

Abstract
Tumor necrosis factor-α (TNF-α)-stimulated gene-6 (TSG-6) is up-regulated by various cytokines and growth factors. TSG-6 binds to hyaluronan in inflamed synovial tissue and forms a complex with a serine protease inter-α-trypsin inhibitor (IαI), increasing the protease inhibitory effect of IαI >100-fold. The TSG-6/IαI complex then blocks serine proteases, including the plasminogen-plasmin activation, probably the most important component in the activation processes of matrix metalloproteinases. To gain insight into the mechanisms of TSG-6 action in arthritis, we have used an autoimmune murine model (proteoglycan-induced arthritis) for systemic, and a monoarticular form of arthritis (antigen-induced arthritis) for local treatment of arthritis with recombinant mouse TSG-6 (rmTSG-6). Intravenous injection of rmTSG-6 induced a dramatic reduction of edema in acutely inflamed joints by immobilizing CD44-bound hyaluronan and, in long-term treatment, protected cartilage from degradation and blocked subchondral and periosteal bone erosion in inflamed joints. The intra-articular injection of a single dose (100 μg) of rmTSG-6 exhibited a strong chondroprotective effect for up to 5 to 7 days, preventing cartilage proteoglycan from metalloproteinase-induced degradation. In contrast, rmTSG-6 did not postpone the onset, nor reduce the incidence of arthritis. We were unable to detect any significant differences between control and rmTSG-6-treated animals when various serum markers (including pro- and anti-inflammatory cytokines, auto- and heteroantibody productions) or antigen-specific T-cell responses were compared, nor when the expressions of numerous cell surface receptors or adhesion molecules were measured. TSG-6 seems to play a critical negative regulatory feed-back function in inflammation, especially in arthritic processes.
Tumor necrosis factor-α (TNF-α)-stimulated gene 6 (TSG-6) was first identified by differential screening of a cDNA library of TNF-α-stimulated human fibroblasts. 1 Although a constitutive expression is low or not detectable, virtually all cell types are able to produce TSG-6 on stimulation with various components either in vitro or in vivo. 2-5
TSG-6 is a 35-kd secreted glycoprotein. 2 The N-terminal half of the amino acid sequence shows homology to the Link module, which is a conservative sequence in hyaluronan (HA)-binding proteins such as CD44, cartilage link protein, and G1 domains of aggrecan and versican. 2,6 The C-terminal half of TSG-6 (CUB domain) shares sequence homologies with the A chain of complement component C1s/C1r, uEGF, a protein involved in development of sea urchin embryos and bone morphogenic protein-1. 7,8 Based on these structural homologies, it is strongly believed that TSG-6 may play a crucial role in extracellular matrix formation, inflammatory cell migration, cell proliferation, and developmental processes. 4,7
Although TSG-6 cannot be detected in synovial fluids or sera of normal individuals, synoviocytes from rheumatoid patients constitutively produce TSG-6 protein and it can be detected in sera of patients with rheumatoid arthritis, or less frequently in osteoarthritis. 3,9,10 Exogenous TNF-α or interleukin (IL)-1 further increases in TSG-6 production by rheumatoid synoviocytes. 3 A 120-kd complex containing TSG-6 was regularly detected in TSG-6 (35 kd)-producing cultures in the presence of serum, but not in serum-free medium. 3,9,11 The protein that bound TSG-6 was purified from human serum and identified as inter-α-inhibitor (IαI). 9,11 IαI is a HA-binding serum protein with serine protease inhibitory activity. 12 The target enzymes include trypsin, chymotrypsin, cathepsin G, leukocyte elastase, acrosin, and plasmin. 8,10,12
Interestingly, when TSG-6 is complexed with IαI, the complex exhibits a more than 100-fold higher protease inhibitory effect against plasmin than the IαI alone. 9,11 This effect of TSG-6 on IαI seems to be specific for plasmin, as no such increase in inhibitory effect was observed on trypsin or neutrophil elastase. 11 To further test the inhibitory effects of the TSG-6/IαI complex on protease network and inflammation, a murine air-pouch model of acute inflammation was used. 11 Simultaneous injection of human recombinant TSG-6 with carrageenan into the air pouch exhibited a potent anti-inflammatory effect. 11 Recently, it was shown that recombinant TSG-6 had a beneficial effect on collagen-induced arthritis (CIA) reducing both the incidence and severity of arthritis. 13 Taken together, TSG-6 induced by proinflammatory cytokines at the site of inflammation, binds tightly to IαI, and dramatically increasing the IαI’s serine protease inhibitory effect. Thus, TSG-6 may be a key component in a negative feedback loop controlling inflammatory response in local arthritic processes.
In this article, we investigated whether TSG-6 plays a role in prevention of cartilage breakdown in two murine models of experimental arthritis. We studied the in vivo role of TSG-6 in a systemic autoimmune disease of proteoglycan-induced arthritis (PGIA), and then in a monoarticular form of inflammatory arthritis by injection of recombinant TSG-6 into the knee joints of animals with antigen-induced arthritis (AIA).
Materials and Methods
Recombinant Mouse TSG-6 (rmTSG-6) and Anti-TSG-6 Antibody
Purified plasmid DNA (Lonza pEE14.1 vector; Lonza Biologics plc., Slough, Berkshire, UK) containing a full-length (1654 bp) mouse TSG-6 cDNA clone 4 was used for transfection of Chinese hamster ovary (CHO-K1; American Type Culture Collection, Rockville, MD) cells by the standard method of CaCl2 precipitation. Transfected cells were cultured in glutamine-free condition in the presence of 25 μmol/L methionine sulfoximine (Sigma Chemical Co., St. Louis, MO) for the selection of stable transfected cell lines. rmTSG-6 was purified from culture media of stable transfected cloned cell line using HA-coupled EAH-Sepharose (Pharmacia Biotech, Piscataway, NJ). HA-bound rmTSG-6 was eluted with 4 mol/L guanidine hydrochloride in 0.1 mol/L Na-acetate buffer (pH 5.8), dialyzed against distilled water, and then lyophilized. Purified rmTSG-6 was tested by Western blotting using a rabbit polyclonal antibody (TSG-6-CR21) raised against a synthetic peptide (135CGGVFTDPKRIFKSPG), located at the end-terminal end of the CUB domain. 4 For quantitative measurement of serum TSG-6 (either measuring serum level in arthritic animals or during the treatments) we used a sandwich enzyme-linked immunosorbent assay (ELISA), confirmed by Western blots. In ELISA, the IgG fraction of rabbit serum to mouse TSG-6 synthetic peptide (36GVYHREARAGRYKL) was used as capture antibody and affinity-purified and biotinylated TSG-6-CR21 antibody for detection of TSG-6 following a protocol described for other serum markers. 14 For organ/tissue distribution and kinetic studies, rmTSG-6 was labeled with 125I using a chloramine-T method.
