

Abstract
Pancreatoblastomas are unusual malignant neoplasms of the pediatric pancreas that may also rarely affect adults. The molecular pathogenesis of pancreatoblastomas is unknown. They are clinicopathologically distinct from adult pancreatic ductal adenocarcinomas, but their occasional occurrence in patients with Beckwith-Wiedemann syndrome and the case presented here of a pancreatoblastoma in an adult patient with familial adenomatous polyposis (FAP) suggests that they might bear a genetic similarity to other infantile embryonal tumors such as hepatoblastomas. We analyzed a series of nine pancreatoblastomas for mutations common to other embryonal malignancies including somatic alterations in the adenomatous polyposis coli (APC)/β-catenin pathway and chromosome 11p, using immunohistochemistry for β-catenin, 5q and 11p allelic loss assays, and direct DNA sequencing of exon 3 of the β-catenin gene and the mutation cluster region of the APC gene. In addition, we analyzed the pancreatoblastomas for alterations found in adult-type pancreatic ductal adenocarcinomas including mutations in the K-ras oncogene and the p53 and DPC4 tumor suppressor genes, using direct DNA sequencing of exon 1 of K-ras and immunohistochemistry for p53 and Dpc4. Allelic loss on chromosome 11p was the most common genetic alteration in pancreatoblastomas, present in 86% (six of seven informative cases). Molecular alterations in the APC/β-catenin pathway were detected in 67% (six of nine), including five neoplasms with activating mutations of the β-catenin oncogene and the one FAP-associated tumor with biallelic APC inactivation (germline truncating mutation combined with loss of the wild-type allele); seven neoplasms showed abnormal nuclear accumulation of β-catenin protein. In contrast, loss of Dpc4 protein expression was present in only two cases (one diffuse and one focal), and no alterations in the K-ras gene or p53 expression were detected. Our findings indicate that pancreatoblastomas are genetically distinct from the more common pancreatic ductal adenocarcinomas, but bear a close molecular pathogenesis to hepatoblastomas. In addition, pancreatoblastoma may represent an extracolonic manifestation of FAP.
Pancreatoblastomas are rare, histologically distinct pancreatic neoplasms, comprising <1% of pancreatic and periampullary tumors. 1 Most commonly encountered in infants and young children, 2-11 these neoplasms pursue a malignant although more indolent course than adult-type ductal adenocarcinomas, and complete excision frequently effects cure. 2,3,9 However, rare cases of pancreatoblastoma have also been reported to occur in adults ranging from 19 to 68 years of age, 2,12-16 among whom the prognosis seems to be more guarded. 2 Histologically, pancreatoblastomas in both the pediatric and adult populations are highly cellular epithelial neoplasms with sheets and nests of polygonal cells, acinar formations, squamoid corpuscles, and a cellular stroma. Acinar differentiation in the form of enzyme production is a consistent feature, and focal endocrine and ductal differentiation may also occur. 2,3,17,18
Although considered by some to be of embryonic origin based on their histological appearance, 19 the molecular events underlying the genesis of these rare neoplasms have been primarily unexplored. A potential molecular association between pancreatoblastoma and the related embryonal tumor hepatoblastoma is suggested by the occurrence of both tumor types in young patients with Beckwith-Wiedemann syndrome, 11,20 raising the possibility that genetic events on chromosome 11p might play a role in pancreatoblastomas as they have been shown to do in hepatoblastomas. 21-25 In addition, one of the pancreatoblastomas included in the series reported here arose in a patient with familial adenomatous polyposis (FAP). Among patients with FAP, hepatoblastomas occur at increased frequency and hepatoblastomas in these patients can be demonstrated to harbor somatic, second-hit mutations of the adenomatous polyposis coli (APC) gene on chromosome 5q. 26-29 This apparently first reported occurrence of a pancreatoblastoma in the setting of FAP further raised the possibility that alterations in the APC/β-catenin pathway might play a role in FAP-associated and sporadic pancreatoblastomas.
We therefore undertook a comprehensive molecular characterization of pancreatoblastomas for genetic alterations found in other pediatric blastomas including allelic loss on chromosome 11p and alterations of the APC/β-catenin pathway, and for mutations in the K-ras oncogene and p53 and DPC4 tumor suppressor genes that characterize the more common adult pancreatic ductal adenocarcinomas.
Materials and Methods
Case Selection
The study population consisted of nine patients with pancreatoblastomas who underwent biopsy and/or surgical resection between 1967 and 2000. One case was from The Johns Hopkins Hospital, four cases were from Memorial Sloan-Kettering Cancer Center, and four cases were from Children’s Hospital and Regional Medical Center in Seattle. In one case, tissues from both biopsy of the pancreatoblastoma and its subsequent resection after chemotherapy were available, and both samples were analyzed separately. Pancreatoblastomas were diagnosed based on characteristic histological features that included varying proportions of sheet-like and acinar growth, as well as squamoid corpuscles, the presence of which serves to distinguish these tumors from acinar cell carcinomas of the pancreas. 2
Immunohistochemistry for β-Catenin, p53, and Dpc4
Immunoperoxidase stains using diaminobenzidine as the chromogen were performed on the Techmate 1000 automatic staining system (BioTek Solutions, Tucson, AZ). Deparaffinized sections of formalin-fixed tissue at 5-μm thickness were stained with β-catenin antibody (1:500 dilution, mouse monoclonal; Becton Dickinson Transduction Laboratories, Lexington, KY), p53 antibody (1:100 dilution, mouse monoclonal clone D07; DAKO, Carpinteria, CA), and Dpc4 antibody (1:100 dilution, monoclonal clone B8; Santa Cruz Biotechnology, Santa Cruz, CA). Heat-induced antigen retrieval using steam for 20 minutes at 80°C was used before incubation with anti-β-catenin, anti-p53, and anti-Dpc4.
