

Abstract
Using angiotensin II (AngII) type 1A receptor-deficient mice [AT1(−/−)], in which we induced protein overload nephropathy, we explored the potential implication of AngII and endothelin-1 (ET-1) in the tubulointerstitial damage because of persistent proteinuria. At day 7, AT1(−/−) showed marked proteinuria to a similar extent to that of wild-type mice (WT). However, at day14, AT1(−/−) had significantly less proteinuria, renal damage, transforming growth factor-β, and matrix mRNA expression and mortality. AT1(−/−) also showed a significant diminution in the activation of the transcriptional factors nuclear factor-κB and AP-1. Unexpectedly, AT1(−/−) had a higher interstitial infiltration than WT. The administration of the angiotensin-converting enzyme inhibitor quinapril to WT caused a marked improvement in proteinuria and renal lesions, resembling that seen in untreated AT1(−/−). However, the interstitial infiltration persisted in AT1(−/−) when treated with quinapril. Because ET-1 may participate in the recruitment of mononuclear cells, we also studied the implication of this peptide. AT1(−/−) had a significantly higher ET-1 expression in tubular epithelial cells than WT. The administration of the dual ETA/ETB antagonist bosentan to AT1(−/−) considerably reduced the interstitial infiltrates. Bosentan also exerted a beneficial effect on proteinuria, renal lesions, and mortality in WT. These data show that in overload nephropathy, proteinuria and renal lesions are, to a large extent, AngII-dependent. The up-regulation of ET-1 in tubular epithelial cells in AT1(−/−), associated with interstitial infiltrates, suggests that the combination of drugs interfering with both vasopeptides may be of therapeutic interest in renal diseases with severe proteinuria and tubulointerstitial damage.
Throughout the last few decades, renoprotective effects of renin-angiotensin system (RAS) blockade with angiotensin-converting enzyme (ACE) inhibitor or angiotensin II type 1 receptor (AT1) antagonists have been confirmed by extensive experimental and clinical studies. 1-7 Early evidence suggested that the beneficial effect was because of the control of glomerular hypertension. 1,8,9 In addition, recent studies have emphasized that locally generated angiotensin II (AngII) modifies resident cell growth and possesses proinflammatory properties, and may therefore participate directly in the pathogenesis of renal injury beyond its hemodynamic effect. 10,11
The prognosis of chronic renal injury, regardless its etiology, is closely correlated to the severity of tubulointerstitial damage. 12,13 Although persistent proteinuria can induce tubulointerstitial fibrosis, 14-16 underlying mechanisms still remain unclear. Previous studies from our group have shown that RAS was activated in tubular cells in animals with persistent proteinuria. 17 Furthermore, ACE inhibitors exert beneficial effects on the tubulointerstitial damage in this disease and these effects were attributed, in part, to an attenuation of the nuclear factor (NF)-κB activation. 18 Protein overload nephropathy is considered an appropriate experimental model to approach the relationship between proteinuria and interstitial damage. The bovine serum albumin (BSA) overload model is frequently used because it is highly reproducible and can induce heterologous as well as autologous proteinuria. 16,19-21
In the kidney, AngII exerts its biological effects mainly through AT1. 22 In rodents, AT1 exists in two isoforms, AT1A and AT1B, encoded by two different genes. The murine AT1A is the predominantly expressed isoform in most tissues. 23 Therefore, the AT1AR-deficient mouse strain [AT1(−/−)] is a powerful model to analyze the effect of AngII blockade. 24 In the present study, we further investigated the contribution of RAS in the pathogenesis of tubulointerstitial damage by persistent proteinuria, using the BSA-overload model in AT1(−/−). Because altered bradykinin synthesis and its metabolism, 25,26 as well as AT2, 27,28 may also be involved in the mechanisms of proteinuria and interstitial cell infiltration, a group of animals was treated with ACE inhibitors. Accumulated evidence has shown that ET-1 also participates in the progression of renal diseases, with or without proteinuria. 18,20,29,30 Moreover, a recent study in rats harboring human renin and angiotensinogen genes suggests that ET-1 might mediate inflammatory processes in AngII-induced tissue damage. 31 In this regard, we also examined the potential role of ET-1 in proteinuric AT1(−/−) and the potential interrelationship between AngII and ET-1 in the tubulointerstitial damage. The data presented here further extend the implication of both peptides in the pathogenesis of interstitial injury because of proteinuria and support the idea that the combination of drugs modulating both peptides may be useful in nephropathies with persistent proteinuria.
Materials and Methods
Animals
We used AT1(−/−) that were generated with a germline chimera derived from TT2 embryonic stem cells with a targeted mutation of the AT1 A gene as previously described. 24 AT1(−/−) were back-crossed for more than six generations with C57BL/6 mice. 32 As their wild-type littermates (WT), C57BL/6 mice were purchased from Harlan Interfauna Ibérica, S.A. (Barcelona, Spain). Only female mice ages 8 to 10 weeks and weighing 18 to 23 g were used.
Experimental Designs
To decide the experimental condition for murine protein-overload nephropathy, WT weighing 20 g were intraperitoneally injected with four different daily doses (0.1, 0.2, 0.4, 1.0 g) of low endotoxin BSA (Sigma, St. Louis, MO) and followed until day 7. According to their effects on the outcome (see Results), protocols with 0.2 and 0.4 g of BSA daily were used as moderate and severe overload nephropathy models, respectively. Groups of AT1(−/−) and WT, injected with saline, were used as controls in all models.
Groups of mice were sacrificed on days 7, 14, and 28 (moderate) and days 7 and 14 (severe overload nephropathy). Four to 16 mice (BSA-injected WT, AT1(−/−), and their controls) were included at each time point. Urinary protein was determined every day by Knight’s method, as previously described. 33 Kidneys of all animals were perfused with cold saline and removed under general anesthesia.
To assess the role of AngII or ET-1 in this disease, we treated animals with moderate and severe nephropathy with the ACE inhibitor quinapril (as powdered hydrochloride salt; Parke-Davis, Barcelona, Spain) or with the dual endothelin type A and B receptor (ETA/B) antagonist bosentan (Ro 47-0203; 4-tert-butyl-N- [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2,2′-bipyrimidin-4-yl]-benzene-sulfonamide; Hoffmann-La Roche Ltd., Basel, Switzerland) or their combination. Quinapril (16 mg/kg in distilled water) and bosentan (100 mg/kg in a dissolution of 5% arabic gum) 18 were given daily by gastric gavage with a cannula from 24 hours before first BSA injection until day 13.
Renal Histopathological Studies
Paraffin-embedded sections (4-μm thick) were prepared and stained with hematoxylin and eosin and Masson’s trichrome for light microscopy. Semiquantification of morphological lesions were evaluated in regard to glomerular lesions (number of glomeruli with hypercellularity or matrix expansion/30 glomeruli scored 0 to 3), tubular lesions (number of tubuli with dilatation or atrophy/×100 high-power fields scored 0 to 4), protein casts (number of casts/×100 high-power fields scored 0 to 4), and interstitial infiltration (number of focus/kidney scored 0 to 4) by three different pathologists in a blind manner.