Immunization Protocol for PGIA, Scoring System, and Treatment with rmTSG-6
High-density cartilage proteoglycan aggrecan (PG) was purified by CsCl gradient centrifugation from human cartilage and depleted of glycosaminoglycan side chains as described. 15 All animal experiments were approved by the Institutional Animal Care and Use Committee (Rush University, Chicago, IL). Female BALB/c mice (National Cancer Institute, Frederick, MD) were immunized intraperitoneally with cartilage PG. 15,16 First antigen injection (100 μg protein) was given in complete Freund’s adjuvant (Difco, MI), and the same doses of antigen were injected as second and third boosters in incomplete Freund’s adjuvant. Typically, BALB/c mice developed swelling and redness in one or more limbs, 7 to 14 days after the third injection of PG. 15,16 This stage of immunized animals (from 7 days after the third injection) is regarded as the prearthritic phase of PGIA. A standard scoring system, based on swelling and redness of paws, was used for the assessment of the severity of arthritis. 17,18 The time of appearance of swelling and redness was recorded as the time of onset of arthritis. Joint swelling was scored (ranging from 0 to 4 of each paw) and expressed as cumulative acute arthritis score resulting in a maximum score of 16. During the treatment period, paw (joint) thicknesses at frontal and sagittal planes of all ankles and wrists were measured daily with a microcaliper. 17 Arthritic animals were injected intraperitoneally or intravenously via the retro-orbicular venous plexus with 100 μl phosphate-buffered saline (PBS), rmTSG-6 (100 μg in PBS), or rmTSG-6 and Pep-1 simultaneously. Pep-1 is a synthetic peptide (NH2-GAHWQFNALTVRGGGS) that shows HA-binding properties 19 and can also bind to the Link module of either CD44 or TSG-6 (our unpublished data). The optimal therapeutic dose, serum/tissue distribution, half-life, and kinetics of injected rmTSG-6 was determined in preliminary experiments. Sera were collected before the injection, or every second hour in kinetic studies, and at the end of the experiment. Spleen cells for in vitro tests were isolated at the end of the experiment and paws from all mice were collected and fixed in 10% formalin containing 5% cetylpirimidium chloride (Sigma). Legs were decalcified in 5% formic acid, paraffin embedded, and sections processed for routine histology or immunohistochemistry. 15,16,20,21
Immunization Protocol for AIA and Experimental Groups
C57BL/6j mice (National Cancer Institute) were immunized with 100 μg of methylated bovine serum albumin (mBSA; Sigma) in 100 μl of PBS emulsified in 100 μl of complete Freund’s adjuvant. Injections were given subcutaneously in the flanks, and into the proximal tail. After 1 week, mice were subcutaneously boosted with 100 μg of mBSA emulsified in 100 μl of incomplete Freund’s adjuvant, and the same boost was repeated 3 weeks later. 21 Two to 3 weeks after the final boost, knees of immunized mice under ketamine/xylazine anesthesia were injected intra-articularly in the following combination: animals of group 1 received a single dose of 60 μg of mBSA in 6 μl of PBS into the right knee, and 6 μl of PBS into the left knee joint. Animals of group 2 received a single dose of 60 μg of mBSA and 100 μg of rmTSG-6 in 6 μl of PBS intra-articularly into the right knee, and 60 μg of mBSA in 6 μl of PBS into the left knee joint. A therapeutic dose of rmTSG-6 injected intra-articularly with mBSA was found to be 25 to 50 μg in preliminary experiments. To avoid repeated intra-articular injections, we used a single double dose (100 μg) of rmTSG-6 throughout this study. Mice (n = 6 to 10 at each time point) were sacrificed on days 1, 3, 5, 7, 12, and 24 after the intra-articular injections. Left and right knees from each mouse were harvested, fixed in 10% formalin containing 5% cetylpirimidium chloride, decalcified, and paraffin embedded. Five- to 7-μm sections were stained with hematoxylin and eosin (H&E), or safranin-O counterstained with fast green. For immunohistochemical analysis, sections were deparaffinized, rehydrated in PBS, and pretreated with chondroitinase ABC (Seikagaku America of Cape Cod, Falmouth, MA) as described. 20 Sections were stained with polyclonal antibodies to neoepitopes -VDIPEN341 [a sequence that is generated by stromelysin-1 (MMP-3) cleavage in the interglobular domain of cartilage aggrecan] and -NITEGE373 (aggrecanase-generated neoepitope) (antibodies were generous gifts from Dr. J. Mort, Shriners Hospital, Montreal, Quebec, Canada and Dr. J. Sandy, Shriners Hospital, Tampa, FL). 21-23 Affinity-purified peroxidase-labeled anti-rabbit immunoglobulin (Accurate Chemical and Scientific Co., Westbury, NY) was used as a second-step antibody and the peroxidase reaction developed by Fast 3,3′-diaminobenzidine (DAB; Sigma) and H2O2 for 5 minutes as described. 20 The DAB reaction was enhanced using DAB Substrate Enhancer (Zymed Laboratories Inc, San Francisco, CA) for 3 minutes. The sections were dehydrated and mounted with Permount.
Measurements of Antigen-Specific Antibodies, T-Cell Responses, and Cytokine Production
MaxiSorp immunoplates (Nunc International, Hanover Park, IL) were coated with human or mouse cartilage PGs (0.1 μg protein/100 μl/well) for ELISAs as described. 24,25 Sera were applied at increasing dilutions and the isotypes of PG-specific antibodies were determined by peroxidase-conjugated rat anti-mouse IgG1 or IgG2a secondary antibodies (Zymed) as described. 24,26 Serum antibody levels were normalized to mouse IgG1 and IgG2a. 24,25
Antigen-specific T-cell responses (ie, IL-2 production and proliferation), were measured in quadruplicate samples of spleen cells (3 × 10 5 cells/well) cultured in the presence of 50 μg PG protein/ml. IL-2 was measured in supernatants harvested on day 2 by the proliferation of the IL-2-dependent CTLL-2 cell line. 24,25 Antigen-specific T-cell proliferation was assessed on day 5 by the incorporation of 3[H]-thymidine. Antigen (PG)-specific interferon-γ (IFN-γ) and IL-4 production by T cells were determined in identical culture conditions as described for T-cell proliferation in 4-day-old conditioned media (2.5 × 10 6 mononuclear cells/ml) using capture ELISAs from R&D Systems (Minneapolis, MN). Serum levels of TNF-α, IL-6, and IL-10 were measured by capture ELISAs (R&D Systems or BD Phar-Mingen, San Diego, CA) and IL-1 by a bioassay using D10S cell line as described. 24,25
Flow Cytometry
Immunostaining of cell-surface molecules and flow cytometry were performed as described previously. 14 Spleen and lymph node cells were stained with monoclonal antibodies (mAbs) to various cell surface molecules. These mAbs, purchased from BD PharMingen, were raised against CD3 (T cell), CD4 (T-helper cell), CD8 (T-suppressor cell), CD11a/b (leukocyte β2 integrin β subunits; LFA-1/Mac-1), CD18 (β2 integrin β subunit; LFA-1), CD19 (B cell), CD29 (β1 integrin subunit), CD25 (IL-2 receptor), CD44 (IM7), CD45/B220 (B cell), CD49d (β1 integrin α4 subunit; VLA-4), CD54 (ICAM-1), CD62L (L-selectin), and CD62P (P-selectin). Isotype-matched irrelevant mAbs were used as controls. Cell surface-bound antibodies were detected by biotinylated goat-anti-rat IgG (Kirkegaard & Perry, Gaithersburg, MD) and streptavidin-phycoerythrin or streptavidin-fluorescein isothiocyanate (Life Technologies; Gaithersburg, MD).
Mouse synovial fibroblast and small venule endothelial cells (SVEC4-10) were digested with streptomyces hyaluronidase (Sigma) for 30 minutes at 37°C to remove endogenous HA. 17 Mouse synovial fibroblast, 17 SVEC4-10, 19,27 and 5/4 T cells, 18 each at 1 × 10 6 in 100 μl, were incubated with fluorescein-labeled HA (5 μg/assay) 14 and rmTSG-6 (0.04 to 5 mg/ml), or unlabeled HA (5 mg/ml), as competitors for 2 hours at 37°C. Cells were washed in PBS, fixed in 2% paraformaldehyde (Sigma) and analyzed using a FACScan instrument and CellQuest software (Becton Dickinson, San Jose, CA).