For β-catenin, immunohistochemical labeling was evaluated for the presence of nuclear, cytoplasmic, and membranous β-catenin accumulation in both the neoplasms and the normal surrounding tissues (including nonneoplastic pancreatic ductal and acinar cells, and hepatic parenchyma or stromal fibroadipose tissue in the case of metastases). Nuclear and cytoplasmic accumulation of β-catenin in the pancreatoblastomas was graded according to the percentage of neoplastic cells with strong immunolabeling. For p53, the percentage of positively labeled nuclei was recorded; we considered strong nuclear labeling in ≥30% of tumor cells as the cutoff for p53 positivity. For Dpc4, neoplasms were classified as showing intact Dpc4 expression if they showed the normal pattern of strong, diffuse cytoplasmic labeling and labeling of scattered nuclei. They were classified as showing loss of Dpc4 expression if they showed no cytoplasmic or nuclear Dpc4 labeling. Negative (normal saline) and positive controls were performed with each labeling. For anti-β-catenin, the positive control consisted of a juvenile nasopharyngeal angiofibroma with intense nuclear β-catenin accumulation (accompanied by a β-catenin gene mutation shown previously by DNA sequencing). For anti-p53 an ovarian serous carcinoma with nuclear p53 accumulation was used, and for anti-Dpc4 a pancreatic ductal adenocarcinoma with Dpc4 loss was used.
DNA Extraction
Microdissection of the pancreatoblastomas for DNA extraction was performed from formalin-fixed, paraffin-embedded specimens. A 271/2-guage-needle tip was used for microdissection of routinely processed, 5-μm hematoxylin and eosin-stained slides under a low-power (×4) objective. Genomic DNA was extracted as described previously. 30 Corresponding normal control DNA was extracted from adjacent nonneoplastic tissue (adjacent pancreatic acini/stroma in four cases, duodenum in one case, lymph node in one case, hepatocytes in two metastatic tumors, and inflamed granulation tissue surrounding a peritoneal implant in one metastatic tumor).
Mutation Analysis of the β-Catenin Gene
Genomic DNA from each sample was amplified by polymerase chain reaction (PCR) using the primer pair: 5′-ATGGAACCAGACAGAAAAGC-3′ (sense) and 5′-GCTA-CTTGTTCTTGAGTGAAG-3′ (antisense). These amplified a 200-bp fragment of exon 3 of the β-catenin gene that encompasses the region for GSK-3β phosphorylation. PCR reactions were performed under standard conditions in a 50-μl volume containing 38 μl of Platinum PCR SuperMix (Life Technologies, Rockville, MD), 5 μl of both 5′ and 3′ oligonucleotides (final concentration of 1 μmol/L), and 2 μl (∼50 ng) of genomic DNA. PCR conditions consisted of an initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, and a final extension at 72°C for 7 minutes. Negative controls using water in place of DNA template were performed with each PCR reaction. PCR products were purified with spin columns using the QIAquick PCR purification kit (Qiagen, Inc., Valencia, CA) before sequencing. Automated sequencing of purified PCR products was performed on an ABI Prism 3700 DNA Analyzer (Applied Biosystems, Inc., Foster City, CA) using the internal primers: 5′-AAAGCGGCTGTTAGTCACTGG-3′ (sense) and 5′-CCTGTTCCCACTCATACAGG-3′ (antisense), and the resulting sequence data were analyzed with the Sequencher analysis program (Gene Codes, Ann Arbor, MI). All mutations were verified in both sense and antisense directions using PCR products from independent reactions.
Base substitutions in codons 32 and 33 were further confirmed by HinfI restriction endonuclease assay (Life Technologies). The 200-bp PCR product for β-catenin contains two HinfI restriction endonuclease sites, yielding 7-bp, 55-bp, and 138-bp DNA fragments after digestion of the wild-type allele. β-catenin mutations in codons 32 and 33 yield only 62-bp and 138-bp fragments after digestion because of ablation of the first HinfI site.
Mutation Analysis of the APC Gene
Mutation analysis of the APC gene was performed on cases that did not show detectable β-catenin mutations. Four sets of oligonucleotide primers (A1: 5′-CAGACTTATTGTGTAGAAGA-3′ and A2: 5′-CTCCTGAAGAAAATTCAACA-3′ for codons 1260 to 1359; B1: 5′-AGGGTTCTAGTTTATCTTCA-3′ and B2: 5′-TCTGCTTGGTGGCATGGTTT-3′ for codons 1339 to 1436; C1: 5′-GGCATTATAAGCCCCAG-TGA-3′ and C2: 5′-AAATGGCTCATCGAGGCTCA-3′ for codons 1417 to 1516; D1: 5′-ACTCCAGATGGATTTTCTTG-3′ and D2: 5′-GGCTGGCTTTTTTGCTTTAC-3′ for codons 1497 to 1596) were used to amplify the mutation cluster region of the APC gene. 31 PCR reactions were performed in 50-μl volumes using the reaction mixture described above. PCR conditions consisted of an initial denaturation step of 94°C for 3 minutes, 40 cycles (94°C for 1 minute, 55°C for 1 minute, and 68°C for 1.5 minutes for APC-B, -C, and -D primer pairs and 94°C for 1 minute, 52°C for 1 minute, and 68°C for 1.5 minutes for APC-A), followed by a final extension at 72°C for 7 minutes. PCR products were purified and sequenced as described above using the same primers as for genomic DNA amplification. Mutations were verified in both sense and antisense directions.
Mutation Analysis of the K-ras Gene
The oligonucleotide primers 5′-GAGAATTCATGACTGAATATAAACTTGT-3′ (sense) and 5′-TCGAATTCCTCTATTGTTGGATCATATTCG-3′ (antisense) were used to amplify a region in exon 1 of K-ras spanning codons 12 and 13. PCR reactions were performed in 50-μl volumes using the reaction mixture described above, with an initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 50°C for 1 minute, and 72°C for 1 minute, and a final extension at 72°C for 7 minutes. PCR products were purified and sequenced as described above using the internal primer 5′-ATTCGTCCACAAAATGAT-3′.