For the immunohistochemistry of ET-1, 29 kidneys were fixed in 4% paraformaldehyde and embedded in paraffin, and 4-μm thick sections were mounted on poly-l-lysine-coated slides. After deparaffinization with graded concentrations of xylene and ethanol, they were quenched in methanol containing 3% H2O2 at 25°C for 30 minutes to block the activity of endogenous peroxidase. These slides were washed and incubated in trypsin (0.1% trypsin CaCl2 wt/v) to expose antigenic sites. They were subsequently incubated with 5% normal swine serum for 30 minutes at room temperature to reduce nonspecific background staining and then incubated overnight at 4°C with rabbit polyclonal anti-ET-1 antibody (Peninsula Laboratories Europe Ltd., Merseyside, UK). Control slides were treated with diluted normal rabbit serum. After being washed, the sections were incubated with biotinylated swine anti-rabbit IgG (DAKO A/S, Glostrup, Denmark), washed again, and incubated with avidin-biotin-peroxidase complex (DAKO A/S) for 30 minutes. The sites of peroxidase activity were visualized with 0.05% 3,3′-diaminobenzidine (DAKO A/S) in 0.01% H2O2 for 10 minutes. Sections were counterstained with Mayer’s hematoxylin (Sigma, St. Louis, MO) for 2 minutes and coverslipped.
The intensity of ET-1 staining was evaluated by a semiautomatic image analysis system with Microimage version 3.0 for Windows (Olympus, Japan). We quantitatively analyzed ET-1 expression in WT and AT1(−/−) by measuring the integrated optical density (OD) of tubular cells and connective perivascular tissue. OD is a logarithmic function of the light transmitted through the stained section. This formula assumes an exponential decay of light inside the transmitting materials, as follows: OD(x, y) = −log [(intensity(x, y) − black)/(incident − black)]. Intensity(x, y) is the intensity at pixel(x, y), black is the intensity generated when no light goes through the material, and incident is the intensity of the incident light. The system was calibrated in a way that the OD of the negative control was 0.
RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Pieces of renal cortex were homogenized, and total RNA was obtained by the acid guanidinium-phenol-chloroform method. 21 One μg of RNA was reverse-transcribed and then amplified with a commercial kit (Promega, Buckinghamshire, UK), with the use of 0.5 μCi [α32P]dCTP (3000 Ci/mmol, Amersham) and 20 pmol of specific primers for rat transforming growth factor (TGF)-β1 (sense: 5′-AATACGTCAGACATTCGGGAAGCA-3′; antisense: 5′-GTCAATGTACAGCTGCCGTACACA-3′; fragment: 498 bp), rat/mouse fibronectin (FN) (sense: 5′-TGCCACTGTTCTCCTACGTG-3′; antisense: 5′-ATGCTTTGACCCT-TACACCG-3′; fragment: 312 bp), mouse tumor necrosis factor (TNF)-α (sense: 5′-GCGACGTGGAA-CTGGCAGAAG-3′; antisense: 5′-GGTACAACCCAT-CGGCTGGCA-3′; fragment: 384 bp), and mouse prepro ET-1 (sense: 5′-TGATCTTCTCTCTGCTGTT-3′; antisense: 5′-TTTAAGCTTTTCTGCATGGT-3′; fragment: 408 bp). The amplifications were performed with annealing temperatures of 62°C (TGF-β), 60°C (FN), 63°C (TNF-α), or 61°C (prepro ET-1). The optimum number of amplification cycles used for semi quantitative RT-PCR (25, 25, 31, and 32, respectively) was chosen on the basis of pilot experiments (data not shown). The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control. Aliquots of each reaction were run on 4% acrylamide-bisacrylamide gels. The gels were dried and exposed to X-OMAT AS films (Eastman Kodak Company, Rochester, NY). Autoradiograms were quantified by scanning densitometry (Molecular Dynamics). The density of each gene was compared after the individual correction by density of GAPDH.
Extraction of Nuclear Proteins and Electrophoretic Mobility Shift Assay
Nuclear extracts were obtained as previously described 18,28 and the activity of transcription factors was evaluated by electrophoretic mobility shift assay. Briefly, frozen kidney pieces were pulverized in a metallic chamber and resuspended in a cold extraction buffer [20 mmol/L Hepes-NaOH, pH 7.6, 20% (v/v) glycerol, 0.35 mol/L NaCl, 5 mmol/L MgCl2, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 1 μg/ml pepstatin A]. The homogenate was vigorously shaken and the insoluble materials precipitated by centrifugation at 40,000 × g for 30 minutes at 4°C. Supernatants were dialyzed overnight against a binding buffer containing 20 mmol/L Hepes-NaOH (pH 7.6), 20% (v/v) glycerol, 0.1 mmol/L NaCl, 5 mmol/L MgCl2, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol, and 0.5 mmol/L phenylmethyl sulfonyl fluoride. These dialysates were cleared by centrifugation at 10,000 × g for 15 minutes at 4°C and stored in aliquots at −80°C until use. Protein concentration was quantified by the bicinchoninic acid method (Bio-Rad Laboratories, Richmond, CA).
NF-κB and AP-1 consensus oligonucleotides (5′-AGTTGAGGGGACTTT-CCCAGGC-3′ and 5′-CGCTTGATGAGTCAGCCGGAA-3′, respectively) were [32P]-end-labeled by incubation for 10 minutes at 37°C with 10 U T4 polynucleotide kinase (Promega) in a reaction containing 10 μCi of [γ-32P]ATP (3000 Ci/mmol; Amersham, Arlington Heights, IL), 70 mmol/L Tris-HCl, 10 mmol/L MgCl2, and 5 mmol/L dithiothreitol. The reaction was stopped by the addition of EDTA to a final concentration of 0.05 mol/L. Nuclear proteins (20 μg NF-κB and 30 μg AP-1) were equilibrated for 10 minutes in a binding buffer containing 4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 50 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 7.5, and 50 μg/ml poly (dI-dC). When competition assays were performed, the cold probe was added to this buffer 10 minutes before the addition of the labeled probe. Labeled probe (0.035 pmol) was added to the reaction and incubated for 20 minutes at room temperature. The reaction was stopped by the addition of gel-loading buffer (250 mmol/L Tris-HCl, 0.2% bromophenol blue, 0.2% xylene cyanol, and 40% glycerol) and run on a nondenaturing, 4% acrylamide gel at 100 V at room temperature in 89 mmol/L Tris-borate, 2 mmol/L EDTA, pH 8.0 (TBE). 18,21 The gel was dried and exposed to X-OMAT AS films (Eastman Kodak Company). Autoradiograms were quantified by scanning densitometry (Molecular Dynamics).
Statistical Analysis
Results are expressed as mean ± SD. Comparisons between groups were made using unpaired Student’s t-test. For statistical analysis of mortality (survival rate), chi-square test using EPI INFO Ver. 6.04 (Centers for Disease Control, Atlanta, GA) was used. Differences were considered as significant if the P value was <0.05.