Statistical Analysis
Statistical analysis was performed using SPSS v7.5 (SPSS, Chicago, IL). The Mann-Whitney and Wilcoxon tests were used for intergroup comparisons. For comparison of results of two groups, Student’s t-test and, for the determination of correlation coefficients, Spearman’s gamma test were used. Significance was set at P < 0.05.
Results
Short-Term Effects of rmTSG-6 Treatment on Joint Swelling in PGIA Mouse Model
PGIA was generated in BALB/c mice by systemic immunization with human cartilage aggrecan. Acutely arthritic animals were injected intraperitoneally with 100 μl of PBS (control) or 50 to 250 μg rmTSG-6 (in 100 μl of PBS) for 3 consecutive days. No effect was observed on joint swelling in either the control or the rmTSG-6-treated mice after three injections (data not shown). To further test the feasibility of TSG-6 treatment, acutely arthritic mice with PGIA were injected intravenously with 100 μg of rmTSG-6. A dramatic reduction in joint edema of each swollen limb was observed within a few hours (Figure 1A) ▶ . Subsequent intravenous injections further reduced joint swelling, whereas the effect was less and less spectacular until the edematous swelling virtually disappeared from acutely inflamed paws (Figure 1A ▶ and Figure 2B ▶ ). Control mice injected with PBS showed no change, or only a slight increase, in joint swelling within this short-term experiment (Figure 1) ▶ .
Figure 1.

Short-term effect of rmTSG-6 on joint swelling in PGIA mice. Animals were randomly grouped 3 to 7 days after the onset of arthritis (onset of swelling and redness) and each experimental group contained a comparable number of inflamed front and hind paws with approximately the same cumulative clinical scores of 8.2 to 8.4 ± 1.2 to 1.4 corresponding to 24.9 to 25.6 ± 1.1 to 1.3 mm cumulative joint diameter. Both PBS-injected (control; filled triangle) and 100 μg of rmTSG-6-injected (filled square) groups contained 16 animals in four independent experiments. Treatments with HA-binding Pep-1 (1 mg) (filled circle) or co-treatment of Pep-1 (10 μg to 1 mg) and rmTSG-6 (100 μg) (symbols of dose combinations are shown in figure) were repeated twice (a total of seven mice in each group). All injections were given intravenously in 100 μl of PBS (arrows). Joints were measured before each injection (early morning) and then every 2 to 3 hours after injections. Ankles and wrists were measured in frontal and sagittal planes, and the cumulative joint thickness 17 of the four paws (eight measurements per animal) are shown.
Figure 2.
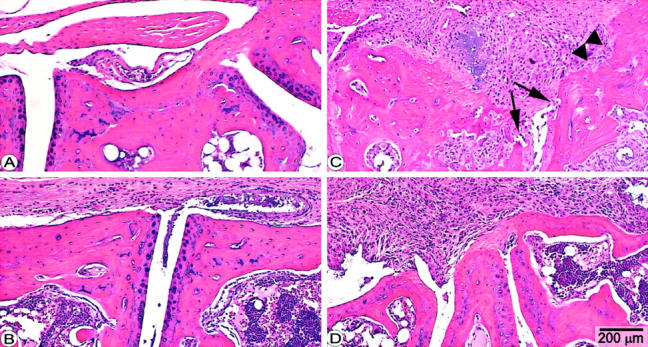
Histopathology of tarsal joints of mice with PGIA. Panels represent sagittal sections of tarsal joints of a noninflamed paw (A) or sections of arthritic tarsal joints from PGIA mice 4 days (B) or 4 weeks after the onset of arthritis (C and D). Arthritic mice were treated with PBS (C) or with rmTSG-6 (B and D). Massive inflammation and synovial cell proliferation were characteristic of both PBS-treated (C) and TSG-6-treated (D) mice, but the articular cartilage remained histologically intact after 4 weeks in TSG-6-treated mice (D) (animals of Figure 3 ▶ ). In contrast, articular cartilage was essentially lost (arrows) from the joint surface and massive bone erosion was also present in nontreated (PBS-injected) mice (C). H&E staining; original magnifications, ×25.
Histopathology of the arthritic joints, except the reduced periarticular edema in TSG-6-treated mice (Figure 2B) ▶ , however, was indistinguishable from those treated with PBS. Because of the lack of any effect of intraperitoneally injected rmTSG-6 versus the dramatic reduction of edema after the intravenous injection, we hypothesized that a free HA-binding capacity of TSG-6 should be critical in function without affecting the inflammatory cell infiltration. Indeed, when rmTSG-6 was added together with either a large dose of HA (data not shown) or with HA-binding peptide Pep-1, the beneficial effect of TSG-6 disappeared proportionally as the dose of Pep-1 was increased (Figure 1B) ▶ .
In Vitro and In Vivo Tests of rmTSG-6 Ability to Compete for HA-Binding Site
The difference between treated animals and untreated controls was the regression of joint swelling because of the loss of edema in the rmTSG-6-treated group. To test if the loss of edema was because of the ability of rmTSG-6 to compete with cell surface receptor CD44 for HA binding, in vitro competition assays were performed using synovial fibroblasts, small venule endothelial cells (SVEC4-10) (both produce high levels of HA), and 5/4 T-cell hybridoma (a non-HA-producing cell line). All these cell types express high amounts of CD44, and hence can bind HA. Cells were incubated with fluorescein-labeled HA and increasing concentrations of rmTSG-6, and the HA binding was measured by fluorescence-activated cell sorting analysis. As shown in Figure 3 ▶ , rmTSG-6 could indeed compete with CD44 and the effect was dose-dependent. If this was the case in vivo, a HA-binding peptide 19 could also reduce joint inflammation and should exhibit an additive or synergistic effect on TSG-6 treatment. As shown in Figure 1B ▶ , Pep-1 alone could not reduce joint swelling, even worse, the co-injection of Pep-1 with rmTSG-6 significantly reduced the anti-inflammatory effect of the TSG-6 (Figure 1B) ▶ . Taken together, TSG-6 had a very strong effect on joint inflammation by reducing tissue edema. This effect was associated with the HA-binding capacity of the TSG-6, which could be competitively inhibited with a synthetic peptide with known HA-binding properties, whereas Pep-1 alone did not exhibit any effect on joint edema.
Figure 3.

Flow cytometry (fluorescence-activated cell sorting) analysis of competition assays for determining the ability of rmTSG-6 to compete with CD44 for HA binding. Mouse synovial fibroblasts and SVEC4-10 endothelial cells were digested with streptomyces hyaluronidase for 30 minutes at 37°C to remove endogenous HA and then cells were washed with PBS. All three cell lines (1 × 106) were incubated with fluorescein-labeled HA (20 μg/ml; 100 μl final volume) and rmTSG-6 (0.04 to 5 mg/ml) or 100 μl of unlabeled HA (5 mg/ml) for 2 hours at 37°C. Cells were washed in PBS and analyzed by fluorescence-activated cell sorting. The y axis shows the cell number and the x axis the fluorescence intensity.