Allelic Loss on Chromosome 5q
Analysis for loss of heterozygosity (LOH) on 5q was performed on cases that did not show detectable β-catenin mutations. LOH was assessed by microsatellite assays using PCR amplification of three microsatellite markers (D5S82, D5S299, and D5S346) as previously described. 32 Briefly, assays were performed in 96-well plates in 10-μl volumes, each containing 5 μl of PCR Master (Boehringer Mannheim, Mannheim, Germany), 3.5 μl of water, 1 μl of genomic DNA, 0.06 μl of 3′ oligonucleotide, and 0.4 μl of end-labeled 5′oligonucleotide. The 5′ oligonucleotide had been end-labeled with [γ-32P]-ATP (Dupont-NEN, Boston, MA) using T4 polynucleotide kinase (New England Biolabs, Beverly, MA). For D5S82 and D5S299, 38 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute were performed, and for D5S342, 38 cycles of 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 1 minute were performed. LOH was considered to be present when there was complete or near-complete disappearance of a heterozygous band as compared with nonneoplastic control tissue in at least one informative marker.
Allelic Loss on Chromosome 11p
LOH on 11p was evaluated for all pancreatoblastomas using the microsatellite markers TH (a tetranucleotide repeat polymorphism on 11p15.5-p15) and D11S1984 (a dinucleotide repeat on 11p15.5). Assays were performed and interpreted as described above using annealing temperatures of 62°C for TH and 55°C for D11S1984.
Results
A summary of the clinicopathological and molecular findings in the nine cases (P1 to P9) is presented in Table 1 ▶ .
Table 1.
Clinicopathological Findings and Genetic Alterations in Pancreatoblastomas
| Case | Age/sex | K-ras | p53 Accumulation | Dpc4 loss | Nuclear β-catenin accumulation, % | β-Catenin | APC alterations | 11p LOH |
|---|---|---|---|---|---|---|---|---|
| P1 | 5 /M | Wild type | − | − | 25 | N32H | N/E | − |
| P2 | 3 /M | N/A | − | − | 15 | Wild type | N/A | N/A |
| P3 | 4 /M | Wild type | − | N/A | 25 | S33F | N/E | N/I |
| P4 | 45 /F | Wild type | − | + (diffuse) | 90 | S37F | N/E | + |
| P5 | 51 /F | Wild type | − | + (focal) | 40 | Wild type | Germline + LOH | + |
| P6 | 6 /M | Wild type | − | − | − | Wild type | Wild type | + |
| P7 | 13 /F | Wild type | − | − | − | Wild type | Wild type | + |
| P8 | 5 /F | Wild type | − | − | 90 | G34R | N/E | + |
| P9 | 3.5 /F | Wild type | − | − | 40 | N32H | N/E | + |
Locations of somatic mutations in β-catenin are shown by codon.
Nuclear β-catenin accumulation was evaluated based on the percentage of strongly staining tumor cell nuclei. Cytoplasmic accumulation was found to mirror nuclear staining in pancreatoblastomas.
LOH, loss of heterozygosity; N/A, DNA did not amplify or immunohistochemistry failed; N/E, not evaluated due to the presence of mutation detected elsewhere; N/I, noninformative for allelic loss assays.
Clinicopathological Characteristics
Seven of the pancreatoblastomas arose in pediatric patients ranging from 3 to 13 years (mean, 5.6 years) and two cases were in adults of ages 45 and 51 years. Five patients (56%) were female and four (44%) were male. One pancreatoblastoma (case P5) arose in a patient with FAP, a 51-year-old Caucasian woman. None of the patients was known to have Beckwith-Wiedemann syndrome.
Alterations in the APC/β-Catenin Pathway
Strong nuclear and cytoplasmic accumulation of β-catenin protein was present in seven of nine (78%) pancreatoblastomas, ranging from 15 to 90% of the neoplastic cells in positive cases. The nuclear and cytoplasmic labeling were highly correlated; neoplastic cells with strong nuclear labeling also showed strong cytoplasmic labeling. In the majority of cases (five of seven) that demonstrated β-catenin accumulation, the labeling was patchy in nature, and was diffusely present in only two cases. Although a firm relationship between β-catenin labeling and morphological pattern of tumor growth was not discernable, we noted a general tendency for areas of acinar differentiation to lack strong nuclear and cytoplasmic β-catenin, and for areas of sheet-like growth and squamoid corpuscles to show β-catenin accumulation in those neoplasms with labeling (Figure 1) ▶ . In addition, stromal cells in fibrous tissue between lobules of neoplastic epithelial cells did not show β-catenin accumulation. Nonneoplastic pancreatic acini and hepatocytes showed the expected membranous and faint cytoplasmic labeling, but no nuclear or strong cytoplasmic β-catenin.
Figure 1.
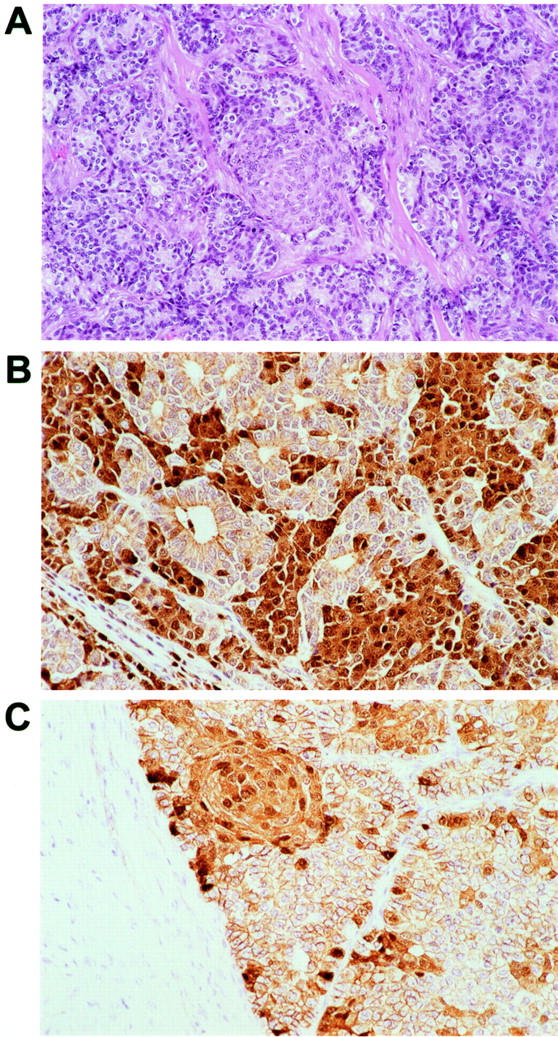
Histopathological appearance and immunohistochemical labeling for β-catenin in pancreatoblastomas. A: Neoplastic epithelial cells show sheet-like and acinar growth patterns and characteristic squamoid corpuscles. Lobules of neoplastic epithelium are divided by bands of fibrous tissue. B: Nuclear and cytoplasmic accumulation of β-catenin is present in this example in foci of sheet-like growth but not in areas with acinar differentiation. C: β-catenin accumulation in this case is present in a squamoid corpuscle and in a patchy distribution at the periphery of solid nests. Stromal cells in the intervening fibrous bands are negative.
