

Abstract
In proteinuric nephropathies tubular atrophy leads to glomerular-tubule disconnection through an unknown mechanism. Here we studied whether proteinuria promoted glomerular-tubule disconnection in individual nephrons and whether this phenomenon was prevented by an angiotensin-converting enzyme (ACE) inhibitor. Passive Heymann nephritis (PHN) and control rats were studied at 4 and 8 months. Two additional groups of PHN rats received lisinopril (40 mg/L) or a calcium channel blocker (lacidipine, 3 mg/kg) from day 7 after surgery to 8 months. At sacrifice, kidneys were serially sectioned to identify glomerular- tubule abnormalities in individual nephrons and changes in interstitial volume. In PHN rats, the time-dependent increase in proteinuria was paralleled by tubular atrophy leading to glomerular-tubule disconnection and interstitial volume enlargement. Marked apoptosis was invariably found in atrophic tubules in contrast to the absent or very mild terminal dUTP nick-end labeling staining in tubules normally connected to glomeruli in PHN animals. Treatment with an ACE inhibitor prevented hypertension, proteinuria, the formation of atrophic tubuli, glomerular-tubule disconnection and limited the fractional interstitial volume expansion. Although lacidipine limited hypertension, it did not reduce proteinuria or prevent tubular atrophy and disconnection. Multivariate analysis showed that the appearance of atubular glomeruli and the increase in interstitial volume were better predicted by proteinuria than blood pressure. This study suggests that ACE inhibitors effectively prevent glomerular-tubule disconnection possibly by their ability of reducing proteinuria, which in turn favors proximal tubular cell apoptosis. Agents that only reduced hypertension but not proteinuria do not affect tubular behavior.
In most renal diseases hemodynamic adaptation of the remaining nephrons, after loss of a critical number of units caused by whatever initial insult, incites a disease progression program ending with uremia. 1-3 Experiments designed to examine the contribution of tubulointerstitial injury to loss of renal function have documented that in rats with renal mass ablation, studied 25 weeks after surgery, progressive renal injury paralleled changes in the normal connections between glomeruli and their proximal tubules. 4 The cause of glomerular-tubule disconnection was not investigated, but the authors speculated that excessive filtration of plasma proteins could be involved. 4 In immune and nonimmune models of renal disease an abnormal accumulation of albumin and IgG in lysosomes of proximal tubular cells has been found in the early stage of the disease. This preceded the interstitial inflammatory reaction, which develops right at the sites of protein overload. 5 Exposure of proximal tubular cells to excess filtered proteins induces the proximal tubular cells to release vasoactive peptides, cytokines, and growth factors that can promote peritubular inflammation and scar formation. 6 Albumin overload of cultured renal epithelial cells to albumin also results in an alteration of the balance between proapoptotic and anti-apoptotic genes in favor of the former. 7 Furthermore, in rats with protein overload proteinuria tubular cell apoptosis is favored over proliferation, 8 which would suggest that heavy and sustained proteinuria itself can cause tubular atrophy and eventual disconnection. Also related to the possible toxicity of reabsorbed proteins on progression, are recent data showing that angiotensin-converting enzyme (ACE) inhibitors, while reducing proteinuria, offer superior protection against declining renal function. 9 Renoprotection afforded by ACE inhibitors has been primarily attributed either to their property of reducing systemic blood pressure or ameliorating the size-selective function of the glomerular membrane by reducing the mean dimensions of large unselective pores, limiting the traffic of macromolecules through the glomerular capillary, and the consequent toxicity to the proximal tubule. 6 The possibility that ACE inhibition also limited the occurrence of glomerular-tubule detachment has not been addressed so far in progressive renal injury.
We have designed the present study to explore: 1) whether proteinuria has any role in tubular atrophy and glomerular-tubule disconnection; and 2) whether treatment with an ACE inhibitor influences tubular atrophy and disconnection independently from the effect on systemic blood pressure control. Studies were performed in rats with passive Heymann nephritis (PHN), an experimental model of human membranous nephropathy.