Long-Term Effects of rmTSG-6 Treatment on Joint Swelling in PGIA Mouse Model
Next, we have monitored the clinical symptoms and histological abnormalities of inflamed joints during repeated (weekly) treatments with rmTSG-6 in PGIA up to 28 days. Arthritic BALB/c mice (n = 14) were injected intravenously with rmTSG-6 (100 μg in PBS) every 24 hours for 7 days and joint/paw swelling measured twice a day (Figure 4) ▶ . As the cumulative joint score (swelling of all paws) almost returned to the pretreatment level after a 7-day treatment-free period (Figure 4A) ▶ , mostly because of the newly inflamed paws, a second treatment cycle was repeated from day 14. As found during the first treatment period, although less impressively, the cumulative joint score in rmTSG-6-treated mice showed a statistically significant regression (P < 0.05) (Figure 4A) ▶ . In contrast, the cumulative joint swelling in the PBS-injected (control) mice remained significantly higher (P < 0.05) up to 3 weeks (Figure 4A) ▶ which was the consequence of the normal progression of the disease. 15,16 Interestingly, additional (new) joints (paws) became inflamed (swollen) in rmTSG-6-treated mice only after weaning of TSG-6 treatment (between days 7 and 14). Therefore, when only the inflamed joint/paw diameters were calculated and shown on Figure 4B, a ▶ less impressive effect of the second-cycle treatment was found. Although the anti-inflammatory (anti-edematous) effect remained significantly low, relative to the joint diameters measured on the zero day of the experimental group, the second-cycle treatment affected essentially only the freshly (acutely) inflamed paws, thus the anti-inflammatory effect of the second treatment period with rmTSG was almost restricted to these new, acutely inflamed joints/paws (Figure 4A) ▶ . Although the cellular infiltration and/or synovial cell proliferation of inflamed joints were very similar in PBS- and TSG-6-treated joints (Figure 2, C and D) ▶ , massive cartilage and bone erosions were found in joints only in PBS-treated animals. In turn, systemic application of rmTSG-6 reduced edema and, more strikingly, prevented articular cartilage from deterioration even in the presence of massive inflammation (Figure 2, C ▶ versus D).
Figure 4.

Long-term effect of repeated treatment with rmTSG-6 on PGIA mice. Arthritic animals were injected intravenously with 100 μl of PBS (n = 14) or 100 μg of rmTSG-6 (n = 15). Each mouse was injected every 24 hours for 7 days as indicated by arrows. Joints were measured before the injections and then twice per day. The treatment and cessation of therapy was repeated for another 14 days. Ankles and wrists were measured in frontal and sagittal planes and the change in cumulative joint thickness of the four paws (eight measurements) are shown. A shows the difference of cumulative joint thickness in mm between the PBS-injected and rmTSG-6-treated group (all paws), whereas B shows the differences of the inflamed joint thickness only (ie, paws were inflamed at the beginning of the TSG-6 treatment). The decrease of joint swelling in the rmTSG-6-treated group was significant within 24 hours (as shown in Figure 1 ▶ ), and the increased joint swelling in the PBS-injected group became significant (*, P < 0.05) throughout the first 10 to 12 days. Changes in joint swelling between PBS-injected and rmTSG-6-treated groups was statistically significant (*, P < 0.05 or smaller) from day 1 of the treatment and remained significant (comparing to day 0) throughout the entire experimental period.
It is important to note here that, although PGIA is progressive and more and more joints are involved, arthritic limbs/joints exhibit a high variability, ie, not all limbs (joints) are inflamed simultaneously, or to the same extent, in an acutely arthritic animal at a single time point. 15,16 In view of the clinical picture of PGIA, we reanalyzed the entire data base of all arthritic (PBS- and rmTSG-treated) mice (Figure 4A) ▶ and compared only those paws that were inflamed before the treatment. The PBS-treated group showed no change during the first 2 weeks, and then the acute inflammatory symptoms moderately regressed and previously inflamed joints became stiff. In contrast, the joint swelling of rmTSG-6-treated groups showed a statistically significant regression (P < 0.05) within a few days (Figure 1A ▶ and Figure 4A ▶ ) that remained unchanged during the 4-week experimental period (Figure 4B) ▶ . In other words, once rmTSG-6 reduced edema and redness, inflammation did not return, not even in the absence of treatment (between days 7 and 14 or 21 and 28) (Figure 4B) ▶ . These results suggest that TSG-6 not only protects arthritic joints from flare-up but also seems to prevent nonarthritic limbs from the clinical symptoms of inflammation determined as redness and swelling.
Effect of rmTSG-6 Treatment on the Development of PGIA
During the intravenous first cycle of rmTSG-6 treatment (Figure 4) ▶ , edema waned in arthritic paws and no additional joints became inflamed. In contrast, previously nonarthritic paws became swollen in PBS-injected animals (Figure 4, A and B) ▶ , and this was also a frequent observation in TSG-6-treated mice after the cessation of the treatment. A significant increase in cumulative paw diameter after day 7 in previously TSG-6-treated animals (Figure 4A) ▶ clearly was the consequence of these newly inflamed joints. Although conventional histopathology did not reveal differences in cellular infiltration (Figure 2) ▶ , the anti-inflammatory effect of TSG-6 was found in CIA 13 and in an air-pouch model. 11 All these observations indicated a preventive effect of TSG-6 on arthritis development. To test this preventive potential of TSG-6 in arthritis, PG-immunized mice (n = 8) in the prearthritic stage (after the third antigen injection) were treated intravenously with a daily dose of 100 μg rmTSG-6 (a dose found to be optimal for the suppression of inflammation) or PBS for 3 weeks. Although the inflammation (cumulative score, 4.2 ± 2.8) was significantly lower (P < 0.05) in rmTSG-6-treated mice than in the PBS group (7.6 ± 1.7), neither the incidence nor the time of onset differed in the two groups (data not shown), ie, TSG-6 had an anti-edematous but not an anti-arthritic effect.
Pathophysiological Markers in rmTSG-6-Treated and Untreated Mice with PGIA
Because we found differences in severity between TSG-6-treated and untreated animals, and then differences in cartilage and bone erosions, it seemed to be prudent to measure serum levels of pro- and anti-inflammatory cytokines and auto- and heteroantibodies, as well as monitor the antigen-specific T-cell responses. Serum samples were collected before the treatment cycles and at 24 hours after the last (seventh) intravenous injection of rmTSG-6, and T-cell responses were measured at the end of experiment. Treatment protocols were the same as shown in Figure 4 ▶ . We could not detect any significant differences between PBS- and rmTSG-6-injected mice measuring serum levels of TNF-α, IL-1, IL-6, IL-10, and auto- and heteroantibodies (including the ratios of IgG1 and IgG2a), or measuring antigen (PG)-specific T-cell proliferation, IL-2 (CTLL-2 assay), IL-4, or IFN-γ production by spleen cells. Similarly, neither the T-cell subsets nor other expression of cell surface receptors (mAbs listed in Materials and Methods) differed when spleen or lymph node cells of the PBS- and rmTSG-6-treated groups were compared (data not shown). Therefore, we concluded that the systemic administration of rmTSG-6 probably has no systemic effect, rather it might be critical at the site of inflammation where tissue destruction occurs.
