

Abstract
The G-protein–coupled receptor GPR74 is a novel candidate gene for body weight regulation. In humans, it is predominantly expressed in brain, heart, and adipose tissue. We report a haplotype in the GPR74 gene, ATAG, with allele frequency ∼4% in Scandinavian cohorts, which was associated with protection against obesity in two samples selected for obese and lean phenotypes (odds ratio for obesity 0.48 and 0.62; nominal P=.0014 and .014; n=1,013 and 1,423, respectively). In a population-based sample, it was associated with lower waist (P=.02) among 3,937 men and with obesity protection (odds ratio 0.36; P=.036) among those selected for obese or lean phenotypes. The ATAG haplotype was associated with increased adipocyte lipid mobilization (lipolysis) in vivo and in vitro. In human fat cells, GPR74 receptor stimulation and inhibition caused a significant and marked decrease and increase, respectively, of lipolysis, which could be linked to catecholamine stimulation of adipocytes through β-adrenergic receptors. These findings suggest that a common haplotype in the GPR74 gene protects against obesity, which, at least in part, is caused by a relief of inhibition of lipid mobilization from adipose tissue. The latter involves a cross-talk between GPR74 and β-adrenoceptor signaling to lipolysis in fat cells.
Although it is generally accepted that heredity has a strong influence on body weight, the resolution of the genetic components underlying variability in BMI (calculated as body weight in kilograms divided by the square of height in meters) among the general population is incomplete. In particular, the genetic factors that protect against obesity are not well understood.1 The G-protein–coupled receptor 74 (GPR74 [MIM 607449]) is a novel candidate gene for regulation of BMI. In humans, the receptor is predominantly expressed in the brain, heart, and adipose tissue.2,3 The true endogenous ligand for the receptor is not known, but GPR74 has a high affinity for neuropeptides, such as neuropeptide FF (NPFF).4 This neuropeptide has been investigated in laboratory animals. Although the opioid modulating effects of NPFF are the best explored,5 NPFF is also involved in cardiovascular regulation6 and response to stress7 or reward.8 As summarized elsewhere,9 NPFF has been reported to have effects on food intake. In addition, NPFF has effects on rodent fat cells, which involve interactions with β-adrenergic receptors.10 Catecholamines are the most important lipolytic hormones in man, and they regulate lipolysis through two major stimulatory adrenoceptors, β1and β2 (β3 is less effective), and through one inhibitory receptor (α2).11 Taken together, these data suggest that GPR74 could be a gene of importance for body weight regulation, maybe by having effects mediated on adipocyte lipolysis. We therefore did a comprehensive analysis of genetic variance in the GPR74 gene, using a large cohort of Swedish subjects with well-defined criteria for either long-standing leanness or severe obesity. The most important polymorphisms were reinvestigated in another large cohort of lean and obese Swedes, and results were confirmed in a large population-based Danish cohort. Finally, studies on lipolysis were performed, first, to find a link between GPR74 polymorphisms and lipid mobilization and, second, to study the mechanisms of action for GPR74 on human fat-cell lipolysis.
Methods
Study Population
Sample 1 was recruited for a study of genes underlying susceptibility to obesity, either from an outpatient center for treatment of obesity or through local advertisement (fig. 1 and table 1). The subjects were carefully selected for a lean or obese phenotype, and were at least 2nd-generation Scandinavians living in Sweden. The obese subjects were either <21 years old with BMI >30 or any age with BMI >40 (morbid obesity). The lean subjects were >45 years old and had never had BMI >25 according to self-report. The aim of selecting subjects with an extreme BMI phenotype in sample 1 was to enrich for a genetic impact on obesity or leanness.12 This was also the purpose of recruiting young obese adults, since early onset of this disorder is believed to have a stronger genetic component because of the reduced time of environmental impact.12 A total of 59 obese subjects had oral treatment for type 2 diabetes, but otherwise the subjects in sample 1 were healthy according to self-report.
Figure 1. .

Schematic presentation of ascertainment schemes for samples 1–3.
Table 1. .