Materials and Methods
Animals and Experimental Design
Male Sprague-Dawley rats (Charles River Italia s.p.a., Calco, Italy) with initial body weights of 250 to 300 g were used in this study. Animal care and treatment were conducted in accordance with the institutional guidelines that are in compliance with national (D.L. n.116, G.U., suppl.40, 18 feb. 1992, Circolare N°8, G.U., 14 Luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJL358-1, December 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996). All animals were housed in a room in which the temperature was kept constant on a 12-hour dark/12-hour light cycle and allowed free access to standard diet containing 20% protein by weight and tap water. PHN was induced in nonanesthetized rats by a single intravenous injection of 0.4 ml/100 g body wt of rabbit anti-Fx1A antibody. Seven days later, when proteinuria was already present, the animals were subjected to right unilateral nephrectomy to accelerate the onset of renal damage. Accelerated PHN is a very severe model of nephrotic syndrome in which ACE inhibitor treatment is not uniformly effective in reducing proteinuria. 10 Animals were divided into four groups: group 1 (n = 4) and group 2 (n = 8): untreated PHN rats followed for 4 or 8 months after PHN induction, group 3 (n = 7) rats given the ACE inhibitor lisinopril (40 mg/L) from day 7 after PHN induction in the drinking water to month 8, group 4 (n = 5) rats treated with the dihydropyridinic calcium antagonist lacidipine (3 mg/kg) by gavage, from day 7 after surgery to month 8. Previous studies have documented that this lacidipine dose is effective in reducing blood pressure throughout a 24-hour observation period. 11 Two additional groups of rats (groups 5 and 6, n = 4), considered as controls, underwent right unilateral nephrectomy and were sacrificed at 4 and 8 months. In all rats the systolic blood pressure, 24-hour urinary protein excretion, and serum creatinine levels were measured at the end of the study. At sacrifice, the kidneys were removed from anesthetized animals for histological and morphological studies. A range of 40 to 46 glomeruli was examined in each animal. One hundred eighty-five glomeruli in group 1, 319 in group 2, 224 in group 3, 225 in group 4, and 180 in group 5 and 6 were studied.
Analyticals
Systolic blood pressure was recorded by tail plethysmography in conscious rats. 12 Twenty-four hour samples were collected using metabolic cages and proteinuria was determined by modified Coomassie blue G dye-binding assay for proteins with bovine serum albumin as standard. 13
Blood was collected from the tail vein of anesthetized animals. Serum was obtained after whole blood clotting and kept frozen at −20°C until assayed. Creatinine was measured by the alkaline picrate method. 14
Renal Histology
The removed kidneys were fixed for 6 hours in Dubosq-Brazil, dehydrated in alcohol, and embedded in paraffin. Sections of renal tissue (3 μm) were stained with Masson’s trichrome, hematoxylin and eosin, and periodic acid-Schiff reagent. Sections including superficial and juxtamedullary glomeruli were evaluated. Tubular (atrophy, casts, and dilatation) and interstitial changes (fibrosis and inflammation) were graded from 0 to 4+ (0, no changes; 1+, changes affecting <25% of the sample; 2+, changes affecting 25 to 50% of the sample; 3+, changes affecting 50 to 75% of the sample; 4+, changes affecting 75 to 100% of the sample). At least 100 glomeruli were examined for each animal and the extent of glomerular damage was expressed as the percentage of glomeruli presenting focal or global sclerotic lesions. All renal biopsies were analyzed by the same pathologist who was unaware of the nature of the experimental groups.
Morphometric Analysis
Kidney samples fixed in Dubosq-Brazil and embedded in paraffin were serially sectioned at 3-μm intervals and three sections per slide were mounted and stained with periodic acid-Schiff reagent. A video camera (Panasonic, Matsushita Elect. Co., Osaka, Japan) connected to a light microscope and a computer was used. The sections under the microscope were simultaneously visualized on the computer display. An average of 78 sections for each rat was examined. Figure 1 ▶ summarizes the procedures for evaluation of glomerular-tubule connections in individual glomeruli and fractional interstitial volume. The section in the middle of the series (39th) was photographed and used as a map of the serial sections. This map served as a guide to light microscopic examination to identify the individual glomeruli that were followed in the other sections. Only those glomeruli contained entirely within the serially sectioned tissue were examined. Glomerular populations ranging from 180 to 319 depending on the animal group were examined through all the sections before and after the map section. Glomeruli were classified as connected to a normal proximal tubule, connected to an atrophic proximal tubule, or without a tubular connection (Figure 1) ▶ . Proximal tubule segments connected to glomeruli were considered atrophic when there was thinning of tubular cells accompanied by loss of brush border and narrowing of the tubular lumen. The findings were expressed as percentage of the three categories of glomeruli over the total number of glomeruli examined. The fractional volume of the cortex occupied by the interstitium was measured by point counting using a 10 × 10 orthogonal grid overlaid on the digital image on the computer monitor at magnification of ×1200. In each case, a minimum of 3000 points in three sections at least 100 μ apart were counted, and the fractional volumes of the interstitium were expressed as the percentages of points falling on these structural components.
Figure 1.

Schematic representation of the procedures for evaluation of glomerular-tubule connections in individual glomeruli and fractional interstitial volume. On the right side of the figure the pictures show the pattern of glomerular- tubule connections.