Histopathology of Knee Joints in AIA Under the Protection of TSG-6
To gain insight into the mechanism of the local chondroprotective effect of TSG-6 (Figure 2D) ▶ in a single joint, a murine model of AIA was used. 21 In this model system, the delayed-type hypersensitivity reaction is localized at a single joint, other joints can be used as controls, and cartilage degradation can be monitored precisely on a daily basis. Intra-articular PBS injection in the left knee joint of mBSA-immunized animals showed no inflammation (Figure 5A) ▶ up to a 24-day period, and no cartilage damage occurred (Figure 5 ▶ ; B to E). The earliest response (lymphocyte infiltration) to the intra-articular injection of mBSA was detected on days 2 to 3 (data not shown) and became more uniformly extensive by day 5 (Figure 5F) ▶ . By this time, the loss of cartilage PG from the superficial middle layers of the articular cartilage became evident (Figure 5 ▶ ; G to J). All mBSA-injected joints exhibited heavy inflammation, independent of whether the mBSA was injected alone (Figure 5F) ▶ or with rmTSG-6 (Figure 5K) ▶ . The loss of PG from cartilage, detected by the loss of safranin-O staining, was moderate on day 3 (not shown), but became extensive by day 5 in mBSA-injected knee joints (Figure 5H) ▶ . In accordance with the loss of safranin-O staining, an intensive accumulation of immunostained -VDIPEN and -NITEGE neoepitopes (the C-terminal cleavage sites of cartilage aggrecan by metalloproteinases and aggrecanase) were detected in the superficial layer of damaged cartilage (Figure 5, I and J) ▶ . By day 7, inflammation further progressed, pannus-like structures eroded bone and cartilage, safranin-O staining significantly decreased down to the calcifying zone, and the expression of -NITEGE neoepitope became slightly reduced (not shown). Knee joints of mBSA-immunized mice were injected with mBSA with or without rmTSG-6. As shown in Figure 5, F and K ▶ , the conventional histopathology of these knee joints (ie, H&E-stained sections) exhibited no differences at any time point. In contrast, safranin-O staining remained intense (compare the superficial layers of cartilage in Figure 5, H ▶ with M) up to 5 to 7 days after the intra-articular administration of mBSA with rmTSG-6. Thus, a single dose of 100 μg of rmTSG-6 (given together with the mBSA) blocked the degradation and loss of aggrecan from cartilage (Figure 5 ▶ ; M to O). However, this chondroprotective effect of TSG-6 in acutely inflamed joints gradually decreased after day 7 and essentially no differences were found between PBS/mBSA and rmTSG-6/mBSA-injected knee joints after day 12, unless additional doses of TSG-6 were introduced locally or systematically (results are not shown).
Figure 5.
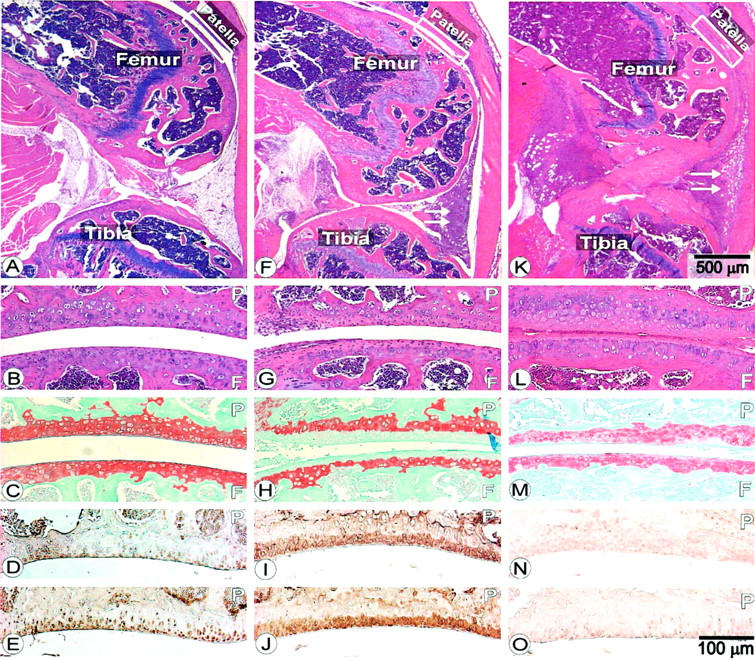
Histology and immunohistochemistry of knee sections from C57BL/6j mice on day 5 of AIA. A–E represent sections from PBS-injected knees, F–J shows sections of knee joints injected intra-articularly with mBSA, and K–O are sections from knee joints injected with mBSA and rmTSG-6 together. A, B, F, G, K, and L are H&E-stained sections. White arrows in F and K show the cellular infiltration. Sections of C, H, and M are stained with safranin-O. D, I, and N are sections stained with antibody to -VDIPEN neoepitope (stromelysin-cleaved product of cartilage aggrecan). E, J, and O are sections stained with antibody to aggrecanase-generated -NITEGE neoepitope. Patella (P) and femoral condyle (F) are indicated. Scale bar on K indicates the scale for A, F, and K, whereas scale bar in O indicates scale for all other panels.
Discussion
TSG-6 can be induced by proinflammatory cytokines (TNF-α, IL-1), growth factors, and various compounds (including lipopolysaccharides), 2,3,5,28 components that in turn activate various transcription factors (nuclear factor-κB, nuclear factor-IL6, or AP-1) and induce the TSG-6 expression in various cell types. 29 In a complex milieu such as the inflamed rheumatoid synovium, among many other components, TSG-6, HA, and CD44 are up-regulated simultaneously, and increased serum levels of these compounds correlate directly with the disease activity of rheumatoid arthritis. 3,8,30-33 To study the pathophysiological relevance of TSG-6 in arthritic processes, and gain insight into the function of this cytokine-induced molecule in inflammatory conditions, we used an autoimmune murine model (PGIA) of rheumatoid arthritis.
The intraperitoneal injection of rmTSG-6 had no effect on joint swelling of mice with acute PGIA, whereas the intravenous injection of rmTSG-6 resulted in a dramatic reduction in joint swelling. The lack of any effect of intraperitoneally injected rmTSG-6 is probably because of the entrapment of rmTSG-6 by the excess amount of HA in the peritoneal cavity occupied by adjuvant-induced granuloma, which is the consequence of intraperitoneal immunization. 34 Indeed, we were unable to detect radiolabeled rmTSG-6 in serum after intraperitoneal injection, whereas the intravenously injected rmTSG-6 preferentially accumulated at the site of inflammation. In this kinetic study, the half life of intravenously injected rmTSG-6 was less than 4 hours, but the serum level of TSG-6 measured by ELISA was almost constant 60 to 90 minutes after the intravenous injection (unpublished data). Thus, although the exogenous TSG-6 (eg, radiolabeled) reached the inflamed joint soon after the intravenous injection, and was detectable in the inflamed joint at least 24 hours, a significant amount of TSG-6 remained in, or returned to, the circulation.
The loss of edema in the presence of massive cellular infiltration indicated that TSG-6 affects cell/tissue-bound HA, rather than the migration of inflammatory cells or the proliferation of synovial cells. The overnight gap in treatment resulted in a plateau in reduction of joint swelling which then further declined after repeated administration of TSG-6 protein (Figure 1A) ▶ . Therefore, we hypothesized that a relatively high dose of rmTSG-6 can competitively occupy the binding sites on HA and displace CD44 in the inflamed tissue. As a result, the CD44-bound HA (and HA-bound water) became diffusible and receded from the tissue. Using in vitro assays with the three different cell lines, SVEC4-10 endothelial cells, mouse synovial fibroblast, and 5/4 T-cell hybridoma, we demonstrated that TSG-6 can indeed compete with CD44 in a dose-dependent manner. This was further confirmed in vivo, when increasing doses of HA-binding peptide Pep-1 19 reciprocally reduced the effect of rmTSG-6 in short-term experiments. However, although this HA-binding synthetic peptide Pep-1 competed effectively with TSG-6 in vivo, the peptide by itself had no effect on joint swelling. This seemingly contradictory observation indicates that Pep-1 was able to occupy free binding sites on HA, but it was unable to displace CD44 bound to HA.