Characteristics of the Cohorts
| Sample, Country, Body Weight Status, and Sex |
n | Agea (years) |
BMIa |
| 1: | |||
| Sweden: | |||
| Obese: | |||
| F | 487 | 42 ± 13 (16–77) | 44 ± 4 (30–64) |
| M | 124 | 45 ± 13 (16–73) | 45 ± 5 (32–58) |
| Lean: | |||
| F | 338 | 50 ± 4 (46–68) | 22 ± 2 (15–25) |
| M | 64 | 57 ± 8 (46–79) | 23 ± 2 (18–25) |
| 2: | |||
| Sweden: | |||
| Obese: | |||
| F | 688 | 44 ± 11 (19–75) | 36 ± 4 (30–58) |
| M | 122 | 45 ± 13 (18–75) | 44 ± 5 (31–66) |
| Lean: | |||
| F | 525 | 39 ± 6 (25–61) | 22 ± 2 (17–25) |
| M | 88 | 36 ± 11 (26–49) | 23 ± 1 (19–25) |
| 3: | |||
| Sweden: | |||
| Nonobese: | |||
| F | 99 | 38 ± 10 (18–60) | 23 ± 2 (18–27) |
| M | 51 | 41 ± 11 (26–77) | 23 ± 2 (19–25) |
| 4: | |||
| Denmark: | |||
| Anyb: | |||
| F | 3,854 | 48 ± 10 (22–88) | 26 ± 5 (15–56) |
| M | 3,937 | 49 ± 10 (19–89) | 27 ± 4 (17–57) |
Values are mean ± SD (range)
Ascertainment was population based.
Sample 2, used for confirmation of genetic associations, was recruited from the same sources but according to less stringent definitions for lean and obese phenotypes than those used for sample 1 (fig. 1 and table 1). The obese subjects had BMI >30 at any age, and the lean subjects were >25 years old and had BMI <25. The reason for the less stringent lean or obese phenotype criteria in sample 2 was practical—it is difficult to find subjects fulfilling the extreme criteria chosen for sample 1. The possibly resulting reduced power was partially overcome by a larger sample size. Sample 2 also comprised subjects who were at least 2nd-generation Scandinavians living in Sweden. Of the obese subjects in sample 2, 197 had hypertension, 116 had type 2 diabetes, and 50 had dyslipidemia, but otherwise the subjects were healthy according to self-report.
Sample 3 comprised healthy, nonobese Swedish subjects who were at least 2nd-generation Scandinavians and did not use any continuous medication. They were recruited through local advertisement with the purpose of studying metabolic regulation in fat cells (fig. 1 and table 1). Those subjects in sample 3 who fulfilled the criteria for inclusion in samples 1 and 2 were also included in samples 1 and 2 (fig. 1 and table 1). In the morning after an overnight fast, a subcutaneous fat biopsy specimen was obtained from the abdominal area by needle biopsy.13
Sample 4 consisted of a population-based Danish cohort of middle-aged people (3,937 men and 3,854 women) recruited as reported elsewhere.14 Clinical details of these subjects have been reported elsewhere.15
All subjects were age 18 years or older and were recruited independently of one another. Thus, there is no known familial relation between recruited subjects. We included both men and women in our analysis because genetic analyses of obesity indicate that the majority of susceptibility alleles are common in both sexes.16,17 Our recruitment strategy for samples 1 and 2 resulted in a higher proportion of women than men in the study. However, in both samples, the ratio of women to men were similar among obese and lean subjects.
All subjects came to the laboratory in the morning after an overnight fast. Height, weight, and, for the majority of subjects, waist circumference (midway between the lower rib-margin and the iliac crest), and hip circumference (widest measure over the great trochanters) were measured. The circumferences were measured in the supine position at the end of gentle expiration. A venous blood sample was obtained for extraction of genomic DNA and for determination of serum insulin, plasma glucose, lipids, glycerol, and free fatty acids by the accredited laboratories of the hospitals. Insulin resistance index (by homeostasis model assessment) (HOMAIR) was calculated as fasting serum insulin (mU/liter) times fasting plasma glucose (mmol/liter) divided by 22.5.18 Measures of serum insulin, plasma glucose, and plasma lipids were available for 1,809–1,977 subjects in samples 1 and 2. In vivo lipolytic activity was available for ∼1,900 subjects in samples 1 and 2 and was determined as plasma glycerol or free fatty acid concentrations divided by total body fat. Total body fat was obtained by a formula based on age, BMI, and sex.19 We compared body fat calculated in this way with the “golden standard,” which is dual x-ray absorptiometric analysis, for 11 nonobese (BMI 16.5–27.7) and 10 obese (BMI 30.2–45.9) subjects. Mean coefficient of variance between the two measures was 4% in nonobese subjects and 3.7% in obese subjects, suggesting that the formula method was sufficiently accurate.