Five of the eight (64%) pancreatoblastomas from patients without FAP contained mutations in exon 3 of the β-catenin gene. All five were 1-bp missense mutations either affecting serine residues at GSK-3β phosphorylation sites encoded in codon 33 (one case) and codon 37 (one case), or in residues around serine/threonine phosphorylation sites at codons 32 and 34 (three cases) (Figure 2) ▶ . All three cases with mutations at codons 32 and 33 demonstrated the expected ablation of the HinfI recognition site (Figure 2) ▶ . The same β-catenin mutation in codon 32 was present in both the separately analyzed initial biopsy and after chemotherapy resection specimens from patient P1. In all pancreatoblastomas with β-catenin gene mutations, a mixture of the wild-type and mutant peaks was present, as would be expected because of the dominant effect of β-catenin gene alterations. The somatic nature of the β-catenin mutations was supported by the presence of only wild-type β-catenin in corresponding normal tissues from these patients.
Figure 2.
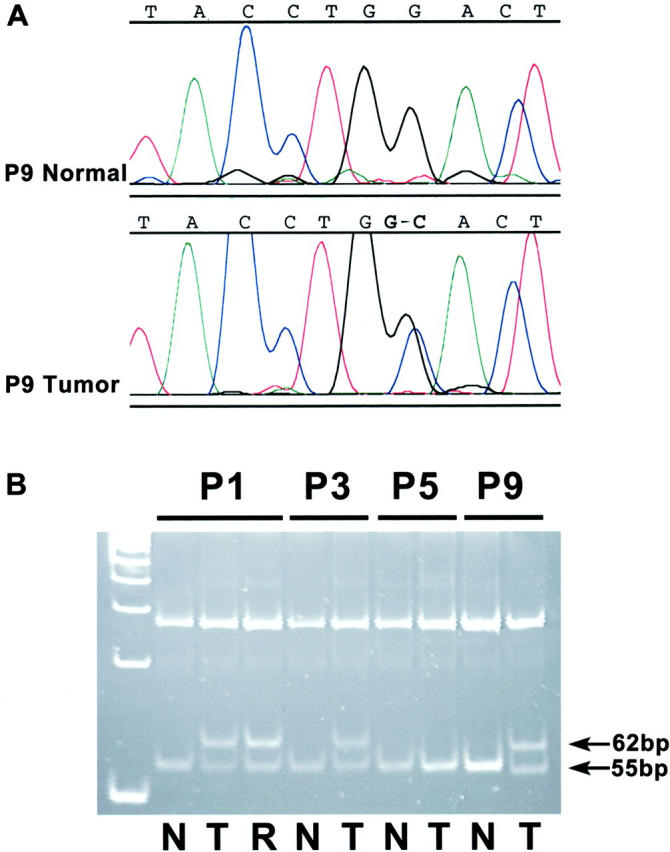
β-catenin mutations in pancreatoblastomas. A: DNA sequencing chromatogram from case P9, showing a GAC (asparagine) 224 CAC (histidine) mutation at codon 32, which is not present in the wild-type β-catenin sequence from this patient. B: HinfI restriction endonuclease assay to verify the presence of the missense mutation in this case and in the other pancreatoblastomas with codon 32 and codon 33 mutations. The normal 200-bp PCR product for β-catenin contains two HinfI restriction endonuclease sites, yielding 7-bp, 55-bp, and 138-bp DNA fragments after digestion of the wild-type allele (the 7-bp fragment is too small to be visualized on the gel). β-catenin mutations in codons 32 (cases P1 and P9) and codons 33 (case P3) yield 138-bp and 62-bp fragments after digestion because of ablation of the first HinfI site. Case P5 did not contain a β-catenin mutation and shows only the expected 138-bp and 55-bp DNA fragments. A molecular weight marker of 50-bp ladder is on the left. N, normal; T, pancreatoblastoma; R, residual pancreatoblastoma after chemotherapy in case P1.
The four pancreatoblastomas that did not show β-catenin gene mutations were further evaluated for APC alterations by sequencing of the APC mutation cluster region and 5q allelic loss assays. Biallelic inactivation of the APC gene was found in one case (case P5), a pancreatoblastoma arising in a patient with FAP (Figure 3) ▶ . Sequencing of normal tissue from this patient revealed a germline truncating 5-bp deletion mutation in the APC mutation cluster region spanning codons 1309 to 1311. Allelic loss on chromosome 5q was also present in the pancreatoblastoma from this patient on LOH assays. DNA sequencing of this neoplasm tissue also showed complete loss of the wild-type DNA sequence in the region of the patient’s germline mutation, confirming that the allelic loss was of the wild-type allele and therefore represented a second-hit APC alteration. No APC mutations or 5q LOH were detected in two other sporadic pancreatoblastomas that did not demonstrate β-catenin mutations (amplifiable DNA for APC analysis was not obtained from the remaining sporadic pancreatoblastoma).