The most unexpected observation after noted excessive reduction in joint swelling in a short-term treatment was the undistinguishable histopathological picture in PBS- and TSG-6-treated animals. Although the reduction of edema was evident within hours (Figure 1A) ▶ , and then visible in paw sections of TSG-6-treated animals (Figure 2B) ▶ , there were no evident differences in cellular infiltration, synovial lining cell proliferation, nor pannus formation for up to 4 weeks of the experimental period. This comparable picture of synovitis was especially in contrast to the lack of cartilage and bone erosions in TSG-6-treated animals, which otherwise is a typical consequence of joint inflammation in RA and in all corresponding animal models. 15,16,20,21,25,35
First, we performed experiments in PGIA. We preferentially use this mouse model for genetic, inflammation, cell migration, and autoimmune studies, rather than CIA, another widely used systemic autoimmune model for human RA. Essentially both inflammatory models show many similarities to RA as indicated by clinical assessment of arthritis and histopathology of the peripheral joints. However, beyond similarities, there are substantial differences between the two autoimmune inflammatory animal models for RA: PGIA shows higher susceptibility (100% in aging female mice), 15-17 the joint inflammation is more progressive leading to complete deterioration of articular cartilage accompanied with massive bone erosions, 15,16,25 the onset of the disease is dictated by a shift from Th2 to Th1 response, 26,36 and it has a recessive inheritance. 24,37 Many of these characteristics are comparable to those in rheumatoid arthritis. In contrast to a recently published study for TSG-6 treatment of CIA, 13 we were unable to find any significant differences in pathophysiological markers (including the serum level of IL-6) between PBS- and TSG-6-treated animals, synovial cell proliferation, or in cell-surface expression of various phenotypic markers. Fluctuation and differences in serum levels of various cytokines clearly associated with the disease state in both PGIA and CIA, rather than the TSG-6 treatment. We could detect the same, occasionally more prominent, differences in serum markers in leflunomide-, 38 IL-4-, 36,39,40 and IL-10-treated 39-41 animals, or during anti-CD44 immunotherapy, 14,17 as found in TSG-6-treated arthritic animals. Although the TSG-6 treatment reduced the severity (edema and cartilage damage) of arthritis, it did not reduce the incidence or delay the onset of arthritis. Therefore, the anti-inflammatory effect of exogenous (human) recombinant TSG-6 associated with increased serum levels of IL-6 measured at a single and late time point in the above-mentioned study 13 may coincide with, but can hardly be the consequence of, the TSG-6 treatment. Rather, it might be the consequence of immunization of DBA/1 mice with a large dose of human TSG-6, or the lipopolysaccharide or IL-1 injection might be used for synchronization of CIA to reach a higher incidence. Although IL-6 locally may stimulate PG synthesis, 35 IL-6 is a key proinflammatory cytokine required for the development of various forms of arthritis, including CIA. 24,25,42-45 In conclusion, it seems to be very likely that a significantly reduced incidence and delayed onset of CIA, 13 without suppression of proinflammatory cytokines, antibodies, or the Th1 response, should not be related to increased levels of IL-6 in TSG-6-treated animals.
To explore the role of TSG-6 during joint inflammation and understand how the morphologically intact cartilage can be preserved in the presence of massive synovial inflammation we extended our studies and used the AIA model. In this model, cartilage damage can be assessed by loss of safranin-O-stained proteoglycan and, simultaneously, we can demonstrate the most characteristic, MMP-generated neoepitopes of cartilage PG (aggrecan), such as the aggrecanase-cleaved -NITEGE and various MMP-generated -VDIPEN sequences. 20,21,23 A single dose of rmTSG-6 administered simultaneously with the antigen challenge into the knee joints showed protection against the loss of cartilage PG. Therefore, the chondroprotective effect of TSG-6 directly, or indirectly, associated with the inhibition of matrix metalloproteinases, enzymes that are also up-regulated by cytokines in inflammatory conditions. It is known that TSG-6 protein can bind to IαI, dramatically increasing the serine protease inhibitory effect of the IαI. 8,9,11 The protease inhibitory effect of IαI/TSG-6 complex was especially evident on plasminogen-plasmin activation, probably the most potent serine protease needed for activation of pro-MMPs. 46 Thus, TSG-6 seems to be a key component of a negative feedback loop controlling extensive tissue damage in inflammatory conditions.
A conclusive evidence that the TSG-6/IαI complex mediates the potent antiplasmin activity will require additional studies in all arthritis models (CIA, PGIA, and AIA), and the isolation of the 120-kd complex. 8,11 Although the serine protease inhibitory activity of IαI family members via their common bikunin domain has been known for a long time, 47 little is known about their functions. However, disease-associated presence in various tissues and fluctuations seen in the serum levels of IαI and IαI-related protein suggest an involvement in pathological processes. Daveau and colleagues 48 reported a distinct pattern of changes in serum concentrations of the different members of the IαI family during acute inflammation. Proteins identical with, or closely related to, the bikunin chain of IαI, have been detected in the stroma and the surrounding connective tissue of malignant tumors, 49 in the brain tissue of patients with Alzheimer’s disease, 50 and in the serum and urine of patients with inflammatory diseases, cancer, and leukemia. 51,52 A link between arthritis and IαI was suggested more than 20 years ago when Becker and Sandson 53 found IαI associated with HA in the synovial fluid of patients with RA. This finding was confirmed to show that IαI associates in vitro with HA isolated from the synovial fluid of healthy patients. 54 In conclusion, TSG-6 stimulated by proinflammatory cytokines may function as an anti-inflammatory component by combining and enhancing the serine protease inhibitory activity of IαI.
Acknowledgments
We thank Dr. Jeffrey M. Otto (Genaissance Pharmaceutical, New Haven, CT), Drs. Vincent C. Hascall and Csaba Fülöp (Cleveland Clinic Foundation, Cleveland, OH), Dr. Aled O. Phillips (University of Wales College of Medicine, Wales, UK), and Drs. Alison Finnegan, Kenneth Roebuck, Gabriella Cs-Szabó, Warren Knudson, and Thomas Schmid (Rush University, Chicago, IL) for helpful comments, discussion, and criticism; Dr. Susan Shott (Rush University) for helpful statistical advice; and David Gerard, Stiliani Christodoulou, and Sonja Velins for expert technical assistance.
Footnotes
Address reprint requests to Tibor T. Glant, M.D., Ph.D., Section of Biochemistry and Molecular Biology, Departments of Orthopedic Surgery and Biochemistry, Rush University, Rush-Presbyterian-St. Luke’s Medical Center, 1653 West Congress Pkwy., Chicago, IL 60612. E-mail: tglant@rush.edu.
Supported by grants from the National Institutes of Health (AR40310, AR45652, and AR47135) and the Coleman Foundation (Chicago, IL).