The study was approved by the local ethics committee at each site, and informed consent was obtained from the subjects.
Studies of Mature Fat Cells
The investigations of mature fat cells were performed for sample 3. Lipolysis in isolated fat cells was investigated as described elsewhere.20,21 Cell suspensions were incubated in the absence or presence of increasing concentrations of noradrenaline, the β1-selective adrenoceptor agonist dobutamine, the β2-adrenoceptor selective agonist terbutaline, and the α2-adrenoceptor selective agonist clonidine. The concentration (log of mol/liter) of agonist causing half the maximum effect was determined. This value was converted to its negative form (pD2), which reflects agonist sensitivity. The maximum effect was determined as glycerol release (lipolysis index) at the maximum effective agonist concentration and was related to the number of incubated fat cells.
Studies of Preadipocytes
Subcutaneous adipose tissue was obtained from patients undergoing cosmetic lipoaspiration (eight women and one man; mean [±SD] age 38 ± 13 years; age range 19–60 years; BMI [±SD] 29 ± 5; BMI range 22–39). Five adipose tissue samples were from the abdominal area; for the other samples, information about the source location was lacking. The isolation and differentiation of preadipocytes was performed as described elsewhere.22
In one type of experiment, preadipocytes were treated with a maximum effective concentration (10−6 mol/liter) of NPFF4 for 3 h or 48 h before lipolysis experiments, which were performed at day 12 or 13 of differentiation. In the second type of experiment, RNA interference was investigated as described elsewhere.23 Different amounts of GPR74 siRNA (1–3 μg) and the transfection reagent RNAiFect/HiPerFect (Qiagen) were titrated to determine the optimal conditions for GPR74 silencing. The preadipocytes were transfected at day 7 of the differentiation process with 2.3 μg of GPR74 siRNA or scrambled (nonsilencing) siRNA. The cells treated with only the transfection agent served as controls. After 24 hours, some cells were lysed for RNA isolation to determine the silencing efficiency. The remaining cells were used for lipolysis and adipocyte differentiation assays.
mRNA levels were determined as described elsewhere.23 Total RNA was extracted and was reverse transcribed to cDNA. Quantitative real-time PCR was performed in an iCycler IQ (BioRad Laboratories). GPR74 and 18S mRNA were quantified using TaqMan kits (Applied Biosystems). Expression of GPR74 mRNA was normalized to the 18S internal control by use of the formula
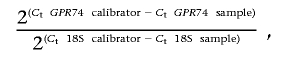 |
where calibrator is a random sample.
Preadipocyte differentiation was measured by quantifying the glycerol-3-phosphate dehydrogenase (GPDH) activity, as described elsewhere.22 Cell proteins were extracted, and GPDH activity and protein concentration were measured spectrophotometrically. Treatment with NPFF or siRNA against GPR74 did not influence preadipocyte differentiation, as evidenced by the GPDH measurements.
Lipolysis experiments on differentiated preadipocytes were performed after the NPFF treatment or siRNA transfection at day 12 or 13 of differentiation, as described elsewhere.22 Cells were incubated for 3 h with or without 10−4 mol/liter of noradrenaline alone or in combination with 10−4 mol/liter of the α2-adrenoceptor selective blocker yohimbine or 10−3 mol/liter of the phosphodiesterase-resistant cyclic AMP analogue, dibutyryl cyclic AMP. Glycerol concentration in the medium was measured and was related to the amount of cellular protein, because cell number cannot be measured in these conditions.
Genotyping
DNA was extracted and purified using a kit procedure (Qiagen). GPR74 genotype information for a region covering from ∼10,000 bp upstream of exon 1 to 10,000 bp downstream of exon 4 was downloaded from the International HapMap Project. Genotype data for the population of individuals of European ancestry were visualized using Haploview. SNPs for genotyping were selected on the basis of the following criteria: (1) SNPs defining haplotypes with frequency >5% and (2) one SNP with frequency >2% every 5,000 bp. In selecting between different SNPs, we prioritized SNPs with Golden-Gate–validated assays or with a high score according to Illumina,24 which indicates that the designed Illumina genotyping assays are likely to work.