Results
Proteinuria and Mortality in WT Depend on the Amount of BSA Injected
The protein overload nephropathy model is well established in rats, but not in mice. Initially, we tested the appropriate amounts of BSA for the murine protein overload model. We used four different daily intraperitoneal doses of BSA and followed urinary protein level and mortality until day 7 (Figure 1) ▶ . Mice weighing 20 g injected with 1 g of BSA daily showed severe proteinuria, and all animals died before day 7. In contrast, 0.1 g of BSA daily was not enough to induce pathological proteinuria. Therefore, we finally used 0.2 and 0.4 g of BSA daily to elicit the moderate and severe overload nephropathy models, respectively.
Figure 1.

Proteinuria and mortality depend on the amount of protein injected into mice. The amount of BSA injected to WT weighing 20 g at four different daily doses is well correlated to peak proteinuria (column) and mortality at day 7 (line).
AT1(−/−) Have Less Proteinuria and Mortality than WT
Moderate Overload Nephropathy
In AT1(−/−) and their WT littermates injected with 0.2 g of BSA, we followed the kinetics of proteinuria for 4 weeks (Figure 2a) ▶ . Peak proteinuria in AT1(−/−) appeared earlier than in WT, although there was no significant difference between them. In AT1(−/−) proteinuria decreased after day 7, whereas in WT it was still elevated until day 14. Both groups did however reveal moderate proteinuria after 3 weeks.
Figure 2.

AT1(−/−) have less proteinuria than WT with moderate (a) and severe (b) overload nephropathy. AT1(−/−) have less proteinuria (moderate overload nephropathy at day 11: 83 ± 80 mg/dl, n = 10, *P < 0.01; severe overload nephropathy at day 7: 121 ± 207, n = 6, **P < 0.05; at day 11: 97 ± 118, n = 6, **P < 0.05) than WT in both models (597 ± 349, n = 8; 1426 ± 1121, n = 5; 1500 ± 1149, n = 4, respectively). Peak proteinuria in AT1(−/−) appeared earlier than that in WT with severe overload nephropathy (at day 1: 1665 ± 563 versus 205 ± 134, n = 9 to 12, *P < 0.01).
Severe Overload Nephropathy
Both groups of animals injected with 0.4 g of BSA daily showed threefold to fourfold higher proteinuria peaks than those of moderate models, but their kinetics were basically similar (Figure 2b) ▶ . AT1(−/−) showed high proteinuria from day 1, returning to the normal limits at approximately day 7. By contrast, severe proteinuria was persistent in WT for several days with an elevated mortality at approximately day 14 (69% animals). AT1(−/−) had a significantly less mortality (22% animals, P < 0.05) at that time.
AT1(−/−) Have Less Renal Lesions, TGF-β, and Matrix mRNA Expression than WT
In both groups of animals with moderate and severe overload nephropathy, we evaluated the renal damage at days 7 and 14. Only data on the severe model are depicted in Figure 3 ▶ . Morphological lesions were significantly less marked in AT1(−/−) than in WT only at day 14 (Figure 3a) ▶ . Differences in the moderate nephropathy were less clear between AT1(−/−) and WT (not shown). Semiquantitative RT-PCR also showed that TGF-β and FN mRNA expression was significantly attenuated in AT1(−/−) in relation to WT at day 14 (Figure 3, b and c) ▶ . At day 7, FN expression in AT1(−/−) was significantly attenuated in both models (moderate model: 1.1 ± 0.2-fold versus 3.4 ± 1.2-fold increase, n = 3, P < 0.05; severe model; Figure 3c ▶ ). On the other hand, we found a significant diminution of TGF-β expression in AT1(−/−) at day 7 in moderate overload nephropathy (1.4 ± 0.1-fold versus 1.8 ± 0.3-fold increase, n = 4, P < 0.05), but not in severe nephropathy (Figure 3b) ▶ . As it is well known, AngII is a strong inducer of TGF-β. To search for another potential candidate that could explain the high expression of TGF-β at day 7 in AT1(−/−), we examined the gene expression of TNF-α one of the most important inflammatory cytokines able to elicit TGF-β expression. 34,35 We noted that TNF-α expression in AT1(−/−) with severe nephropathy at day 7 increased as in WT in relation to controls (3.5 ± 0.5-fold versus 3.8 ± 1.4-fold increase, n = 4 to 5, P = 0.76).
Figure 3.
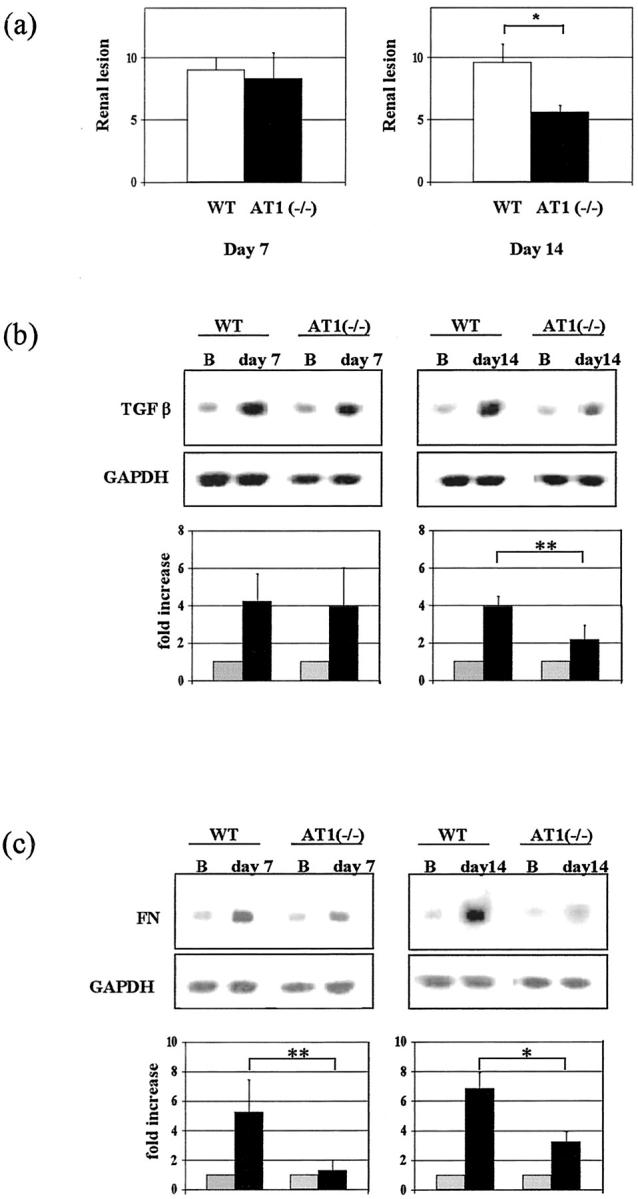
Diminution of renal lesions (a), TGF-β (b), and matrix (c) mRNA expression in AT1(−/−) with severe overload nephropathy. Morphological lesions (at day 7: 8.3 ± 2.1 versus 9.0 ± 1.0, n = 5, P = 0.63; at day 14: 5.6 ± 0.5 versus 9.6 ± 1.5, n = 5, *P < 0.01) (a) and TGF-β mRNA expression (at day 7: 3.9 ± 2.1-fold versus 4.3 ± 1.5-fold increase, n = 3, P = 0.84; at day 14: 2.1 ± 0.8 versus 3.9 ± 0.5, n = 4, **P < 0.05) (b) were significantly attenuated in AT1(−/−) with severe overload nephropathy at day 14, whereas FN mRNA expression was diminished during the whole disease course (at day 7: 1.3 ± 0.7 versus 5.3 ± 2.2, n = 4, **P < 0.05; at day 14: 3.2 ± 0.7 versus 6.8 ± 1.1, n = 3, *P < 0.01) (c).