Figure 3.

Biallelic inactivation of the APC gene in the pancreatoblastoma from a patient with FAP (case P5). A: DNA-sequencing chromatogram of normal tissue from this patient demonstrates a germline truncating 5-bp deletion mutation in APC spanning codons 1309 to 1311. The pancreatoblastoma shows complete loss of the wild-type DNA sequence in this region, indicating loss of the wild-type allele. B: Allelic loss assays show corresponding LOH with 5q microsatellite markers. N, normal; T, pancreatoblastoma.
Overall, molecular alterations in the APC/β-catenin pathway were detected in six of nine (67%) pancreatoblastomas, including five cases with activating β-catenin mutations and one case with biallelic APC inactivation. Good correlation was found between abnormal nuclear and cytoplasmic accumulation of β-catenin protein by immunostaining and the presence of detectable APC/β-catenin gene alterations; both of the two pancreatoblastomas without β-catenin immunostaining (P6 and P7) also lacked β-catenin and APC mutations. Only one neoplasm with β-catenin immunostaining failed to show an associated molecular alteration; this case (P2, a tumor block prepared several decades previously) contained primarily degraded DNA that failed to amplify for APC sequencing and allelic loss assays.
Allelic Loss on 11p
Allelic loss on 11p15.5 was present in six of seven (86%) pancreatoblastomas that contained amplifiable DNA and were informative in one or both 11p microsatellite markers (Figure 4) ▶ . All three cases that were informative at both TH and D11S1984 showed LOH on both markers in the pancreatoblastomas. Because parental tissue was not available in any of the cases, we could not determine the parental origin of the lost allele in those pancreatoblastomas with 11p LOH.
Figure 4.

Allelic loss on 11p15.5 in pancreatoblastomas. LOH is present with both the D11S1984 and TH1 microsatellite markers for case P5, which was informative for both markers. Cases P4 and P6 show LOH in the marker for which each was informative. N, normal; T, pancreatoblastoma.
Alterations in the DPC4, K-ras, and p53 Genes
Infrequent (two of nine cases, 22%) loss of Dpc4 protein expression was present in the pancreatoblastomas. In one pancreatoblastoma, Dpc4 expression was diffusely lost throughout all neoplastic epithelial cells, whereas the intervening fibrous bands between neoplastic lobules retained the normal pattern of cytoplasmic and nuclear Dpc4 immunolabeling (Figure 5) ▶ . In the other case, focal loss of Dpc4 was present in the neoplastic epithelial component, and the fibrous stroma again retained normal cellular Dpc4. All of the remaining pancreatoblastomas demonstrated normal Dpc4 labeling of epithelial and stromal cells.
Figure 5.
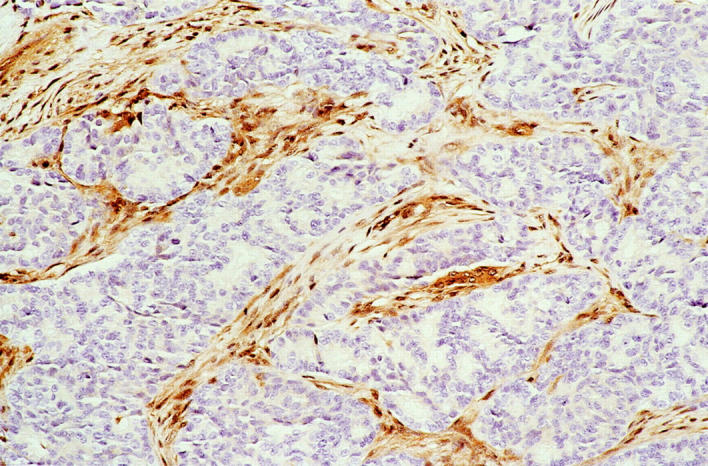
Loss of normal Dpc4 protein expression in case P4. Dpc4 expression is diffusely lost throughout all neoplastic epithelial cells from this pancreatoblastoma. The intervening fibrous bands show the normal pattern of cytoplasmic and nuclear Dpc4 immunolabeling.
No K-ras gene mutations were detected by sequencing of exon 1 encompassing codons 12 and 13 in eight pancreatoblastomas from which amplifiable DNA was obtained. No significant p53 accumulation was detected by immunohistochemistry.
Discussion
The rarity of pancreatoblastomas has previously prevented detailed investigations of their molecular pathogenesis. K-ras oncogene mutations are commonly present (>90%) in adult pancreatic ductal adenocarcinomas, but the single pancreatoblastoma analyzed in the literature was wild type for K-ras. 15 Aside from this single case report, molecular alterations characterizing pancreatoblastomas have remained obscure. Several clinicopathological features of pancreatoblastomas led us to hypothesize that the molecular pathogenesis of these neoplasms might on the one hand be similar to another embryonal tumor, hepatoblastoma, and on the other hand, be distinct from the more common but prognostically, clinically, and histopathologically disparate adult pancreatic ductal adenocarcinomas. We found a high frequency of allelic loss on chromosome 11p and somatic mutations in the APC/β-catenin pathway in pancreatoblastomas, genetic alterations that are also common to hepatoblastomas. In contrast, alterations in the K-ras and p53 genes that characterize most pancreatic ductal adenocarcinomas were not detected in pancreatoblastomas, and loss of Dpc4 expression was only infrequently present.
Eighty-six percent of pancreatoblastomas in this series (six of seven informative cases) demonstrated LOH for TH and D11S1984, microsatellite markers near the WT-2 locus on chromosome 11p15.5. Several cases of pancreatoblastoma have previously been reported to occur in young patients in association with Beckwith-Wiedemann syndrome, 11,20 a maldevelopmental syndrome characterized by tissue overgrowth and organomegaly and an increased risk for embryonal tumors including Wilms’ tumor, hepatoblastoma, and rhabdomyosarcoma. 33,34 The heterogeneous congenital and acquired manifestations of Beckwith-Wiedemann syndrome involve dysregulation of growth- and cell cycle-regulatory genes on a heavily imprinted region of chromosome 11p15.5. 33,34 Previous studies of hepatoblastomas have shown frequent allelic loss on 11p ranging from 25 to 75% of sporadic cases, 21-25 and in particular, selective loss of the maternal allele. 21 The high rate of 11p allelic loss in pancreatoblastomas in our series provides the first molecular evidence for a common genetic relationship between pancreatoblastomas, Beckwith-Wiedemann syndrome, and related embryonal malignancies. Because parental tissue was not available for analysis in our patients, evaluation of the parental origin of allelic loss in pancreatoblastomas was not possible.