Subjects in sample 1 and 3 were genotyped using Illumina.24 Samples 2 and 4 were genotyped using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (SEQUENOM) as described elsewhere.25 For both genotyping platforms, the overall genotype call rate was 96% and the accuracy was 99.99%, according to duplicate analysis of, on average, 2% of the total genotypes. Primers are available on request. Two independent scorers confirmed all genotypes. Hardy-Weinberg calculations were performed to ensure that each marker was in population equilibrium.
Sequencing
To identify promoter or coding SNPs carried by the ATAG haplotype, we sequenced all GPR74 exons, the exon-intron borders, and 1,730 bp 5′ of exon 1 in five subjects homozygous for ATAG and in three subjects who did not carry ATAG. We failed to sequence further 5′ because of a repeat region. Staden software was used for sequence assembly. All sequences were scored manually for the presence of SNPs.
Statistical Analysis
Haploview was employed to calculate Hardy-Weinberg P values and linkage disequilibrium, to infer haplotypes, and to test for allelic and haplotype association with obesity. Linkage disequilibrium was calculated as D′. For defining haploblocks, 95% confidence bounds on D′ were generated, and each comparison was classified as “strong LD,” “inconclusive,” or “strong recombination.” A block was created when 95% of informative (i.e., not inconclusive) comparisons showed “strong LD.” This method, by default, ignores markers with minor-allele frequency <0.05. An accelerated expectation-maximization algorithm similar to the partition/ligation method described by Qin et al.26 was used in Haploview to estimate haplotypes. This creates highly accurate population-frequency estimates of the phased haplotypes on the basis of the maximum likelihood as determined from the unphased input. In single-locus and multimarker haplotype-association tests, the χ2 and P value for the allele frequencies in cases versus controls were calculated. The haplotype analysis counts for the association analysis were obtained by summing the fractional likelihoods of each individual for each haplotype. Individuals with >50% missing genotypes were excluded from analysis. It is not possible in Haploview to visualize the probabilities of each individual’s estimated haplotypes. Therefore, SNPHAP, which uses a method similar to Haploview to infer haplotypes, was used to assign probabilities to each individual’s estimated haplotypes. Odds ratios (ORs) were calculated using the Finetti software (Hardy-Weinberg equilibrium Web site). We did not adjust for hypertension, type 2 diabetes, or dyslipidemia because, in obese subjects, these phenotypes are usually the consequence of obesity.
In samples 1 and 2, which were ascertained on the basis of obesity status, waist circumference was analyzed by Mann-Whitney U test (Statview). In sample 4, which was population-based and thus not ascertained on the basis of analyzed phenotypes, we applied an analysis of the quantitative trait waist or BMI by using analysis of covariance (ANCOVA) with age as covariate. HOMAIR, plasma lipid, plasma glucose, and serum insulin are dependent on adiposity and age. In samples 1 and 2, these phenotypes were analyzed by ANCOVA, with stratification for obesity status and with age as covariate. In sample 4, these phenotypes were analyzed by ANCOVA with age and BMI as covariates. To study in vivo lipolysis, which was already corrected for body fat, we employed ANCOVA with age as covariate. In vitro lipolysis phenotypes in the nonobese sample 3 were analyzed by ANCOVA with age and BMI as covariates. Sex has impact on the analyzed phenotypes; therefore, we repeated all ANCOVA analyses with stratification for sex. We report nonadjusted results of ANOVA because the different adjustments of P values described above did not, in any case, make an important change in the P values, which usually were identical with the P values from ANOVA. All reported P values are nominal, unless otherwise stated. A permutation test with 10,000 permutations was used to adjust significance in the initial analysis of association between the ATAG haplotype and obesity. For confirmation, when one hypothesis was tested, no correction for multiple comparisons was used. Bonferroni correction for the number of comparisons was applied when multiple phenotypes were analyzed—for example, in analysis of the adipocyte lipolysis data.
Results
The recruitment criteria for sample 1, which aimed to recruit subjects with extreme phenotypes and with high or no predisposition to obesity, resulted in obese subjects that were younger (mean age 44 years) than the lean subjects (mean age 52 years) (table 1). In addition, less stringent BMI recruitment criteria for sample 2 resulted in phenotypic differences between samples 1 and 2 (table 1). In sample 2, the mean age among obese subjects (44 years) was similar to that in sample 1, whereas the lean subjects were younger (mean age 39 years). The obese subjects in sample 1 had a mean BMI of 45, and, in sample 2, the obese subjects had a mean BMI of 38. In summary, sample 4 comprised 775 obese women and 793 obese men with BMI >30 (mean age 51 ± 9 years; BMI 34 ± 4) and 3,079 nonobese women and 3,144 nonobese men (mean age 48 ± 10 years; BMI 25 ± 3) (table 1). Thus, the subjects in the Danish sample 4 were older and less obese than those in Swedish samples 1 and 2.