Different Kinetics in the Activation of the Transcription Factors NF-κB and AP-1 between AT1(−/−) and WT
Several groups have previously demonstrated that overload proteinuria induces NF-κB activation in tubular epithelial cells both in vivo and in vitro. 18,31,36,37 To further approach the underlying mechanisms by which AngII participates at transcriptional levels, we examined NF-κB and AP-1 activation in both groups of animals with severe overload nephropathy. Electrophoretic mobility shift assay showed that NF-κB in AT1(−/−) was activated like that in WT at day 7, but significantly attenuated at day 14 (Figure 4) ▶ . NF-κB activation was correlated with the urinary protein levels. Although AT1(−/−) revealed high proteinuria before day 7, AP-1 activation in those mice was significantly attenuated in relation to WT along the disease course (Figure 4) ▶ .
Figure 4.

Different kinetics of NF-κB and AP-1 activation between WT and AT1(−/−). Electrophoretic mobility shift assay showed that NF-κB activation in AT1(−/−) was significantly attenuated only at day 14 (at day 7: 2.6 ± 0.7-fold versus 2.7 ± 1.9-fold increase, n = 4, P = 0.97; at day 14: 2.2 ± 0.7 versus 5.2 ± 1.7, n = 4, *P < 0.05), whereas AP-1 activation was absent in AT1(−/−) during the disease outcome (at day 7: 1.3 ± 0.1 versus 2.4 ± 0.7, n = 4, *P < 0.05; at day 14: 1.3 ± 0.3 versus 2.2 ± 0.2, n = 4, **P < 0.01). Lane C denotes co-incubation with NF-κB or AP-1 cold oligos.
The ACE Inhibitor Quinapril Diminishes Proteinuria in Moderate and Severe Overload Nephropathy in WT
To further confirm whether the attenuation of overload nephropathy in AT1(−/−) was because of the absence of AT1 stimulation, a group of animals was treated with the ACE inhibitor quinapril. AT1(−/−) with moderate overload nephropathy showed, regardless of quinapril, the same kinetics of urinary protein excretion (Figure 5a) ▶ , but the early peak of proteinuria in AT1(−/−) with severe overload nephropathy was delayed by quinapril (Figure 5b) ▶ . WT with moderate or severe overload nephropathy and treated with quinapril had a significant reduction in proteinuria with a pattern very similar to that of untreated AT1(−/−). Mortality in treated WT with severe nephropathy was also decreased (20% versus 69% in untreated WT; n = 5 to 13, P = 0.117).
Figure 5.

The ACE inhibitor quinapril improves persistent proteinuria in WT with moderate (a) and severe (b) overload nephropathy. WT treated with quinapril (WT Tx) were significantly protected from persistent proteinuria in moderate overload nephropathy (at day 7: 62 ± 6 versus 515 ± 411 mg/dl, n = 3 to 5, *P < 0.05; at day 11: 88 ± 42 versus 776 ± 366, n = 3 to 5, **P < 0.01) (a). In severe overload nephropathy, treated WT reproduced the same beneficial effects on persistent proteinuria as AT1(−/−) (at day 7: 550 ± 209 versus 2492 ± 394, n = 4, **P < 0.01; at day 11: 105 ± 69 versus 1420 ± 1183, n = 5, ***P < 0.05) (b). Early peak of proteinuria in AT1(−/−) was significantly delayed by quinapril in severe overload model (at day 1: 92 ± 48 versus 1268 ± 634, n = 4 to 5, *P < 0.01).
The ETA/ETB Antagonist Bosentan Decreases Severe Proteinuria and Mortality in WT
Because we have previously demonstrated that ET-1 also participates in this disease in rats, 18,29 we used the dual ETA/ETB antagonist bosentan in this murine model. In WT with severe overload nephropathy, bosentan significantly decreased severe proteinuria (Figure 6a) ▶ , renal lesions (6.3 ± 1.0 versus 11.7 ± 2.3, n = 3 to 4, P < 0.01), and improved survival rate (Figure 6b) ▶ . Although we could not find statistical significance, WT treated with the combination of bosentan and quinapril showed an additional improvement in relation to those treated only with bosentan in proteinuria (Figure 6a) ▶ , renal lesions (5.5 ± 0.6, n = 4, versus WT, P < 0.01), and survival rate (Figure 6b) ▶ . In WT with moderate nephropathy, both bosentan and the combined treatment with quinapril also had a certain beneficial effect on proteinuria (data not shown). However, in AT1(−/−) treated only with bosentan, the improvement in proteinuria and survival rate at day14 was less marked (data not shown).
Figure 6.

The ET-1 A/B antagonist bosentan and the combination with quinapril and bosentan decrease severe proteinuria and mortality in WT. WT with severe overload nephropathy treated with bosentan (WT B/Tx) or quinapril and bosentan (WT QB/Tx) had less proteinuria (at day 7: 953 ± 942* versus 324 ± 282** versus 1997 ± 119 mg/dl, n = 6, *P < 0.05, **P < 0.01) and survived longer than WT (33 versus 63* versus 86** %, n = 7 to 9, versus WT *P = 0.34, **P = 0.06).
The ETA/ETB Antagonist Bosentan Attenuates Interstitial Infiltration in AT1(−/−)
AT1(−/−) were protected from persistent proteinuria and glomerular and tubular lesions, but in moderate nephropathy they showed a significantly higher interstitial infiltration than WT at day 14, mainly located around small vessels in cortex (Figure 7) ▶ . In severe nephropathy, we failed to find a significantly marked cell infiltration in AT1(−/−), because of that WT also showed an important infiltration.
Figure 7.

The ETA/ETB antagonist bosentan attenuates interstitial infiltration in AT1(−/−). AT(−/−) with moderate overload nephropathy showed a significantly higher interstitial cell infiltration than WT at day 14 (2.8 ± 0.9 versus 0.7 ± 0.7, n = 9, *P < 0.01), which was mainly located around small vessels, and was significantly attenuated by bosentan (1.2 ± 0.5, n = 5, **P < 0.05). H&E stain; original magnification, ×100.