Terminal Deoxynucleotidyl (TdT)-Mediated dUTP Nick-End Labeling (TUNEL) Assay
TUNEL assay for the detection of apoptosis was performed on Dubosq-Brazil-fixed and paraffin-embedded renal sections. Apoptosis was evaluated in three sections at least 9 μ apart in all PHN animals at 8 months. Briefly, 3-μm sections were deparaffinized and treated with 20 μg/ml of proteinase K for 10 minutes at 37°C. After two washes with phosphate-buffered saline (PBS) endogenous peroxidase activity of kidney sections was blocked by incubation with 0.3% H2O2 in PBS for 15 minutes. Then, sections were incubated with the TUNEL reaction mixture (Roche Diagnostic, Mannheim, Germany) containing terminal deoxynucleotidyl transferase and fluorescein isothiocyanate-labeled nucleotide mixture for 60 minutes at 37°C in the dark. Sheep anti-fluorescein antibody conjugated with peroxidase (TUNEL Pod; Boehringer-Mannheim, Mannheim, Germany) was added for 30 minutes at 37°C and finally developed with diaminobenzidine. Sections were then counterstained with Harris hematoxylin (Biooptica, Milan, Italy). Negative control was obtained by omitting terminal deoxynucleotidyl transferase in TUNEL reaction mixture while positive control with the addition of DNase for 15 minutes at 37°C.
Statistical Analysis
Food and water intake, body and kidney weight, and blood pressure data are given as mean ± SD. All of the other data are reported as median value (interquartile range). One way-analysis of variance followed by Tukey test for multiple comparisons was used to compare experimental groups. Morphometric data were analyzed by the nonparametric Kruskal-Wallis test. Pearson correlation coefficient was used to assess the relationship between urinary protein excretion or systolic blood pressure and morphometric parameters (percent atubular glomeruli and fractional interstitial volume) in all groups of rats. Multivariate analysis was performed using multiple linear regression. 15 The statistical significance level was defined as P < 0.05.
Results
Systemic and Renal Parameters
All animals in the experimental groups survived the duration of the study. Total food intake was comparable in all PHN and control rats for the entire study period. The average food intake at sacrifice was: PHN 4 months: 24.3 ± 7.8; PHN 8 months: 25 ± 2.2; PHN 8 months lisinopril: 26.8 ± 4.0; PHN 8 months lacidipine: 25 ± 2.9; controls 4 months: 20.8 ± 2.6; 8 months: 26 ± 2.5 g/day. Water intake was 55 ± 13 ml/day in PHN at 4 months. At month 8 all PHN animals had an increase in water intake over controls independently of treatment (PHN, 66 ± 15.1; PHN and lisinopril, 59 ± 12.5; PHN and lacidipine, 70 ± 12.2 versus controls 4 months, 43.8 ± 9.5; 8 months, 40 ± 10 ml/day). Rats with PHN gained weight along the study; however, at 8 months body weight was numerically, but not significantly, lower than controls (PHN 8 months, 614 ± 53; controls, 686 ± 73 g). Treatments of PHN rats did not affect the weight gain (PHN 8 months lisinopril, 622 ± 49; PHN 8 months lacidipine, 593 ± 80 g). Hypertrophy of the left kidney was observed already at 4 months in PHN rats (PHN 4 months, 4.1 ± 1 g) and persisted at month 8 (4.2 ± 1.1 versus controls, 3.0 ± 0.4 g). Lisinopril, but not lacidipine, decreased left kidney hypertrophy to a significant extent (PHN 8 months lisinopril, 3.6 ± 0.6 g; P < 0.05 versus PHN 8 months, PHN 8 months lacidipine, 4.3 ± 0.5 g). As shown in Table 1 ▶ , in untreated PHN rats systolic blood pressure increased throughout time reaching values at month 8 that were significantly higher than controls (P < 0.05). Both lisinopril and lacidipine prevented (P < 0.05) the increase in systolic blood pressure. Lisinopril and lacidipine maintained systolic blood pressure at levels that were not significantly different from those of controls (Table 1) ▶ . Values of urinary protein excretion are given in Table 1 ▶ . PHN rats showed significant proteinuria at month 4 (P < 0.05 versus controls). Urinary protein excretion further rose at month 8 in untreated PHN rats in respect to controls (P < 0.05). The median value of proteinuria in the lisinopril-treated group was significantly (P < 0.05) lower than that of untreated PHN, the ACE inhibitor being effective in significantly limiting proteinuria in five out of seven PHN animals. In contrast, the calcium antagonist did not affect the development of proteinuria. Urinary protein levels in lacidipine-treated animals were comparable to untreated PHN rats. PHN rats developed a mild renal insufficiency at month 4. Renal function was significantly impaired starting from month 8 (P < 0.05 versus controls). In PHN animals given lisinopril, serum creatinine was numerically lower than in untreated PHN, the difference, however, did not reach statistical significance because of the presence of two animals that did not respond to ACE inhibition. Furthermore, PHN rats given lacidipine exhibited high levels of serum creatinine comparable to those of untreated PHN rats. The results of renal histology are reported in Table 1 ▶ . The percentage of sclerotic glomeruli and the score of interstitial damage were significantly increased in untreated PHN rats at months 4 and 8 in respect to controls (P < 0.05). The value of tubular damage was significantly increased (P < 0.05) over controls at month 8. Lisinopril strikingly protected animals from the development of glomerular and tubular structural injury as indicated by a median value of glomerulosclerosis of 9% and the score for tubulointerstitial damage similar to that of controls. By contrast, lacidipine did not affect the development of glomerulosclerosis and tubulointerstitial lesions.