We initially investigated sample 1, which had the most-rigorous selection criteria for a lean or obese phenotype, for SNPs covering the GPR74 gene selected from the International HapMap Project (fig. 2). Of the 25 SNPs tested in genotyping assays, 3 failed to show association (12%), which is close to the expected 10% failure rate, and 3 were nonpolymorphic (table 2). Of the remaining 19 GPR74 SNPs, 9 were nominally associated with obesity in sample 1—2 of these, rs9637554 and rs9291171, were more strongly associated, with P values of .0058 and .0004, respectively (table 3). rs6446796 displayed a low call rate and was therefore excluded from analysis of allelic association. Single SNPs associated with obesity with P values <.01, as well as SNPs that defined obesity-associated haplotypes, were genotyped in a second cohort of lean or obese Swedes, sample 2, which had less rigorous selection criteria for leanness (age >25 years and BMI <25) and obesity (BMI >30 at any age) than those of sample 1. In sample 2, no allelic difference between the lean and obese groups was observed (table 3). Since 80%–85% of subjects in samples 1 and 2 were women, no sex-specific analysis was performed.
Figure 2. .

GPR74 SNPs and haplotypes. Bold font indicates SNPs with a call rate >90% and a frequency >5% that were used for inferring the haplotypes. Allelic frequency of specific GPR74 haplotypes in obese case patients (n=611) and lean control subjects (n=402) in sample 1 analyzed with Haploview. Haplotype P = mean ± SD (lower value of range) of probabilities by which haplotypes have been assigned to individuals. P values were calculated by χ2 test for significance of the difference in haplotype frequency between cases and controls. An asterisk (*) indicates significant difference in ATAG haplotype frequency between cases and control s in joint analysis of samples 1 and 2; P=.0014.
Table 2. .
GPR74 SNPs
| SNP | Region | Positiona | Alleles | Allele Frequencyb (%) |
Call Rate (%) |
HWE P |
| rs6817845 | 5′ | 73259416 | … | NP | 97.7 | … |
| rs6856651 | 5′ | 73261492 | A→T | 29.0 | 100.0 | .34 |
| rs6446796 | Intron 1 | 73264126 | T→C | 30.2 | 76.8 | .021c |
| rs6824342 | Intron 1 | 73268217 | T→C | 30.0 | 100.0 | .39 |
| rs13150414 | Intron 1 | 73270871 | G→T | 30.2 | 99.7 | .47 |
| rs9637554 | Intron 1 | 73278192 | C→G | 1.6 | 94.7 | .42 |
| rs4694453 | Intron 1 | 73284045 | C→A | .9 | 100.0 | .19 |
| rs11938755 | Intron 1 | 73286432 | G→A | 1.7 | 98.5 | .51 |
| rs6446809 | Intron 1 | 73290529 | T→G | .1 | 100.0 | 1 |
| rs4353870 | Intron 1 | 73297930 | A→T | 1.7 | 99.9 | .5 |
| rs4694462 | Intron 1 | 73302773 | C→G | 1.7 | 100.0 | .5 |
| rs12642985 | Intron 1 | 73312934 | C→T | 30.2 | 98.8 | .38 |
| rs9993413 | Intron 1 | 73319113 | C→T | .2 | 99.9 | 1 |
| rs12510838 | Intron 1 | 73326573 | A→G | 17.3 | 99.5 | .78 |
| rs4129733 | Intron 1 | 73328055 | T→G | 35.7 | 98.0 | .42 |
| rs7662933 | Intron 1 | 73335910 | … | NP | 98.0 | … |
| rs13107347 | Intron 1 | 73339783 | … | … | .0 | … |
| rs4264803 | Intron 1 | 73343505 | … | … | .0 | … |
| rs9291171 | Intron 1 | 73346661 | A→G | 28.9 | 92.1 | .22 |
| rs11940192 | Intron 1 | 73356942 | … | … | .0 | … |
| rs7679840 | Intron 2 | 73362944 | C→T | .1 | 100.0 | 1 |
| rs12650900 | Intron 2 | 73367493 | … | NP | 98.0 | … |
| rs11940196 | Intron 2 | 73368604 | A→G | 38.7 | 97.6 | .27 |
| rs6826854 | Intron 3 | 73373464 | C→T | .1 | 99.9 | 1 |
| rs17775309 | 3′ | 73382993 | T→G | 32.4 | 87.9 | .13 |
Position on chromosome 4, genome build 35.