Although bosentan did not afford additional anti-proteinuric effect on AT1(−/−), interstitial cell infiltration was significantly attenuated (Figure 7) ▶ . We also examined the effect of quinapril on the interstitial infiltration, but could not find any additional protective effect, as it was the case in bosentan-treated mice (data not shown).
AT1(−/−) Show a Higher ET-1 Protein Expression than WT in Tubular Epithelial Cells
To approach a potential explanation for the cell infiltration observed in AT1(−/−), we examined ET-1 by immunohistochemistry and gene expression. AT1(−/−) with moderate overload nephropathy showed at day14 higher ET-1 expression in tubular epithelial cells and in the connective tissue around the vessels than WT (146 ± 98 versus 17 ± 23 OD, n = 5, *P < 0.01) (Figure 8) ▶ . We also examined the prepro ET-1 mRNA expression in whole kidney from both animals. Semiquantification studies revealed that AT1(−/−) had a higher expression than WT, but without statistical significance (at day 7: 1.4 ± 0.1-fold versus 1.0 ± 0.4-fold increase, n = 3, P = 0.14; at day 14: 1.8 ± 0.3-fold versus 1.3 ± 0.4-fold, n = 4, P = 0.09). However, there were no significant differences between AT1(−/−) and WT with severe overload nephropathy.
Figure 8.

AT1(−/−) show higher ET-1 protein expression than WT in tubular epithelial cells and connective tissue around small vessels. AT1(−/−) with moderate overload nephropathy at day 14 showed a significantly higher expression of ET-1 in tubular epithelial cells (A, control; B, nephropathy) and also in connective tissue around small vessels (C) than WT (D, control; E, nephropathy). The computer analyses of the data are shown in F (117 ± 67 versus 19 ± 29 OD, n = 6, *P < 0.01). Immunohistochemistry; original magnifications, ×100.
Discussion
Establishment of a Murine Model of Overload Nephropathy
BSA-induced overload nephropathy has already been established in rats, but not in mice. Only one study on chronic murine model has been recently published. 38 Therefore, we initially examined the experimental conditions to have a reproducible model in mice. Although the adequate range of daily BSA to induce intense proteinuria and renal damage was very narrow, we were able to define two different pictures of overload nephropathy that, in accordance with the intensity, were named moderate or severe. The obtained model in mice resembles to that seen in rats with regards to proteinuria, and glomerular and tubulointerstitial damage. Based on these findings, we induced this model in AT1(−/−) to study the contribution of RAS to the pathogenesis of tubulointerstitial damage because of persistent proteinuria. In addition, we compared both models (moderate and severe) to clarify whether the involved mechanisms are affected by the intensity of proteinuria, which is an important clinical parameter for the efficacy of RAS blockade. 5
Overload Nephropathy Is Attenuated in AT1(−/−)
The present study shows that renal injury and mortality at day 14 in overload nephropathy were significantly decreased in AT1(−/−). These data are consistent with the hypothesis that RAS participates in the pathogenesis of tubulointerstitial injury because of persistent proteinuria. 17,18
Because the intensity of the early peak of proteinuria was the same in WT and AT1(−/−), with both moderate and severe overload nephropathy, initial damage in glomerular epithelial cells should be the same in both types of mice. 19 Consequently, tubular epithelial cells must be exposed to considerable amounts of urinary proteins before day 7. It is well known that persistent proteinuria is a key factor to accelerate the tubulointerstitial damage in human and experimental renal diseases. 14,15 We have previously shown that persistent proteinuria elicited RAS activation in tubular epithelial cells. 17 Because AngII has been considered a potent inflammatory mediator, 11 the increased local AngII generation might directly contribute to the disease progression. As AngII causes an augmentation in TGF-β and FN, it was not surprising that AT1(−/−) with moderate overload nephropathy had significantly less TGF-β and FN mRNA expression. The importance of AngII in the synthesis of TGF-β is supported by several studies showing its marked reduction (∼45%) in animals with various models of renal injury treated with ACE inhibitors or AT1 antagonists. 39,40 However, in AT1(−/−) with severe overload nephropathy, TGF-β was only partially decreased, suggesting that other inflammatory mediators, besides AngII, could be implicated. In fact, animals at day 7 showed high TNF-α mRNA expression in relation to controls. 34,35 Moreover, NF-κB, a regulator of TNF-α, 41 was similarly activated at day 7, but attenuated at day 14 concomitantly with the absence of persistent proteinuria, therefore suggesting that proteinuria may directly cause NF-κB activation and subsequent inflammation in an AT1 unrelated manner. By contrast, AT1(−/−) had significantly less AP-1 along the disease, even in the severe nephropathy, suggesting that the activation of this transcription factor is not linked to persistent proteinuria but dependent on the stimulation of AT1. In fact, in vascular smooth muscle cells, AngII activates AP-1 through AT1 by a signaling mechanism dependent on protein kinase C and protein tyrosine kinase. 42 Also, stretch activation of mesangial cells causes FN up-regulation that requires AP-1 activation. 43 In addition, AngII-induced FN mRNA expression in fibroblasts is regulated by a transcriptional control on AP-1 site and a posttranscriptional control based on mRNA stabilization by TGF-β. 44 In this context, the reduction of FN expression associated with the attenuation of AP-1, but not NF-κB, activation in AT1(−/−) is compatible with these data.
Contrary to that observed in the early stage of the disease, after day 7 AT1(−/−) showed significantly less proteinuria in both moderate and severe nephropathy, and consequently less tubulointerstitial damage, that coincided with a significant attenuation of NF-κB activation. It is well known that RAS blockade by ACE inhibitors or AT1 antagonists decreases proteinuria in human and experimental renal diseases. 1-7 Although the anti-proteinuric effect of ACE inhibitors was related to the enhancement of kinin activity, 45,46 other reports have shown that these effects were independent. 47 Alternatively, it has also been shown that ACE inhibitors and AT1 antagonists were equally effective in reducing proteinuria. 48 In our models, urinary protein kinetics was the same in WT receiving ACE inhibitors and AT1(−/−), suggesting that the anti-proteinuric action after day 7 was mainly dependent on AT1 blockade. In fact, the anti-proteinuric effect of RAS blockade has been attributed mostly to the interference with glomerular hemodynamics and intrinsic permeability of the glomerular membrane. 40 Interestingly, AT1(−/−) had the proteinuria peak earlier than WT, especially in the severe overload model, indicating that AngII may functionally participate in the regulation of glomerular hemodynamics in the early phase of this disease. Studies in rats with the amino-acid infusion model demonstrated that AngII regulates glomerular hemodynamics through the NO synthesis. 49 A similar mechanism might be involved in the early phase of protein overloading. Because AT1(−/−), with or without quinapril treatment, showed the same kinetics of urinary proteins, these functional alterations might be mainly because of an AT1-mediated mechanism. However, we found a different kinetics in treated and untreated animals with severe overload nephropathy, suggesting that kinins may have a role in this alteration.