R. V. K. and T. B. were equally involved in this study and both regarded as first author.
References
- 1.Lee TH, Lee GW, Ziff EB, Vilcek J: Isolation and characterization of eight tumor necrosis factor-induced gene sequences from human fibroblasts. Mol Cell Biol 1990, 10:1982-1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee TH, Wisniewski H-G, Vilcek J: A novel secretory tumor necrosis factor-inducible protein (TSG-6) is a member of the family of hyaluronate binding proteins, closely related to the adhesion receptor CD44. J Cell Biol 1992, 116:545-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wisniewski H-G, Maier R, Lotz M, Lee S, Klampfer L, Lee TH, Vilcek J: TSG-6: a TNF-, IL-1-, and LPS-inducible secreted glycoprotein associated with arthritis. J Immunol 1993, 151:6593-6601 [PubMed] [Google Scholar]
- 4.Fülöp C, Kamath RV, Li Y, Otto JM, Salustri A, Olsen BR, Glant TT, Hascall VC: Coding sequence, exon-intron structure and chromosomal localization of murine TNF-stimulated gene 6 that is specifically expressed by expanding cumulus cell-oocyte complexes. Gene 1997, 202:95-102 [DOI] [PubMed] [Google Scholar]
- 5.Bayliss MT, Howat SLT, Dudhia J, Murphy JM, Barry FP, Edwards JCW, Day AJ: Up-regulation and differential expression of the hyaluronan-binding protein TSG-6 in cartilage and synovium in rheumatoid arthritis and osteoarthritis. Osteoarthr Cart 2001, 9:42-48 [DOI] [PubMed] [Google Scholar]
- 6.Kohda D, Morton CJ, Parkar AA, Hatanaka H, Inagaka FM, Campbell ID, Day AJ: Solution structure of the link module: a hyaluronan-binding domain involved in extracellular matrix stability and cell migration. Cell 1996, 86:767-775 [DOI] [PubMed] [Google Scholar]
- 7.Bork P, Beckmann G: A widespread module in developmentally regulated proteins. J Mol Biol 1993, 231:539-545 [DOI] [PubMed] [Google Scholar]
- 8.Wisniewski HG, Vilcek J: TSG-6: an IL-1/TNF-inducible protein with anti-inflammatory activity. Cytokine Growth Factor Rev 1997, 8:143-156 [DOI] [PubMed] [Google Scholar]
- 9.Wisniewski H-G, Burgess WH, Oppenhein JD, Vilcek J: TSG-6, an arthritis-associated hyaluronan binding protein, forms a stable complex with the serum protein inter-α-inhibitor. Biochemistry 1994, 33:7423-7429 [DOI] [PubMed] [Google Scholar]
- 10.Ronday HK, Smits HH, van Muijen GNP, Pruszczynski MSM, Dolhain RJEM, van Langelaan EJ, Breedveld FC, Verheijen JH: Difference in expression of the plasminogen activation system in synovial tissue of patients with rheumatoid arthritis and osteoarthritis. Br J Rheumatol 1996, 35:416-423 [DOI] [PubMed] [Google Scholar]
- 11.Wisniewski H-G, Hua J-C, Poppers DM, Naime D, Vilcek J, Cronstein BN: TNF/IL-1-inducible protein TSG-6 potentiates plasmin inhibition by inter-α-inhibitor and exerts a strong anti-inflammatory effect in vivo. J Immunol 1996, 156:1609-1615 [PubMed] [Google Scholar]
- 12.Bost F, Diarra-Mehrpour M, Martin JP: Inter-α-trypsin inhibitor proteoglycan family—a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem 1998, 252:339-346 [DOI] [PubMed] [Google Scholar]
- 13.Mindrescu C, Thorbecke GJ, Klein MJ, Vilcek J, Wisniewski H-G: Amelioration of collagen-induced arthritis in DBA/1J mice by recombinant TSG-6, a tumor necrosis factor/interleukin-1-inducible protein. Arthritis Rheum 2000, 43:2668-2677 [DOI] [PubMed] [Google Scholar]
- 14.Mikecz K, Dennis K, Shi M, Kim JH: Modulation of hyaluronan receptor (CD44) function in vivo in a murine model of rheumatoid arthritis. Arthritis Rheum 1999, 42:659-668 [DOI] [PubMed] [Google Scholar]
- 15.Glant TT, Cs-Szabó G, Nagase H, Jacobs JJ, Mikecz K: Progressive polyarthritis induced in BALB/c mice by aggrecan from human osteoarthritic cartilage. Arthritis Rheum 1998, 41:1007-1018 [DOI] [PubMed] [Google Scholar]
- 16.Glant TT, Mikecz K, Arzoumanian A, Poole AR: Proteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathology. Arthritis Rheum 1987, 30:201-212 [DOI] [PubMed] [Google Scholar]
- 17.Mikecz K, Brennan FR, Kim JH, Glant TT: Anti-CD44 treatment abrogates tissue edema and leukocyte infiltration in murine arthritis. Nat Med 1995, 1:558-563 [DOI] [PubMed] [Google Scholar]
- 18.Buzás EI, Brennan FR, Mikecz K, Garzó M, Negroiu G, Holló K, Cs-Szabó G, Pintye É, Glant TT: A proteoglycan (aggrecan)-specific T cell hybridoma induces arthritis in BALB/c mice. J Immunol 1995, 155:2679-2687 [PubMed] [Google Scholar]
- 19.Mummert ME, Mohamadzadeh M, Mummert DI, Mizumoto N, Takashima A: Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med 2000, 192:769-779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singer II, Kawka DW, Bayne EK, Donatelli SA, Weidner JR, Williams HA, Mumford RA, Lark MW, Glant TT, Nabozny GH, David CS: VDIPEN, a metalloproteinase-generated neoepitope, is induced and immunolocalized in articular cartilage during inflammatory arthritis. J Clin Invest 1995, 95:2178-2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Meurs JBJ, van Lent PLEM, Holthuysen AEM, Singer II, Bayne EK, van den Berg WB: Kinetics of aggrecanase- and metalloproteinase-induced neoepitopes in various stages of cartilage destruction in murine arthritis. Arthritis Rheum 1999, 42:1128-1139 [DOI] [PubMed] [Google Scholar]
- 22.Flannery CR, Lark MW, Sandy JD: Identification of a stromelysin cleavage site within the interglobular domain of human aggrecan. Evidence for proteolysis at this site in vivo in human articular cartilage. J Biol Chem 1992, 267:1008-1014 [PubMed] [Google Scholar]
- 23.Hughes CE, Caterson B, Fosang AJ, Roughley PJ, Mort JS: Monoclonal antibodies that specifically recognize neoepitope sequences generated by ‘aggrecanase’ and matrix metalloproteinase cleavage of aggrecan: application to catabolism in situ and in vitro. Biochem J 1995, 305:799-804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto JM, Chandrasekaran R, Vermes C, Mikecz K, Finnegan A, Rickert SE, Enders JT, Glant TT: A genome scan using a novel genetic cross identifies new susceptibility loci and traits in a mouse model of rheumatoid arthritis. J Immunol 2000, 165:5278-5286 [DOI] [PubMed] [Google Scholar]
- 25.Glant TT, Bárdos T, Vermes C, Chandrasekaran R, Valdéz JC, Otto JM, Gerard D, Velins S, Lovász G, Zhang J, Mikecz K, Finnegan A: Variations in susceptibility to proteoglycan-induced arthritis and spondylitis among C3H substrains of mice. Evidence of genetically acquired resistance to autoimmune disease. Arthritis Rheum 2001, 44:682-692 [DOI] [PubMed] [Google Scholar]