NP = nonpolymorphic.
HWE among controls.
Table 3. .
Association of GPR74 SNPs with Obesity in Samples 1 and 2
| No. of Chromosomes |
|||||||
| Cases |
Controls |
Allele Frequency |
|||||
| Sample and Marker |
Allele 1 | Allele 2 | Allele 1 | Allele 2 | Cases | Controls | P |
| 1: | |||||||
| rs6856651 | 365 | 813 | 206 | 592 | .31 | .26 | .013 |
| rs6824342 | 377 | 801 | 216 | 582 | .32 | .27 | .019 |
| rs13150414 | 378 | 794 | 217 | 581 | .32 | .27 | .016 |
| rs9637554 | 1,076 | 10 | 766 | 20 | .99 | .98 | .0058 |
| rs4694453 | 1,168 | 10 | 788 | 10 | .99 | .99 | .38 |
| rs11938755 | 1,140 | 14 | 772 | 20 | .99 | .98 | .03 |
| rs6446809 | 1,177 | 1 | 797 | 1 | .999 | .999 | .78 |
| rs4353870 | 1,162 | 14 | 778 | 20 | .99 | .98 | .027 |
| rs4694462 | 1,164 | 14 | 778 | 20 | .99 | .98 | .027 |
| rs12642985 | 372 | 782 | 217 | 581 | .32 | .27 | .017 |
| rs9993413 | 1,174 | 2 | 796 | 2 | .998 | .997 | .7 |
| rs12510838 | 216 | 956 | 124 | 670 | .18 | .16 | .11 |
| rs4129733 | 737 | 403 | 508 | 288 | .65 | .64 | .7076 |
| rs9291171 | 336 | 700 | 190 | 594 | .32 | .24 | .0004 |
| rs7679840 | 2 | 1176 | 0 | 798 | .002 | .000 | .24 |
| rs11940196 | 714 | 424 | 467 | 323 | .63 | .59 | .11 |
| rs6826854 | 1,175 | 1 | 776 | 0 | .00 | .00 | .41 |
| rs17775309 | 663 | 299 | 510 | 264 | .69 | .66 | .18 |
| 2: | |||||||
| rs6856651 | 456 | 1138 | 354 | 862 | .29 | .29 | .77 |
| rs9637554 | 1,581 | 21 | 1,202 | 20 | .99 | .98 | .47 |
| rs12510838 | 293 | 1315 | 228 | 992 | .18 | .19 | .75 |
| rs4129733 | 1,035 | 575 | 825 | 395 | .64 | .68 | .064 |
| rs9291171 | 460 | 1148 | 351 | 863 | .29 | .29 | .86 |
| rs11940196 | 1,014 | 582 | 795 | 423 | .64 | .65 | .34 |
In general, there was strong LD between SNPs in the GPR74 gene (fig. 3). The nine GPR74 SNPs with frequency >5% were used to infer haplotypes. Because haplotypes inferred from unrelated subjects have uncertainty, we also present mean ± SD values (and the lower value of the range) for the probability by which haplotypes have been assigned to individual chromosomes in sample 1 (fig. 2). In sample 1, three haplotypes differed in frequency between lean and obese groups, with P values .0076–.022 (fig. 2). Two of these haplotypes shared a common ATAG motif at SNPs rs12510838, rs4129733, rs9291171, and rs11940196. Haplotypes with the ATAG motif were inferred with high confidence, with haplotype probability mean 1.00 (lower value of range 0.95) (fig. 2). In sample 1, the ATAG haplotype was associated with obesity, with nominal P value .0014, and the OR for obesity was 0.48 (95% CI 0.30–0.76) (table 4). The permutated P value was .0070. The association between the ATAG haplotype and obesity was confirmed in sample 2, with nominal P value .014 and OR 0.62 (95% CI 0.42–0.90) (table 4). Pooling samples 1 and 2 gave a P value of .000089. The ORs for obesity of 0.48 and 0.62 for samples 1 and 2, respectively, suggest that ATAG protects against obesity. ATAG was not associated with obesity in the initial analysis of the Danish sample 4. However, when sample 4 was enriched for subjects at either end of the BMI distribution, including only elderly lean subjects (BMI <25; age >45 years) or severely obese subjects (BMI >35) in the analysis, ATAG was associated with protection against obesity in men, with nominal P value .036 and OR 0.36 (95% CI 0.14–0.94). The other haplotype associated with obesity in sample 1, defined by alleles ATGA at SNPs rs12510838, rs4129733, rs9291171, and rs11940196, displayed no association with obesity in sample 2 and weak opposite association in sample 4 and was not further investigated.