Interstitial Cell Infiltration Is Higher in AT1(−/−) than in WT with Overload Nephropathy
By semiquantitative evaluation of glomerular and tubular lesions, protein casts and interstitial infiltration, we noted an attenuation of renal damage in AT1(−/−). However, taking into account the chemotactic properties of AngII via AT1, 50 it was surprising to note that AT1(−/−) still have a marked interstitial cell infiltration mainly detected around the small vessels and in a focal manner. Those higher infiltrations in AT1(−/−) were conspicuous in moderate nephropathy, because WT with severe nephropathy finally showed severe interstitial infiltration and thus higher scores than AT1(−/−). In this sense, quinapril-treated rats in chronic phase also showed a certain cell infiltration although there was attenuation in the total renal lesion score. Recent data suggest that AT2 can also be implicated in the recruitment of mononuclear cells. 28,51 In fact, this receptor has also been involved in NF-κB activation 28 and in the expression of some chemokines such as RANTES. 51 The absence of a marked reduction of the interstitial cells in quinapril-treated AT1(−/−), could be because of the inability of those drugs to completely inhibit the angiotensin II generation. In this regard, further experiments are needed (eg, the treatment with AT2 antagonist) to solve this issue. In addition, other molecules and intracellular pathways could be implicated in the mononuclear cell infiltration observed in overload nephropathy.
Beneficial Effects of ETA/ETB Blockade
In several models of renal damage characterized by intense proteinuria a number of studies have revealed overexpression of ET-1, mainly located in tubular epithelial cells and, to a lesser extent, in glomeruli. 20,48 ET-1 may induce NF-κB activation in tubular epithelial cells via ETA and ETB receptors. 18 The present study shows that in WT with severe overload nephropathy, ET-1 blockade with bosentan exerted significant beneficial effects on proteinuria, renal lesions, and mortality. Although ET-1 has a proinflammatory effect, 48 findings in double-transgenic rats (dTGR) harboring human renin and angiotensinogen genes 31 suggest that ET-1 may be secondarily activated by AngII. In fact, bosentan significantly reduced AngII-induced NF-κB activation, adhesion molecule expression, and macrophage infiltration in a blood pressure-independent manner in these animals. 31 Moreover, several studies on angiotensin-mediated hypertension 52 and hypertrophy 53 also suggest the existence of an interrelationship between AngII and ET-1 (Figure 9 ▶ , pathway 2).
Figure 9.

Potential interrelation between AngII and ET-1 in tubulointerstitial nephritis. Persistent intense proteinuria may cause an overproduction of AngII and ET-1 in proximal tubular cells, and both peptides, directly or through the induction of each other, may participate in the tubulointerstitial injury in this model. AT1(−/−) animals and WT treated with the ACE inhibitor quinapril had less renal damage probably because of the interference with pathway 1, and pathways 1 and 2, respectively. The beneficial effect of the ETA/ETB antagonist bosentan could be because of the blockade of pathways 2 and 3. The combined therapy of quinapril and bosentan may afford a synergistic effect because they block the pathways 1, 2, and 3.
However, in certain conditions, bosentan significantly improved the interstitial infiltration both in AT1(−/−) and in rats treated with quinapril, further suggesting that ET-1 may play a role in the interstitial cell infiltration, 20 in an AT1-stimulation-independent manner (Figure 9 ▶ , pathway 3). AT1(−/−) did indeed show a higher ET-1 expression in tubular epithelial cells than WT, despite the absence of persistent proteinuria. In this regard, it is uncertain if the same occurs in tubular epithelial cells, but AngII elicits ET-1 synthesis via AT1 in vascular smooth muscle cells (VSMC). 54,55 Furthermore, although we failed to find statistical significance, WT with quinapril and bosentan showed a further improvement in proteinuria, renal lesions, and mortality than those treated with quinapril or bosentan alone. In agreement, the combined therapy was also superior to each drug alone in rats with chronic overload nephropathy. 18 Those findings further support the idea that both peptides contribute in parallel to the pathogenesis of this disease (Figure 9 ▶ , pathways 1 and 3) as previously suggested. 56,57 In addition, the fact that proteinuric AT1(−/−) had ET-1 overexpression and a marked interstitial cell infiltration postulates their interrelationship (Figure 9 ▶ , pathway 2) and does suggest not only an additive, but also a synergistic benefit of the combined therapy.
On the whole, we have shown that the renal tubulointerstitial damage caused by persistent proteinuria is attenuated in mice strain lacking AT1. The up-regulation of ET-1, mainly in tubular cells, as well as the major beneficial effect of the combined therapy with ACE inhibitors and ETA/ETB antagonists, suggest the implication of AngII and ET-1 in the pathogenesis. The present study further indicates that this combination may afford a synergistic therapeutic effect in renal diseases with severe proteinuria and tubulointerstitial damage.
Acknowledgments
We thank Yasuhiko Tomino, M.D. for his support to Y.S.; Covadonga Alonso, M.D. and José Fortes M.D. for helping us with pathological evaluation; Marta Ruiz-Ortega, Ph.D., for helpful discussion and advice; Dr. Juan José Granizo Martínez for helping us with statistical analyses; Lise Lotte Gulliksen for secretarial assistance; and Belen Huesa for technical assistance.
Footnotes
Address reprint requests to Jesús Egido, M.D., Ph.D., Renal and Vascular Laboratory, Fundación Jiménez Díaz., Avda.de los Reyes Católicos, 2, 28040-Madrid, Spain. E-mail. jegido@fjd.es.
Supported by the Comunidad Autónoma de Madrid (grants 08.9/0002.1/2000, 08.4/0019.1/2000), the Fondo de Investigación Sanitaria (grants 99/0425, 00/0111), the Ministerio de Educación y Ciencia (grant PM 97/85), EU Concerted Action Grant (BMH 4-CT98-3631, DG 12-SSMI), and the Japan Health Science Foundation (to Y. S.).
This work was partly presented as oral communication in the meeting of American Society of Nephrology (Toronto, Canada, October 2000). O. L. F., N. T., and R.B. are fellows from Fundación Iñigo Álvarez de Toledo, Fundación Conchita Rábago and Fundación Fernández-Cruz, respectively.