- 26.Holló K, Glant TT, Garzó M, Finnegan A, Mikecz K, Buzás EI: Complex pattern of Th1 and Th2 activation with a preferential increase of autoreactive Th1 cells in BALB/c mice with proteoglycan (aggrecan)-induced arthritis. Clin Exp Immunol 2000, 120:167-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohamadzadeh M, DeGrendele H, Arizpe H, Estess P, Siegelman M: Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44/HA-dependent primary adhesion. J Clin Invest 1998, 101:97-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye L, Mora R, Akhayani N, Haudenschild CC, Liau G: Growth factor and cytokine-regulated hyaluronan-binding protein TSG-6 is localized to the injury-induced rat neointima and confers enhanced growth in vascular smooth muscle cells. Circ Res 1997, 81:289-296 [DOI] [PubMed] [Google Scholar]
- 29.Klampfer L, Chen-Kiang S, Vilcek J: Activation of the TSG-6 gene by NF-IL6 requires two adjacent NF-IL6 binding sites. J Biol Chem 1995, 270:3677-3682 [DOI] [PubMed] [Google Scholar]
- 30.Balazs EA, Watson D, Duff IF, Roseman S: Hyaluronic acid in synovial fluid. I. Molecular parameters of hyaluronic acid in normal and arthritic human fluids. Arthritis Rheum 1967, 10:357-376 [DOI] [PubMed] [Google Scholar]
- 31.Emlen W, Niebur J, Flanders G, Rutledge J: Measurement of serum hyaluronic acid in patients with rheumatoid arthritis: correlation with disease activity. J Rheumatol 1996, 23:974-978 [PubMed] [Google Scholar]
- 32.Haynes BF, Hale LP, Patton KL, Martin ME, McCallum RM: Measurement of an adhesion molecule as an indicator of inflammatory disease activity. Up-regulation of the receptor for hyaluronate (CD44) in rheumatoid arthritis. Arthritis Rheum 1991, 34:1434-1443 [DOI] [PubMed] [Google Scholar]
- 33.Goldberg RL, Doughty J, Spirito S, Klien M, Peppard J, Crowl RM, Hu S-I: Elevated TSG-6 as a biochemical marker of rheumatoid arthritis. Trans ORS 1998, 44:215(abstract) [Google Scholar]
- 34.Bekierkunst A, Levij IS, Yarkoni E, Vilkas E, Adam A, Ledered E: Granuloma formation induced in mice by chemically defined mycobacterial fractions. J Bacteriol 1969, 100:95-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van de Loo FAJ, Joosten LAB, van Lent PLEM, Arntz OJ, van den Berg WB: Role of interleukin-1, tumor necrosis factor α, and interleukin-6 in cartilage proteoglycan metabolism and destruction. Effect in situ blocking in murine antigen- and zymosan-induced arthritis. Arthritis Rheum 1995, 38:164-172 [DOI] [PubMed] [Google Scholar]
- 36.Finnegan A, Mikecz K, Tao P, Glant TT: Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol 1999, 163:5383-5390 [PubMed] [Google Scholar]
- 37.Otto JM, Cs-Szabó G, Gallagher J, Velins S, Mikecz K, Buzás EI, Enders JT, Li Y, Olsen BR, Glant TT: Identification of multiple loci linked to inflammation and autoantibody production by a genome scan of a murine model of rheumatoid arthritis. Arthritis Rheum 1999, 42:2524-2531 [DOI] [PubMed] [Google Scholar]
- 38.Glant TT, Mikecz K, Bartlett RR, Deák F, Thonar EJ-MA, Williams JM, Mattar T, Kuettner KE, Schleyerbach R: Immunomodulation of proteoglycan-induced progressive polyarthritis by leflunomide. Immunopharmacology 1992, 23:105-116 [DOI] [PubMed] [Google Scholar]
- 39.Finnegan A, Tao P, Mikecz K, Glant TT: IL-4 and IL-10 treatment of proteoglycan-induced arthritis. Arthritis Rheum 1998, 41:S100(abstract) [Google Scholar]
- 40.Joosten LAB, Lubberts E, Durez P, Helsen MMA, Jacobs MJM, Goldman M, van den Berg WB: Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum 1997, 40:249-260 [DOI] [PubMed] [Google Scholar]
- 41.Persson S, Mikulowska A, Narula S, O’Garra A, Holmdahl R: Interleukin-10 suppresses the development of collagen type II-induced arthritis and ameliorates sustained arthritis in rats. Scand J Immunol 1996, 44:607-614 [DOI] [PubMed] [Google Scholar]
- 42.Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, de Benedetti F, Poli V, Ciliberto G: Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 1998, 187:461-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M, Kishimoto T: Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA 1998, 95:8222-8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasai M, Saeki Y, Ohshima S, Nishioka K, Mima T, Tanaka T, Katada Y, Yoshizaki K, Suemura M, Kishimoto T: Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum 1999, 42:1635-1643 [DOI] [PubMed] [Google Scholar]
- 45.Boe A, Baiocchi M, Carbonatto M, Papoian R, Serlupi-Crescenzi O: Interleukin 6 knock-out mice are resistant to antigen-induced experimental arthritis. Cytokine 1999, 11:1057-1064 [DOI] [PubMed] [Google Scholar]
- 46.Murphy G, Stanton H, Cowell S, Butler G, Knauper V, Atkinson S, Gavrilovic J: Mechanisms for pro matrix metalloproteinase activation. APMIS 1999, 107:38-44 [DOI] [PubMed] [Google Scholar]
- 47.Meyer JF, Bieth J, Metais P: On the inhibition of elastase by serum. Some distinguishing properties of alpha1-antitrypsin and alpha2-macroglobulin. Clin Chim Acta 1975, 62:43-53 [DOI] [PubMed] [Google Scholar]
- 48.Daveau M, Rouet P, Scotte M, Faye L, Hiron M, Lebreton JP, Salier JP: Human inter-alpha-inhibitor family in inflammation: simultaneous synthesis of positive and negative acute-phase proteins. Biochem J 1993, 292:485-492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida E, Sumi H, Maruyama M, Tsushima H, Matsuoka Y, Sugiki M, Mihara H: Distribution of acid stable trypsin inhibitor immunoreactivity in normal and malignant human tissues. Cancer 1989, 64:860-869 [DOI] [PubMed] [Google Scholar]
- 50.Yoshida E, Yoshimura M, Mihara H: Demonstration of an active component of inter-alpha-trypsin inhibitor in the brains of Alzheimer type dementia. Biochem Biophys Res Commun 1991, 174:1015-1021 [DOI] [PubMed] [Google Scholar]
- 51.Chawla RK, Lawson DH, Travis J: Plasma inter-alpha-trypsin inhibitor-related urinary glycoprotein EDC1 inhibits the growth of a Burkitt’s lymphoma cell line. J Cell Biochem 1990, 42:207-217 [DOI] [PubMed] [Google Scholar]
- 52.Franck C, Pedersen JZ: Trypsin-inhibitory activities of acid-stable fragments of the inter-alpha-trypsin inhibitor in inflammatory and uraemic conditions. Scand J Clin Lab Invest 1983, 43:151-155 [PubMed] [Google Scholar]
- 53.Becker A, Sandson J: The source of the inter-alpha trypsin inhibitor in pathologic hyaluronateprotein. Arthritis Rheum 1971, 14:764-766 [DOI] [PubMed] [Google Scholar]
- 54.Hutadilok N, Ghosh P, Brooks PM: Binding of haptoglobin, inter-α-trypsin inhibitor, and α1 proteinase inhibitor to synovial fluid hyaluronate and the influence of these proteins on its degradation by oxygen derived free radicals. Ann Rheum Dis 1988, 47:377-385 [DOI] [PMC free article] [PubMed] [Google Scholar]