The second most common genetic alteration in pancreatoblastomas in this study— mutations involving the APC/β-catenin pathway—is of particular interest because this pathway has also been implicated frequently in the development of hepatoblastomas. 25,29,35-40 Among patients with FAP (characterized by germline mutation in the APC gene on chromosome 5q and an increased rate of development of colorectal and extra-colonic malignancies), the risk of hepatoblastoma is markedly increased (∼1000-fold more than that of the general population), even though most hepatoblastomas represent sporadic, non-FAP and non-Beckwith-Wiedemann-associated tumors. 26,27 One hepatoblastoma arising in association with germline APC mutation has been demonstrated to have biallelic inactivation of the APC gene, 29 and in sporadic hepatoblastomas APC alterations including truncating mutations and 5q allelic loss have been reported at widely varying frequencies from 0 to 69%. 29,35,37-39
Unlike hepatoblastomas, pancreatoblastomas have not previously been known to arise in association with FAP, and our patient with FAP and a pancreatoblastoma represents the first such occurrence reported to date. The demonstration of biallelic APC inactivation in this patient’s pancreatoblastoma (germline-truncating mutation with loss of the wild-type allele) suggests that the relationship is not coincidental and that alterations in the APC pathway played an etiological role in the development of this patient’s pancreatoblastoma. One of the functions of APC tumor suppressor protein involves regulation of the cellular level of β-catenin, which acts in part as a downstream transcriptional activator in the Wnt signaling pathway. 41,42 APC, along with glycogen synthase kinase-3β (GSK-3β) and AXIN1, promotes phosphorylation of serine/threonine residues encoded in exon 3 of the β-catenin gene. 41,43,44 Phosphorylation is followed by ubiquitin-mediated degradation of β-catenin protein. 45,46 Loss of β-catenin regulatory activity resulting in abnormal accumulation of β-catenin protein can occur either by truncating APC mutations or by stabilizing β-catenin mutations at GSK-3β phosphorylation sites. 44,47,48 This pathway can therefore be targeted by either inactivating APC tumor suppressor gene mutations or by activating mutations of the β-catenin oncogene. 49,50
Among eight sporadic pancreatoblastomas, we identified mutations in exon 3 of the β-catenin gene in five (62%) cases. All were 1-bp missense mutations, located either at serine GSK-3β phosphorylation sites in codons 33 and 37 (two cases) or around serine/threonine phosphorylation sites in codons 32 and 34 (three cases). Those cases containing exon 3 mutations in codons 32 and 34 did not involve loss of a phosphorylation site but may still be expected to interfere with normal degradation of the β-catenin gene product. 50 The strong correlation between the presence of abnormal β-catenin protein accumulation by immunohistochemistry and the presence of β-catenin or APC gene alterations in our pancreatoblastomas supports this association.
Overall, somatic alterations of the APC/β-catenin pathway were identified in 67% (six of nine) pancreatoblastomas, indicating an important role for this pathway in pancreatoblastomas in addition to other embryonal malignancies. 51 Interestingly, we were able to demonstrate APC inactivation only in the pancreatoblastoma arising in association with FAP, whereas the sporadic pancreatoblastomas instead showed a predominance of β-catenin mutations. Among hepatoblastomas, only two studies of neoplasms from Asian patients have reported APC alterations in sporadic hepatoblastomas, 35,37 whereas other studies of Western and Taiwanese patients have failed to detect APC alterations and have instead shown frequent involvement of the β-catenin gene in the genesis of these tumors. 25,36,38-40 Both geographic/ethnic differences as well as the relative susceptibility of neoplastic cells to somatically acquire a single β-catenin hit as opposed to the two hits required for APC inactivation may underlie some of this discrepant genetic constitution in the FAP-associated and sporadic tumors.
In contrast to the frequent involvement of the APC/β-catenin pathway and chromosome 11p losses in pancreatoblastomas, we found no evidence for K-ras oncogene mutations by DNA sequencing or p53 tumor suppressor gene alterations by immunohistochemistry in these neoplasms. Our findings are in agreement with and expand those of Hoorens and colleagues, 15 who found neither K-ras mutation nor p53 overexpression in one case of pancreatoblastoma arising in an adult. Although our sequencing for K-ras was designed to detect mutations only in codons 12 or 13 of the K-ras oncogene, there is little evidence that mutations in K-ras codon 61 play a significant role in pancreatic carcinomas. Previous investigations of adult pancreatic ductal adenocarcinomas, for example, have demonstrated that K-ras mutations occur almost exclusively in codon 12; codon 12 base substitutions were detected in 28 of 30 cases, 18 of 18 cases, and 35 of 35 cases, respectively, in the studies of Smit and colleagues, 52 Tada and colleagues, 53 and Luttges and colleagues. 54 Among these adult pancreatic adenocarcinomas, K-ras gene activation occurs early in the neoplastic progression of almost all tumors, 55,56 whereas p53 inactivation is a relatively late event in neoplastic progression, but can be demonstrated in up to 70% of invasive pancreatic adenocarcinomas. 55,57-59 Loss of Dpc4 tumor suppressor protein, present diffusely in only one of our pancreatoblastomas and focally in a second case (interestingly, both neoplasms with Dpc4 loss occurred in adult patients), also contrasts with adult ductal adenocarcinomas, among which slightly more than one-half have DPC4 inactivation. 60,61 This pattern of genetic alterations in pancreatoblastomas, unique among the pancreatic malignancies studied to date, underscores the distinctive morphological, epidemiological, and clinical manifestations of these rare neoplasms.