References
- 1.Anderson S, Rennke HG, Brenner BM: Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 1986, 77:1993-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Remuzzi A, Perico N, Amuchastegui CS, Malanchini B, Mazerska M, Battaglia C, Bertani T, Remuzzi G: Short- and long-term effect of angiotensin II receptor blockade in rats with experimental diabetes. J Am Soc Nephrol 1993, 4:40-49 [DOI] [PubMed] [Google Scholar]
- 3.Remuzzi A, Fassi A, Sangalli F, Malanchini B, Mohamed EI, Bertani T, Remuzzi G: Prevention of renal injury in diabetic MWF rats by angiotensin II antagonism. Exp Nephrol 1998, 6:28-38 [DOI] [PubMed] [Google Scholar]
- 4.Kasiske BL, Kalil RS, Ma JZ, Liao M, Keane WF: Effect of antihypertensive therapy on the kidney in patients with diabetes: a meta-regression analysis. Ann Intern Med 1993, 118:129-138 [DOI] [PubMed] [Google Scholar]
- 5.: The Gruppo Italiano di Studi Epidemiologici in Nefrologia Group: Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 1997, 349:1857-1863 [PubMed] [Google Scholar]
- 6.Salvetti A, Mattei P, Sudano I: Renal protection and antihypertensive drugs: current status. Drugs 1999, 57:665-693 [DOI] [PubMed] [Google Scholar]
- 7.Burnier M, Brunner HR: Angiotensin II receptor antagonists. Lancet 2000, 355:637-645 [DOI] [PubMed] [Google Scholar]
- 8.Anderson S, Diamond JR, Karnovsky MJ, Brenner BM: Mechanisms underlying transition from acute glomerular injury to late glomerular sclerosis in a rat model of nephrotic syndrome. J Clin Invest 1988, 82:1757-1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer TW, Anderson S, Rennke HG, Brenner BM: Reversing glomerular hypertension stabilizes established glomerular injury. Kidney Int 1987, 31:752-759 [DOI] [PubMed] [Google Scholar]
- 10.Egido J: Vasoactive hormones and renal sclerosis. Kidney Int 1996, 49:578-597 [DOI] [PubMed] [Google Scholar]
- 11.Suzuki Y, Ruiz-Ortega M, Egido J: Angiotensin II: a double-edged sword in inflammation. J Nephrol 2000, 13(Suppl 3):S101-S110 [PubMed] [Google Scholar]
- 12.Eddy AA: Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol 1996, 7:2495-2508 [DOI] [PubMed] [Google Scholar]
- 13.Becker GJ, Hewitson TD: The role of tubulointerstitial injury in chronic renal failure. Curr Opin Nephrol Hypertens 2000, 9:133-138 [DOI] [PubMed] [Google Scholar]
- 14.Remuzzi G, Ruggenenti P, Benigni A: Understanding the nature of renal disease progression. Kidney Int 1997, 51:2-15 [DOI] [PubMed] [Google Scholar]
- 15.Burton C, Harris KP: The role of proteinuria in the progression of chronic renal failure. Am J Kidney Dis 1996, 27:765-775 [DOI] [PubMed] [Google Scholar]
- 16.Eddy AA: Interstitial nephritis induced by protein-overload proteinuria. Am J Pathol 1989, 135:719-733 [PMC free article] [PubMed] [Google Scholar]
- 17.Largo R, Gomez-Garre D, Soto K, Marron B, Blanco J, Gazapo RM, Plaza JJ, Egido J: Angiotensin-converting enzyme is upregulated in the proximal tubules of rats with intense proteinuria. Hypertension 1999, 33:732-739 [DOI] [PubMed] [Google Scholar]
- 18.Gomez-Garre D, Largo R, Tejera N, Fortes J, Manzarbeitia F, Egido J: Activation of NFkB in tubular epithelial cells of rats with intense proteinuria. Role of angiotensin II and endothelin-1. Hypertension 2001, 37:1171-1178 [DOI] [PubMed] [Google Scholar]
- 19.Weening JJ, Van Guldener C, Daha MR, Klar N, van der Wal A, Prins FA: The pathophysiology of protein-overload proteinuria. Am J Pathol 1987, 129:64-73 [PMC free article] [PubMed] [Google Scholar]
- 20.Zoja C, Benigni A, Remuzzi G: Protein overload activates proximal tubular cells to release vasoactive and inflammatory mediators. Exp Nephrol 1999, 7:420-428 [DOI] [PubMed] [Google Scholar]
- 21.Largo R, Gomez-Garre D, Santos S, Penaranda C, Blanco J, Esbrit P, Egido J: Renal expression of parathyroid hormone-related protein (PTHrP) and PTH/PTHrP receptor in a rat model of tubulointerstitial damage. Kidney Int 1999, 55:82-90 [DOI] [PubMed] [Google Scholar]
- 22.Zhuo J, Song K, Harris PJ, Mendelsohn FA: In vitro autoradiography reveals predominantly AT1 angiotensin II receptors in rat kidney. Renal Physiol Biochem 1992, 15:231-239 [DOI] [PubMed] [Google Scholar]
- 23.Llorens-Cortes C, Greenberg B, Huang H, Corvol P: Tissular expression and regulation of type 1 angiotensin II receptor subtypes by quantitative reverse transcriptase-polymerase chain reaction analysis. Hypertension 1994, 24:538-548 [DOI] [PubMed] [Google Scholar]
- 24.Sugaya T, Nishimatsu S, Tanimoto K, Takimoto E, Yamagishi T, Imamura K, Goto S, Imaizumi K, Hisada Y, Otsuka A, Uchida H, Sugiura M, Fukuta K, Fukamizu A, Murakami K: Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem 1995, 270:18719-18722 [DOI] [PubMed] [Google Scholar]
- 25.Campbell DJ, Kladis A, Duncan AM: Effects of converting enzyme inhibitors on angiotensin and bradykinin peptides. Hypertension 1994, 23:439-449 [DOI] [PubMed] [Google Scholar]
- 26.Gansevoort RT, de Zeeuw D, de Jong PE: Is the antiproteinuric effect of ACE inhibition mediated by interference in the renin-angiotensin system? Kidney Int 1994, 45:861-867 [DOI] [PubMed] [Google Scholar]
- 27.Matsubara H: Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 1998, 83:1182-1191 [DOI] [PubMed] [Google Scholar]
- 28.Ruiz-Ortega M, Lorenzo O, Ruperez M, Blanco J, Egido J: Angiotensin II activates nuclear factor-kB via AT1 and AT2 receptors in the kidney. Am J Pathol 2001, 158:1743-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez-Garre D, Largo R, Liu XH, Gutierrez S, Lopez-Armada MJ, Palacios I, Egido J: An orally active ETA/ETB receptor antagonist ameliorates proteinuria and glomerular lesions in rats with proliferative nephritis. Kidney Int 1996, 50:962-972 [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Ortega M, Gomez-Garre D, Liu XH, Blanco J, Largo R, Egido J: Quinapril decreases renal endothelin-1 expression and synthesis in a normotensive model of immune-complex nephritis. J Am Soc Nephrol 1997, 8:756-768 [DOI] [PubMed] [Google Scholar]