Figure 3. .

Linkage disequilibrium (D′) between GPR74 SNPs in sample 1, according to Haploview analysis
Table 4. .
Association of GPR74 Haplotypes with Obesity
| Cases |
Controls |
Haplotype Frequency |
||||||
| Sample and Haplotypea |
No. of ATAG or ATGA Chromosomes | No. of Non-ATAG or Non-ATGA Chromosomes | No. of ATAG or ATGA Chromosomes | No. of Non-ATAG or Non-ATGA Chromosomes | Cases | Controls | P | OR (95% CI) |
| 1: | ||||||||
| ATGA | 152 | 1,024 | 70 | 728 | .13 | .09 | .0042 | |
| ATAG | 32 | 1,144 | 44 | 754 | .027 | .055 | .0014 | .48 (.30–.76) |
| 2: | ||||||||
| ATGA | 170 | 1,444 | 125 | 1,093 | .1 | .1 | .83 | |
| ATAG | 51 | 1,563 | 61 | 1,157 | .032 | .05 | .014 | .62 (.42–.90) |
| 1 and 2: | ||||||||
| ATGA | 320 | 2,466 | 195 | 1,819 | .11 | .097 | .043 | |
| ATAG | 83 | 2,703 | 105 | 1,909 | .03 | .052 | .000089 | .55 (.29–.87) |
| 4: | ||||||||
| ATGA | 296 | 2,794 | 1,332 | 10,936 | .096 | .109 | .04 | |
| ATAG | 108 | 2,982 | 460 | 11,809 | .035 | .037 | .51 | .93 (.87–1.33) |
Haplotype for rs12510838, rs4129733, rs9291171, and rs11940196.
The influence of the ATAG haplotype on body composition and metabolic phenotypes was subsequently examined. A joint analysis of men in samples 1 and 2 was performed because of the small number of men in these samples. In samples 1 and 2, both men and women carrying ATAG had smaller waists than did those who carried two other haplotypes (fig. 4A and 4B). In sample 4, men but not women carrying ATAG had significantly smaller waists than did noncarriers (fig. 4). There was no effect of ATAG on plasma glucose, serum insulin, HOMAIR, or plasma lipids among men or women in sample 1, 2, or 4 (data not shown).
Figure 4. .

Association between carriage of GPR74 ATAG haplotype and waist circumference in men (A) and women (B). The blackened bar represents subjects carrying the ATAG haplotype; the unblackened bar represents subjects not carrying ATAG. The frequencies of haplotypes were as follows: in men in samples 1 and 2, ATAG = 19 and non-ATAG = 327; in men in sample 4, ATAG = 276 and non-ATAG = 3,615; in women in sample 1, ATAG = 55 and non-ATAG = 667; in women in sample 2, ATAG = 78 and non-ATAG = 995; in women in sample 4, ATAG = 262 and non-ATAG = 3,550. Samples 1 and 2 were analyzed by the Mann-Whitney U test, and sample 4 was analyzed using ANCOVA, with age as covariate.
The relationship between the ATAG haplotype and lipolysis was investigated next (fig. 5). Plasma levels of glycerol and free fatty acids, adjusted for total body fat, were significantly elevated among subjects carrying ATAG (fig. 5A and 5B). With regard to in vitro data on human fat cells in sample 4, the lipolytic sensitivity (pD2) of noradrenaline was increased 10-fold among nonobese subjects carrying ATAG, compared with nonobese subjects who were noncarriers (fig. 5C). The increased noradrenaline sensitivity could be attributed to a 10-fold increase in β2-adrenoceptor sensitivity (pD2 for terbutaline) (fig. 5C). These results remained significant after Bonferroni correction for analysis of four adrenergic drugs. There was no effect of ATAG on β1-adrenoceptor sensitivity (pD2 for dobutamine) or on α2-adrenoceptor sensitivity (pD2 for clonidine) (fig. 5C). Likewise, the spontaneous or basal lipolytic activity and the maximum lipolytic effect of lipolysis stimulation were not altered in ATAG-carrying subjects (results not shown).