Table 1.
Systemic and Renal Functional Parameters in Control Rats and Rats with Accelerated PHN
| Controls 4 months | PHN 4 months | Controls 8 months | PHN 8 months | PHN + Lisinopril | PHN + Lacidipine | |
|---|---|---|---|---|---|---|
| Systolic blood pressure, mmHg | 119± 7 | 143± 28 | 130± 4 | 160 ± 13* | 110± 19† | 100± 6† |
| Proteinuria, mg/day | 32 (28–42) | 663* (658–665) | 47 (37–56) | 742* (664–875) | 155*† (63–707) | 831.4* (807.3–860.1) |
| Serum creatinine, mg/dl | 0.7 (0.7–0.8) | 1.0 (0.9–1.1) | 0.8 (0.7–0.8) | 1.4* (1.2–1.5) | 0.9 (0.8–1.4) | 1.3 (1.1–1.6) |
| Glomeruli with sclerosis, % | 0.5 (0–1) | 35* (24–54.5) | 4 (3.5–4.5) | 65* (55–74) | 9† (6.0–52.0) | 80* (71–89) |
| Interstitial damage, score | 0 (0–0) | 2.0* (1.5–2.0) | 0 (0–0.5) | 2.5* (2.0–3.0) | 1.0† (0–2.0) | 2.0* (2.0–3.0) |
| Tubular damage, score | 0 (0–0.5) | 2.0 (1.5–2.0) | 1.0 (1.0–1.0) | 2.9* (2.4–3.0) | 1.0† (0–3.0) | 2.0* (2.0–3.0) |
All data are expressed as median and interquartile range except the blood pressure (mean ± SD).
*P < 0.05 versus controls at corresponding time.
†P < 0.05 versus PHN 8 months.
Morphometric Studies
Kidneys of PHN rats at month 4 showed tubular injury characterized by mild dilation, atrophy, and luminal casts. Tubular changes were associated with interstitial mononuclear cell accumulation and fibrosis. As shown in Table 2 ▶ , by month 4 there was a remarkable increase in the prevalence of atubular glomeruli and glomeruli connected with atrophic tubuli with reduced lumen size, thickened basement membrane, and without brush border compared to controls. Stenosis of tubular neck was similar to that described in a radiation model of chronic renal failure. 16 At month 8, tubulointerstitial changes consisted of severe tubular atrophy, interstitial fibrosis, and inflammation, were associated with the presence of large proteinaceous casts in tubular lumens. Within the glomerular population, the number of glomeruli connected with a normal tubule significantly (P < 0.01) decreased in PHN rats at month 8 over month 4, both values were significantly different from those of respective controls (PHN 4 months, P < 0.05; PHN 8 months, P < 0.01; Table 2 ▶ ). Concomitantly, a further and significant (P < 0.01) increase in the percentage of atubular glomeruli and glomeruli connected with atrophic tubules was observed at month 8 in respect to month 4. Altogether, almost three quarters of the glomerular population were no longer connected to normal proximal tubules. Lisinopril prevented the disconnection of glomeruli from tubules and the appearance of atrophic tubules. The median value of atubular glomeruli in lisinopril-treated PHN rats was comparable to the values of control animals. In the two PHN rats that did not respond to the ACE inhibitor, atubular glomeruli and glomeruli with an atrophic tubule were observed. Lacidipine did not affect the percentage of atubular glomeruli and glomeruli connected with atrophic tubules with respect to untreated PHN animals at month 8. Fractional interstitial volume significantly increased in PHN rats at month 4 with respect to controls (P < 0.05), and further rose at month 8 (P < 0.01 versus PHN 4 months and controls). Median fractional interstitial volume was significantly reduced in the lisinopril-treated groups (P < 0.05). Interstitial volume was even normalized in animals whose proteinuria was effectively lowered by the ACE inhibitor. In contrast, lacidipine treatment did not affect fractional interstitial volume, which was not statistically different from PHN at month 8.
Table 2.
Summary of Morphometric Studies
| Controls 4 months | PHN 4 months | Controls 8 months | PHN 8 months | PHN +Lisinopril | PHN +Lacidipine | |
|---|---|---|---|---|---|---|
| Glomeruli with a normal tubule, % | 98.5 (96–99.5) | 81* (70–86) | 98 (98–99) | 32†‡ (31–39) | 95§ (45–98) | 47* (43–57) |
| Glomeruli with an atrophic tubule, % | 1 (0–2) | 11* (8–20) | 0.5 (0–1.5) | 41†‡ (35–43) | 5* (2–32) | 34* (23–37) |
| Atubular glomeruli, % | 0.5 (0–2.5) | 9* (8–11) | 0.5 (0–2) | 27†‡ (24–30) | 0§ (0–23) | 20* (19–20) |
| Fractional interstitial volume, % | 12.5 (11–13) | 21* (20–23) | 9.5 (9–10.5) | 39†‡ (29–49) | 16*§ (11–30) | 26* (20–26) |
All data are expressed as median and interquartile range.