- 31.Muller DN, Mervaala EM, Schmidt F, Park JK, Dechend R, Genersch E, Breu V, Loffler BM, Ganten D, Schneider W, Haller H, Luft FC: Effect of bosentan on NF-kappaB, inflammation, and tissue factor in angiotensin II-induced end-organ damage. Hypertension 2000, 36:282-290 [DOI] [PubMed] [Google Scholar]
- 32.Hisada Y, Sugaya T, Yamanouchi M, Uchida H, Fujimura H, Sakurai H, Fukamizu A, Murakami K: Angiotensin II plays a pathogenic role in immune-mediated renal injury in mice. J Clin Invest 1999, 103:627-635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C: Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int 1998, 54:1166-1174 [DOI] [PubMed] [Google Scholar]
- 34.Phillips AO, Topley N, Steadman R, Morrisey K, Williams JD: Induction of TGF-beta 1 synthesis in D-glucose primed human proximal tubular cells by IL-1 beta and TNF alpha. Kidney Int 1996, 50:1546-1554 [DOI] [PubMed] [Google Scholar]
- 35.Pawluczyk IZ, Harris KP: Cytokine interactions promote synergistic fibronectin accumulation by mesangial cells. Kidney Int 1998, 54:62-70 [DOI] [PubMed] [Google Scholar]
- 36.Donadelli R, Abbate M, Zanchi C, Corna D, Tomasoni S, Benigni A, Remuzzi G, Zoja C: Protein traffic activates NF-kB gene signaling and promotes MCP-1-dependent interstitial inflammation. Am J Kidney Dis 2000, 36:1226-1241 [DOI] [PubMed] [Google Scholar]
- 37.Rangan GK, Wang Y, Tay YC, Harris DC: Inhibition of nuclear factor-kappaB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int 1999, 56:118-134 [DOI] [PubMed] [Google Scholar]
- 38.Eddy A, Kim H, López-Guisa J, Oda T, Soloway P: Interstitial fibrosis in mice with overload proteinuria: deficiency of TIMP-1 is not protective. Kidney Int 2000, 58:618-628 [DOI] [PubMed] [Google Scholar]
- 39.Peters H, Border WA, Noble NA: Targeting TGF-beta overexpression in renal disease: maximizing the antifibrotic action of angiotensin II blockade. Kidney Int 1998, 54:1570-1580 [DOI] [PubMed] [Google Scholar]
- 40.Benigni A, Remuzzi G: Mechanism of progression of renal disease: growth factors and related mechanisms. J Hypertens 1998, 16(Suppl):S9-S12 [PubMed] [Google Scholar]
- 41.Guijarro C, Egido J: Transcriptional factor-kB (NF-kB) and renal disease. Kidney Int 2001, 59:415-424 [DOI] [PubMed] [Google Scholar]
- 42.Tamura K, Nyui N, Tamura N, Fujita T, Kihara M, Toya Y, Takasaki I, Takagi N, Ishii M, Oda K, Horiuchi M, Umemura S: Mechanism of angiotensin II-mediated regulation of fibronectin gene in rat vascular smooth muscle cells. J Biol Chem 1998, 273:26487-26496 [DOI] [PubMed] [Google Scholar]
- 43.Ingram AJ, James L, Ly H, Thai K, Scholey JW: Stretch activation of jun N-terminal kinase/stress-activated protein kinase in mesangial cells. Kidney Int 2000, 58:1431-1439 [DOI] [PubMed] [Google Scholar]
- 44.Moriguchi Y, Matsubara H, Mori Y, Murasawa S, Masaki H, Maruyama K, Tsutsumi Y, Shibasaki Y, Tanaka Y, Nakajima T, Oda K, Iwasaka T: Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ Res 1999, 84:1073-1084 [DOI] [PubMed] [Google Scholar]
- 45.Gainer JV, Morrow JD, Loveland A, King DJ, Brown NJ: Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med 1998, 339:1285-1292 [DOI] [PubMed] [Google Scholar]
- 46.Hutchison FN, Cui X, Webster SK: The antiproteinuric action of angiotensin-converting enzyme is dependent on kinin. J Am Soc Nephrol 1995, 6:1216-1222 [DOI] [PubMed] [Google Scholar]
- 47.Wapstra FH, Navis G, de Jong PE, de Zeeuw D: Chronic angiotensin II infusion but not bradykinin blockade abolishes the antiproteinuric response to angiotensin-converting enzyme inhibition in established adriamycin nephrosis. J Am Soc Nephrol 2000, 11:490-496 [DOI] [PubMed] [Google Scholar]
- 48.Zoja C, Donadelli R, Corna D, Testa D, Facchinetti D, Maffi R, Luzzana E, Colosio V, Bertani T, Remuzzi G: The renoprotective properties of angiotensin-converting enzyme inhibitors in a chronic model of membranous nephropathy are solely due to the inhibition of angiotensin II: evidence based on comparative studies with a receptor antagonist. Am J Kidney Dis 1997, 29:254-264 [DOI] [PubMed] [Google Scholar]
- 49.Gabbai FB, De Nicola L, Garcia GE, Blantz RC: Role of angiotensin in the regulation of renal response to proteins. Semin Nephrol 1995, 15:396-404 [PubMed] [Google Scholar]
- 50.Wolf G, Schneider A, Helmchen U, Stahl RA: AT1-receptor antagonists abolish glomerular MCP-1 expression in a model of mesangial proliferative glomerulonephritis. Exp Nephrol 1998, 6:112-120 [DOI] [PubMed] [Google Scholar]
- 51.Wolf G, Ziyadeh FN, Thaiss F, Tomaszewski J, Caron RJ, Wenzel U, Zahner G, Helmchen U, Stahl RA: Angiotensin II stimulates expression of the chemokine RANTES in rat glomerular endothelial cells. Role of the angiotensin type 2 receptor. J Clin Invest 1997, 100:1047-1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, Giaid A, Harrison DG: Role of endothelin-1 in angiotensin II-mediated hypertension. Hypertension 1997, 30:29-34 [DOI] [PubMed] [Google Scholar]
- 53.Moreau P, d’Uscio LV, Shaw S, Takase H, Barton M, Luscher TF: Angiotensin II increases tissue endothelin and induces vascular hypertrophy: reversal by ET(A)-receptor antagonist. Circulation 1997, 96:1593-1597 [DOI] [PubMed] [Google Scholar]
- 54.Imai T, Hirata Y, Emori T, Yanagisawa M, Masaki T, Marumo F: Induction of endothelin-1 gene by angiotensin and vasopressin in endothelial cells. Hypertension 1992, 19:753-757 [DOI] [PubMed] [Google Scholar]
- 55.Sung CP, Arleth AJ, Storer BL, Ohlstein EH: Angiotensin type 1 receptors mediate smooth muscle proliferation and endothelin biosynthesis in rat vascular smooth muscle. J Pharmacol Exp Ther 1994, 271:429-437 [PubMed] [Google Scholar]
- 56.Li JS, Knafo L, Turgeon A, Garcia R, Schiffrin EL: Effect of endothelin antagonism on blood pressure and vascular structure in renovascular hypertensive rats. Am J Physiol 1996, 271:H88-H93 [DOI] [PubMed] [Google Scholar]
- 57.Rossi GP, Sacchetto A, Rizzoni D, Bova S, Porteri E, Mazzocchi G, Belloni AS, Bahcelioglu M, Nussdorfer GG, Pessina AC: Blockade of angiotensin II type 1 receptor and not of endothelin receptor prevents hypertension and cardiovascular disease in transgenic (mREN2)27 rats via adrenocortical steroid-independent mechanisms. Arterioscler Thromb Vasc Biol 2000, 20:949-956 [DOI] [PubMed] [Google Scholar]