Acknowledgments
We thank Dr. Murray F. Brennan, who performed the pancreatoblastoma resections in the Department of Surgery, Memorial Sloan-Kettering Cancer Center, New York, NY.
Footnotes
Address reprint requests to Susan Abraham, M.D., Division of GI/Liver Pathology, Dept. of Pathology, Ross Building, Room 632, The Johns Hopkins University School of Medicine, 720 Rutland Ave., Baltimore, MD 21205-2196. E-mail: sabraham@jhmi.edu.
Supported by a National Cancer Institute SPORE grant in gastrointestinal cancer (P50-CA62924).
References
- 1.Serio G, Bortolasi L, Iacono C, Montresor E: Non ductal adenocarcinoma neoplasms of the pancreas. Chir Ital 1999, 51:181-188 [PubMed] [Google Scholar]
- 2.Klimstra DS, Wenig BM, Adair CF, Heffess CS: Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol 1995, 19:1371-1389 [DOI] [PubMed] [Google Scholar]
- 3.Kissane JM: Pancreatoblastoma and solid and cystic papillary tumor: two tumors related to pancreatic ontogeny. Semin Diagn Pathol 1994, 11:152-164 [PubMed] [Google Scholar]
- 4.Ogawa T, Okinaga K, Obana K, Nakamura K, Hattori T, Ito T, Yanagawa Y, Tanaka F, Imamura T: Pancreatoblastoma treated by delayed operation after effective chemotherapy. J Pediatr Surg 2000, 35:1663-1665 [DOI] [PubMed] [Google Scholar]
- 5.Hua C, Shu XK, Lei C: Pancreatoblastoma: a histochemical and immunohistochemical analysis. J Clin Pathol 1996, 49:952-954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchino JJ, Castello FM, Nagaraj HS: Pancreatoblastoma. A histochemical and ultrastructural analysis. Cancer 1984, 53:963-969 [DOI] [PubMed] [Google Scholar]
- 7.Wiley J, Posekany K, Riley R, Holbrook T, Silverman J, Joshi V, Bowyer S: Cytogenetic and flow cytometric analysis of a pancreatoblastoma. Cancer Genet Cytogenet 1995, 79:115-118 [DOI] [PubMed] [Google Scholar]
- 8.Nagashima Y, Misugi K, Tanaka Y, Ijiri R, Nishihira H, Nishi T, Kigasawa H, Kato K: Pancreatoblastoma: a second report on cytogenetic findings. Cancer Genet Cytogenet 1999, 109:178-179 [DOI] [PubMed] [Google Scholar]
- 9.Chun Y, Kim W, Park K, Lee S, Jung S: Pancreatoblastoma. J Pediatr Surg 1997, 32:1612-1615 [DOI] [PubMed] [Google Scholar]
- 10.Murakami T, Ueki K, Kawakami H, Gondo T, Kuga T, Esato K, Furukawa S: Pancreatoblastoma: case report and review of treatment in the literature. Med Pediatr Oncol 1996, 27:193-197 [DOI] [PubMed] [Google Scholar]
- 11.Koh TH, Cooper JE, Newman CL, Walker TM, Kiely EM, Hoffmann EB: Pancreatoblastoma in a neonate with Wiedemann-Beckwith syndrome. Eur J Pediatr 1986, 145:435-438 [DOI] [PubMed] [Google Scholar]
- 12.Robin E, Terris B, Valverde A, Molas G, Belghiti J, Bernades P, Ruszniewski P: Pancreatoblastoma in adults. Gastroenterol Clin Biol 1997, 21:880-883 [PubMed] [Google Scholar]
- 13.Palosaari D, Clayton F, Seaman J: Pancreatoblastoma in an adult. Arch Pathol Lab Med 1986, 110:650-652 [PubMed] [Google Scholar]
- 14.Dunn JL, Longnecker DS: Pancreatoblastoma in an older adult. Arch Pathol Lab Med 1995, 119:547-551 [PubMed] [Google Scholar]
- 15.Hoorens A, Gebhard F, Kraft K, Lemoine NR, Kloppel G: Pancreatoblastoma in an adult: its separation from acinar cell carcinoma. Virchows Arch 1994, 424:485-490 [DOI] [PubMed] [Google Scholar]
- 16.Levey JM, Banner BF: Adult pancreatoblastoma: a case report and review of the literature. Am J Gastroenterol 1996, 91:1841-1844 [PubMed] [Google Scholar]
- 17.Klimstra DS, Rosai J, Heffess CS: Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol 1994, 18:765-778 [DOI] [PubMed] [Google Scholar]
- 18.Kloppel G: Mixed exocrine-endocrine tumors of the pancreas. Semin Diagn Pathol 2000, 17:104-108 [PubMed] [Google Scholar]
- 19.Vossen S, Goretzki PE, Goebel U, Willnow U: Therapeutic management of rare malignant pancreatic tumors in children. World J Surg 1998, 22:879-882 [DOI] [PubMed] [Google Scholar]
- 20.Kohda E, Iseki M, Ikawa H, Endoh M, Yokoyama J, Mukai M, Hata J, Yamazaki H, Miyauchi J, Saeki M: Pancreatoblastoma. Three original cases and review of the literature. Acta Radiol 2000, 41:334-337 [DOI] [PubMed] [Google Scholar]
- 21.Albrecht S, von Schweinitz D, Waha A, Kraus JA, von Deimling A, Pietsch T: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 1994, 54:5041-5044 [PubMed] [Google Scholar]
- 22.Byrne JA, Simms LA, Little MH, Algar EM, Smith PJ: Three non-overlapping regions of chromosome arm 11p allele loss identified in infantile tumors of adrenal and liver. Genes Chromosom Cancer 1993, 8:104-111 [DOI] [PubMed] [Google Scholar]
- 23.Kiechle-Schwarz M, Scherer G, Kovacs G: Cytogenetic and molecular studies on six sporadic hepatoblastomas. Cancer Genet Cytogenet 1989, 41:286(abstract) [Google Scholar]
- 24.Koufos A, Hansen MF, Copeland NG, Jenkins NA, Lampkin BC, Cavenee WK: Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature 1985, 316:330-334 [DOI] [PubMed] [Google Scholar]
- 25.Blaker H, Hofmann WJ, Reiker RJ, Penzel R, Graf M, Otto HF: Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosom Cancer 1999, 25:399-402 [PubMed] [Google Scholar]
- 26.Giardiello FM, Petersen GM, Brensinger JD, Luce MC, Cayouette MC, Bacon J, Booker SV, Hamilton SR: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1996, 39:867-869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes LJ, Michels VV: Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet 1992, 43:1023-1025 [DOI] [PubMed] [Google Scholar]
- 28.Cetta F, Montalto G, Petracci M: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1997, 41:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurahashi H, Takami K, Oue T, Kusafuka T, Okada A, Tawa A, Okada S, Nishisho I: Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res 1995, 55:5007-5011 [PubMed] [Google Scholar]