*P < 0.05 and †P < 0.01 versus controls at correspondent time.
‡P < 0.01 versus PHN 4 months; §P < 0.05 versus PHN 8 months.
The percentage of atubular glomeruli or the fractional interstitial volume had a significant positive correlation with urinary protein excretion, as well as with systolic blood pressure (Table 3) ▶ . The Pearson coefficients were higher for the correlations with proteinuria than with blood pressure. Multivariate analysis showed that proteinuria predicted better renal structural changes than systolic blood pressure (Table 3) ▶ .
Table 3.
Univariate and Multivariate Analysis of the Correlation between Urinary Protein Excretion or Systolic Blood Pressure and Morphometric Results
| Parameters | Atubular glomeruli | Fractional interstitial volume | ||||
|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||
| r Coefficient | P value | P value | r Coefficient | P value | P value | |
| Urinary protein excretion | 0.88 | <0.0001 | <0.0001 | 0.79 | <0.0001 | <0.0001 |
| Systolic blood pressure | 0.46 | 0.008 | 0.07 | 0.51 | 0.003 | 0.03 |
Evaluation of Apoptosis in Normal and Atrophic Tubules of PHN Rats
Evidence that albumin overload in vitro and in vivo results in marked apoptosis of tubular epithelial cells, 7,8 together with the observation of the presence of albumin and IgG reabsorption droplets in proximal tubular cells of PHN rats, 5 prompted us to evaluate apoptosis in glomeruli connected to a normal or an atrophic tubule in all PHN rats at 8 months. Three sections of each animal were evaluated. No positive or a very mild TUNEL staining in occasionally few cells was observed in tubuli normally connected to the glomerulus. By contrast, a strong TUNEL staining was invariably detected in tubular cells of all atrophic tubules that also showed a flattened appearance, scarce cytoplasm, and no recognizable brush border. This latter findings are in line with a very recent observation of positively stained nuclei of apoptotic cells in atrophic tubules of rats with subtotal nephrectomy. 17 Figure 2 ▶ shows representative photomicrographs of proximal tubules normally connected to a glomerulus and an atrophic tubule.
Figure 2.
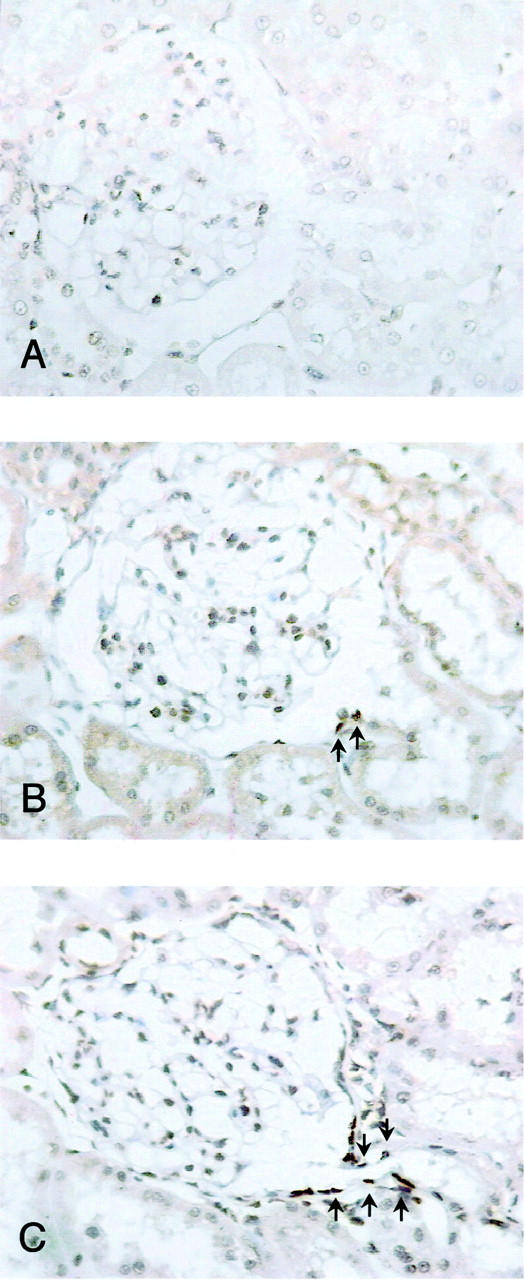
Representative TUNEL staining of proximal tubules normally connected to a glomerulus (A and B) and an atrophic tubule (C) of PHN rats at month 8. A: The absence of TUNEL staining observed in the majority of tubuli normally connected to a glomerulus is shown. B: The staining (arrows) occasionally detected in a few cells of tubuli normally connected to a glomerulus is shown. Original magnifications, ×300.
