google Scholar]
- 29.Borthwick N, Akbar A, Maccormac L, Lowdell M, Craigen J, Hassan I, Grundy J, Salmon M, Yong K: Selective migration of highly differentiated primed T cells, defined by low expression of CD45RB, across human umbilical vein endothelial cells: effects of viral transfection on transmigration. Immunology 1997, 90:272-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao X, Zhang W, He L, Xie Z, Ma S, Tao Q, Yu Y, Hamada H, Wang J: Lymphotactin gene-modified bone marrow dendritic cells act as more potent adjuvans for peptide delivery to induce specific antitumor immunity. J Immunol 1998, 161:6238-6244 [PubMed] [Google Scholar]
- 31.Zhang W, He L, Yuan Z, Xie Z, Wang J, Hamada H, Cao X: Enhanced therapeutic efficacy of tumor RNA-pulsed dendritic cells after genetic modification with lymphotactin. Hum Gene Ther 1999, 10:1151-1161 [DOI] [PubMed] [Google Scholar]