- 30.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 31.Yashima K, Nakamori S, Murakami Y, Yamaguchi A, Hayashi K, Ishikawa O, Konishi Y, Sekiya T: Mutations of the adenomatous polyposis coli gene in the mutation cluster region: comparison of human pancreatic and colorectal cancers. Int J Cancer 1994, 59:43-47 [DOI] [PubMed] [Google Scholar]
- 32.Wu TT, Watanabe T, Heitmiller R, Zahrak M, Forastiere AA, Hamilton SR: Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998, 153:287-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Squire JA, Weksberg R: Molecular genetics of Beckwith-Wiedemann syndrome. Curr Opin Pediatr 1997, 9:623-629 [DOI] [PubMed] [Google Scholar]
- 34.Li M, Squire JA, Weksberg R: Molecular genetics of Wiedemann-Beckwith syndrome. Am J Med Genet 1998, 79:253-259 [PubMed] [Google Scholar]
- 35.Oda H, Imai Y, Nakatsuru Y, Hata J, Ishikawa T: Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res 1996, 56:3320-3323 [PubMed] [Google Scholar]
- 36.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999, 59:269-273 [PubMed] [Google Scholar]
- 37.Sang Park W, Ra Oh R, Young Park J, Joon Kim P, Sun Shin M, Heun Lee J, Sug Kimx H, Hyung Lee S, Young Kim S, Gyu Park Y, Gun An W, Seung Kim H, June Jang J, Jin Yoo N, Young Lee J: Nuclear localization of beta-catenin is an important prognostic factor in hepatoblastoma. J Pathol 2001, 193:483-490 [DOI] [PubMed] [Google Scholar]
- 38.Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA: Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene 2000, 19:498-504 [DOI] [PubMed] [Google Scholar]
- 39.Jeng YM, Wu MZ, Mao TL, Chang MH, Hsu HC: Somatic mutations of beat-catenin play a crucial role in the tumorigenesis of sporadic hepatoblastoma. Cancer Lett 2000, 152:45-51 [DOI] [PubMed] [Google Scholar]
- 40.Takayasu H, Horie H, Hiyama E, Matsunaga T, Hayashi Y, Watanabe Y, Suita S, Kaneko M, Sasaki F, Hashizume K, Ozaki T, Furuuchi K, Tada M, Ohnuma N, Nakagawara A: Frequent deletions and mutations of the beta-catenin gene are associated with overexpression of cyclin D1 and fibronectin and poorly differentiated histology in childhood hepatoblastoma. Clin Cancer Res 2001, 7:901-908 [PubMed] [Google Scholar]
- 41.Barth AI, Nathke IS, Nelson WJ: Cadherins, catenins, and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 1997, 9:683-690 [DOI] [PubMed] [Google Scholar]
- 42.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W: Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature 1996, 382:638-642 [DOI] [PubMed] [Google Scholar]
- 43.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P: Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996, 272:1023-1026 [DOI] [PubMed] [Google Scholar]
- 44.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P: Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA 1995, 92:3046-3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R: β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997, 16:3797-3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW: Serine phosphorylation-regulated ubiquitination and degradation of β-catenin. J Biol Chem 1997, 272:24735-24738 [DOI] [PubMed] [Google Scholar]
- 47.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancers by mutations in β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 48.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler W, Vogelstein B, Clevers H: Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon cancer. Science 1997, 275:1784-1787 [DOI] [PubMed] [Google Scholar]
- 49.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 50.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998, 58:1130-1134 [PubMed] [Google Scholar]
- 51.Koesters R, Ridder R, Kopp-Schneider A, Betts D, Adams V, Niggli F, Briner J, von Knebel Doeberitz M: Mutational activation of the β-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res 1999, 59:3880-3882 [PubMed] [Google Scholar]
- 52.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL: KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988, 16:7773-7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tada M, Yokosuka O, Omata M, Ohto M, Isono K: Analysis of ras gene mutations in biliary and pancreatic tumors by polymerase chain reaction and direct sequencing. Cancer 1990, 66:930-935 [DOI] [PubMed] [Google Scholar]
- 54.Luttges J, Schlehe B, Menke MA, Vogel I, Henne-Bruns D, Kloppel G: The K-ras mutation pattern in pancreatic ductal adenocarcinoma is usually identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer 1999, 85:1703-1710 [PubMed] [Google Scholar]
- 55.Hruban RH, Goggins M, Parsons J, Kern SE: Progression model for pancreatic cancer. Clin Cancer Res 2000, 6:2969-2972 [PubMed] [Google Scholar]
- 56.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53:549-554 [DOI] [PubMed] [Google Scholar]
- 57.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 58.DiGiuseppi JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allsion DC, Cameron JL, Offerhaus GJA: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 59.Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A: Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001, 84:253-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA, Jr, Meltzer PS, Hahn SA, Kern SE: DPC4 gene in various tumor types. Cancer Res 1996, 56:2527-2530 [PubMed] [Google Scholar]
- 61.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]