Discussion
In the accelerated PHN model, used in this study, a time-dependent increase in proteinuria was found during the 8-month follow-up, associated with tubulointerstitial injury and signs of renal dysfunction. This was paralleled by findings of atrophic tubules and of glomeruli no longer connected to their proximal tubules.
Glomerular-tubule disconnection has been reported in several experimental models such as radiation nephropathy 18 or nephropathy induced by tubular toxic substances such as lithium 19 and cisplatin 20 and also in patients with chronic allograft rejection. 21 In lithium-induced nephropathy of newborn rats, the noxious agent was administered during the developing phase of the kidney, 19 which is obviously more vulnerable to the toxic substances. In the model of cisplatin toxicity of adult rats, damage of specific segments of the tubules clearly preceded tubular atrophy, which in turn determined glomerular-tubule disconnection. 20 In a model of renal mass ablation, Gandhi and colleagues 4 also reported that declining renal function with time paralleled tubular atrophy and disconnection and suggested that the process of protein handling by the kidney could play a role. This possibility is entirely consistent with previous findings, that in the remnant kidney, renal injury correlated more closely with proteinuria than with the fraction of renal mass removed. 22,23
Here, in a model that mimics chronic proteinuric diseases in humans 10 much more closely than the remnant kidney, we found a highly significant correlation between the amount of protein excreted in the urine and the percentage of atubular glomeruli. After excessive glomerular ultrafiltration of plasma proteins, tubule over-reabsorption activates an intracellular program of up-regulation of vasoactive and inflammatory genes, which has been extensively studied in the past 10 years in the context of interstitial inflammation and fibrosis. 5,6,24,25 It is possible that the above changes promote a progressive obliteration of peritubular capillaries, which in turn serve to disconnect proximal tubuli from their corresponding glomeruli. 23,24,26,27 This is in harmony with human data that show a positive correlation between the volume fraction of the interstitium and the percentage of proximal tubuli without connection to a glomerulus. 28 An alternative mechanism is that tubular protein reabsorption can directly induce proapoptotic gene activation, thus favoring tubular atrophy and disconnection. This possibility has received a strong support by findings that albumin overload dose and time-dependently induced apoptosis of cultured proximal tubular cells and resulted in up-regulation of pro-apoptotic Fas and Fas-associated protein with death domain and activation of caspase 8. 7 Data in rats with overload proteinuria are also in favor of the above possibility, to the extent that significantly more apoptotic particles were found in cortical and medullary tubular cells, as compared to control rats. 8 The present findings of a strong TUNEL staining in atrophic tubules of PHN rats, but not in tubuli normally connected to glomeruli, which parallels tubular protein accumulation, indicate that indeed protein handling may favor apoptosis and subsequent injury.
When rats with PHN were given lisinopril, which limits proteinuria by restoring the size-selective properties of the glomerular filter, 29 tubular atrophy and disconnection were remarkably prevented. PHN animals, which did not respond to ACE inhibition in terms of reduction of proteinuria, had a glomerular population consisting mainly of atubular glomeruli and glomeruli connected with atrophic tubules, which again would seem consistent with the possibility that excessive protein handling favors disconnection. The present results are at variance to those of a previous study showing that reduction in AII activity did not prevent tubule loss during the early and late phase of recovery from acute ischemia despite AII blockade primarily prevented the development of proteinuria. 30 Differences between the animal models as for the type of glomerular injury, which in this latter study was secondary to acute ischemia, and treatment mode could account for opposing results. On the other hand, a protective effect of ACE inhibitors was been recently described in radiation nephropathy, characterized by glomeruli with stenosis of glomerulotubular neck and atubular glomeruli. 18 In this study although captopril did not influence the absolute fraction of necks that were stenotic, it did prevent the evolution of glomeruli with necks to atubular glomeruli. The ACE inhibitors’ renoprotection has been attributed to their systemic antihypertensive effect that translates to the reduction of glomerular capillary hypertension. Here, we formally compared the effect of lisinopril with that of the dihydropyridinic calcium antagonist lacidipine, which is equally effective on systemic blood pressure but does not affect urinary protein excretion rate. The results indicated that lacidipine-treated animals had a high percentage of atubular glomeruli and glomeruli connected with atrophic tubules. Multivariate analysis of all of the available data found that tubular injury and disconnection were better predicted by proteinuria than by blood pressure. Whether the observed tubular changes were the consequence of a diverse effect of the two study drugs on glomerular pressure could not be excluded. In this context, conflicting results on the effect of calcium channel blocker on intrarenal hemodynamics derived from micropuncture studies. Data in uninephrectomized diabetic rats has shown that fosinopril, but not nifedipine, normalized glomerular capillary pressure and increased Kf. 31 However, studies in the remnant kidney model indicated that verapamil was also effective. 32,33 In the present study, micropuncture experiments were not performed for the lack of superficial glomeruli in the animal strain. Further studies are needed to definitely address the different effect of ACE inhibitor and calcium channel blocker on glomerular capillary pressure in PHN rats.
The fractional volume of the cortex occupied by interstitium was markedly increased in PHN rats, and lisinopril reduced both interstitial expansion and disconnection in those rats whose protein excretion was reduced by ACE inhibitors, an effect not shared by lacidipine. Taken together, the present data reinforce the possibility that the extent of interstitial inflammation played a role in the process of disconnection.
In summary, the present study has documented that in the accelerated PHN model 1) a time-dependent increase in urinary protein excretion parallels the emergence of atrophic tubules and glomerular-tubule disconnection and the expansion of fractional volume of the cortex occupied by the interstitium; 2) atrophic tubuli invariably showed signs of cell apoptosis, which precedes and may favor disconnection; 3) ACE inhibition effectively limits proteinuria and prevents the formation of atrophic tubules, glomerular-tubule disconnection, and the fractional interstitial volume expansion; 4) lacidipine, that does not reduce proteinuria despite an excellent effect on systemic blood pressure, failed to prevent the appearance of atrophic tubules or atubular glomeruli.
The speculation raised by previous studies that proteinuria might be a factor in tubular atrophy and disconnection is suggested by our present results. Findings that an ACE inhibitor, but not lacidipine, prevents glomerular-tubule disconnection disclose a novel mechanism of renoprotection of this class of compounds and may have implications in human renoprotective medicine.
Acknowledgments
We thank Dr. Carla Zoja and Dr. Mauro Abbate for invaluable help and fruitful discussion; Fabio Sangalli for computer assistance; Dr. Annalisa Perna for performing the statistical analysis; and Laura Arioli for help in the preparation of the manuscript.
Footnotes
Address reprint requests to Elena Gagliardini, Biol. Sci.D., “Mario Negri” Institute for Pharmacological Research, Via Gavazzeni, 11, 24125 Bergamo, Italy. E-mail: gagliardini@marionegri.it.
E. G. is the recipient of a fellowship in memory of Dott. Luigi and Tilde Bianchi from the “Associazione Ricerca Malattie Rare” (ARMR), Bergamo, Italy.
References
- 1.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM: Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol 1981, 241:F85-F93 [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Meyer TW, Rennke HG, Brenner BM: Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J Clin Invest 1985, 76:612-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner BM, Meyer TW, Hostetter TH: Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 1982, 307:652-659 [DOI] [PubMed] [Google Scholar]
- 4.Gandhi M, Olson JL, Meyer TW: Contribution of tubular injury to loss of remnant kidney function. Kidney Int 1998, 54:1157-1165 [DOI] [PubMed] [Google Scholar]
- 5.Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G: In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol 1998, 9:1213-1224 [DOI] [PubMed] [Google Scholar]
- 6.Remuzzi G, Ruggenenti P, Benigni A: Understanding the nature of renal disease progression. Kidney Int 1997, 51:2-15 [DOI] [PubMed] [Google Scholar]
- 7.Erkan E, De Leon M, Devarajan P: Albumin overload induces apoptosis in LLC-PK1 cells. Am J Physiol 2001, 280:F1107-F1114 [DOI] [PubMed] [Google Scholar]
- 8.Thomas ME, Brunskill NJ, Harris KPG, Bailey E, Pringle JH, Furness PN, Walls J: Proteinuria induces tubular cell turnover: a potential mechanism for tubular atrophy. Kidney Int 1999, 55:890-898 [DOI] [PubMed] [Google Scholar]
- 9.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 1998, 339:1448-1456 [DOI] [PubMed] [Google Scholar]
- 10.Benigni A, Corna D, Maffi R, Benedetti G, Zoja C, Remuzzi G: Renoprotective effect of contemporary blocking of angiotensin II and endothelin-1 in rats with membranous nephropathy. Kidney Int 1998, 54:353-359 [DOI] [PubMed] [Google Scholar]
- 11.Perico N, Amuchastegui CS, Malanchini B, Bertani T, Remuzzi G: Angiotensin-converting enzyme inhibition and calcium channel blockade both normalize early hyperfiltration in experimental diabetes, but only the former prevents late renal structural damage. Exp Nephrol 1994, 2:220-228 [PubMed] [Google Scholar]
- 12.Pfeffer JM, Pfeffer MA, Frohlich ED: Validity of an indirect tail-cuff method for determining systolic arterial pressure in unanesthetized normotensive and spontaneously hypertensive rats. J Lab Clin Med 1971, 78:957-962 [PubMed] [Google Scholar]
- 13.Read SM, Northcote DH: Minimization of variation in the response to different proteins of the Coomassie blue G dye-binding assay for protein. Anal Biochem 1981, 116:53-64 [DOI] [PubMed] [Google Scholar]
- 14.Bonsnes RW, Tausski HA: The colorimetric determination of creatinine of the Jaffé reaction. J Biol Chem 1945, 158:581 [Google Scholar]
- 15.Draper N, Smith H: Applied Regression Analysis. 1981. J. Wiley and Sons Inc., New York
- 16.Cohen EP, Robbins MEC, Whitehouse E, Hopewell JW: Stenosis of tubular neck: a possible mechanism for progressive renal failure. J Lab Clin Med 1997, 129:567-573 [DOI] [PubMed] [Google Scholar]
- 17.Yang B, Johnson TS, Thomas GL, Watson PF, Wagner B, Skill NJ, Haylor JL, Al Nahas AM: Expression of apoptosis-related genes and proteins in experimental chronic renal scarring. J Am Soc Nephrol 2001, 12:275-288 [DOI] [PubMed] [Google Scholar]
- 18.Cohen EP, Regner K, Fish BL, Moulder JE: Stenotic glomerulotubular necks in radiation nephropathy. J Pathol 2000, 190:484-488 [DOI] [PubMed] [Google Scholar]
- 19.Marcussen N, Ottosen PD, Christensen S, Olsen TS: Atubular glomeruli in lithium-induced chronic nephropathy in rats. Lab Invest 1989, 61:295-301 [PubMed] [Google Scholar]
- 20.Marcussen N: Atubular glomeruli in cisplatin induced chronic interstitial nephropathy: an experimental stereologic investigation. APMIS 1992, 98:1087-1097 [DOI] [PubMed] [Google Scholar]
- 21.Pagtalunan ME, Oberbauer R, Haas M, Barlan M, Mayer G, Olson J, Meyer TW: Atubular glomeruli in patients with chronic allograft rejection. Transplantation 1996, 61:1166-1171 [DOI] [PubMed] [Google Scholar]
- 22.Gretz N, Waldherr R, Stauch M: The remnant kidney model. Gretz N Strauch M eds. Rat Model of Chronic Renal Failure. 1992, :pp 1-28 Karger, Basel [Google Scholar]
- 23.Griffin KA, Picken M, Bidani AK: Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J Am Soc Nephrol 1994, 4:2023-2031 [DOI] [PubMed] [Google Scholar]
- 24.Eddy AA: Experimental insights into the tubulointerstitial disease accompanying primary glomerular lesions. J Am Soc Nephrol 1994, 5:1273-1287 [DOI] [PubMed] [Google Scholar]
- 25.Zoja C, Donadelli R, Colleoni S, Figliuzzi M, Bonazzola S, Morigi M, Remuzzi G: Protein overload stimulates RANTES production by proximal tubular cells depending on NF-kB activation. Kidney Int 1998, 53:1608-1615 [DOI] [PubMed] [Google Scholar]
- 26.Mackensen-Haen S, Bader R, Ground K, Bohle A: Correlations between renal cortical interstitial fibrosis, atrophy of proximal tubules and impairment of glomerular filtration rate. Clin Nephrol 1981, 15:167-171 [PubMed] [Google Scholar]
- 27.Bohle A, Gise H, Mackensen-Haen S, Stark-Jacob B: The obliteration of the postglomerular capillaries and its influence upon the function of both glomeruli and tubuli. Klin Wschwr 1981, 59:1043-1051 [DOI] [PubMed] [Google Scholar]
- 28.Marcussen N, Olsen TS: Atubular glomeruli in patients with chronic pyelonephritis. Lab Invest 1990, 62:467-473 [PubMed] [Google Scholar]
- 29.Remuzzi A, Puntorieri S, Battaglia C, Bertani T, Remuzzi G: Angiotensin converting enzyme inhibition ameliorates glomerular filtration of macromolecules and water and lessens glomerular injury in the rat. J Clin Invest 1990, 85:541-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pagtalunan ME, Olson JL, Meyer TW: Contribution of angiotensin II to late renal injury after acute ischemia. J Am Soc Nephrol 2000, 11:1278-1286 [DOI] [PubMed] [Google Scholar]
- 31.Anderson S, Rennke HG, Brenner BM, Zayas MA, Lafferty HM, Troy JL, Sandstrom DJ: Nifedipine versus fosinopril in uninephrectomized diabetic rat. Kidney Int 1992, 41:891-897 [DOI] [PubMed] [Google Scholar]
- 32.Anderson S: Renal hemodynamic effects of calcium antagonists in rats with reduced renal mass. Hypertension 1991, 17:288-295 [DOI] [PubMed] [Google Scholar]
- 33.Yoshioka T, Shiraga H, Yoshida Y, Fogo A, Glick AD, Deen WM, Hoyer JR, Ichikawa I: “Intact nephrons” as the primary origin of proteinuria in chronic renal disease. Study in the rat model of subtotal nephrectomy. J Clin Invest 1988, 82:1614-1623 [DOI] [PMC free article] [PubMed] [Google Scholar]