Figure 5. .

A, Association between carriage of the ATAG haplotype and in vivo lipolysis measured as plasma glycerol corrected for body fat. The blackened bar represents the 124 subjects carrying ATAG; the unblackened bar represents the 1,771 subjects not carrying ATAG. B, Association between carriage of the ATAG haplotype and in vivo lipolysis measured as plasma free fatty acids corrected for body fat. The blackened bar represents the 100 subjects carrying ATAG; the unblackened bar represents the 1,494 subjects not carrying ATAG. C, Association between carriage of the ATAG haplotype and adipocyte lipolytic sensitivity (pD2) of different adrenergic agonists. The blackened bar represents the 14 subjects carrying ATAG; the unblackened bar represents the 136 subjects not carrying ATAG. Analyzed subjects came from samples 1 and 2. Groups were compared by ANCOVA, with age as covariate in panel A and with age and BMI as covariates in panel B.
The role of GPR74 in regulation of human adipocyte lipolysis was investigated using a natural ligand with high affinity for GPR74, NPFF (fig. 6). Short- or long-term exposure of human fat cells in primary culture to NPFF, which was added at a maximum effective concentration for adipocytes, showed that the neuropeptide markedly counteracted the lipolytic effect of noradrenaline alone and together with a high concentration of yohimbine, which blocks the α2-receptor–mediated effects of noradrenaline (fig. 6A). However, the lipolytic effect of a cyclic AMP analogue, which stimulates lipolysis distal to β-adrenoceptor signaling, was not influenced by NPFF treatment (fig. 6A). Treatment of fat cells with siRNA oligonucleotides against GPR74 caused a specific decrease of GPR74 mRNA levels by ∼70% (fig. 6B) and a specific 2.5-fold increase in noradrenaline induced lipolysis (fig. 6C); there was no effect of a random (scrambled) oligonucleotide.
Figure 6. .

A, Effect on human preadipocytes differentiated into adipocytes of the GPR74 agonist NPFF on lipolysis measured as plasma glycerol (five experiments). The white bar represents controls, the blackened bar represents NPFF for 3 h, and the striped bar represents NPFF for 48 h. B, Effect of siRNA against GPR74 on expression of GPR74 mRNA (four experiments). C, Effect of siRNA against GPR74 on lipolysis. The white bar represents no oligo, the gray shaded bar represents scrambled oligo, and the blackened bar represents GPR74 siRNA. Groups were compared by ANOVA.
No SNP was detected for the ATAG haplotype in the sequencing of all GPR74 exons, exon-intron borders, and 1,730 bp upstream of exon 1 in five subjects homozygous for ATAG and in three subjects not carrying ATAG. It was sufficient to investigate eight subjects because we specifically searched for polymorphism linked to the ATAG haplotype.
Discussion
In this study, the genetic variance in GPR74 has been investigated, to our knowledge, for the first time. Our results, which were generated with the use of three large cohorts, suggest that GPR74 is in control of BMI. In two Swedish samples carefully selected for leanness and obesity, we found a common haplotype, ATAG, with allele frequency 4%, that displayed a strong negative association with obesity. We also found a strong association between ATAG and waist circumference in the two Swedish cohorts. Both men and women carriers of ATAG had smaller waists than did noncarriers. The allele frequency of ATAG was similar in the population-based Danish sample 4. In sample 4, ATAG was associated with protection against obesity in analysis of lean and severely obese men and with waist circumference among men.
The strength of a true causal association between obesity and single markers or haplotypes can depend on phenotypic distributions for age and BMI. The different ascertainment schemes used for samples 1 and 2, which resulted in different age and BMI distributions, could explain the inability to confirm single-marker association in sample 2. The phenotypic differences between samples 1 and 2 may also explain why the association between ATAG and obesity status was weaker in sample 2. The somewhat different results obtained in analysis of ATAG in sample 4, compared with in samples 1 and 2, may be because the Danish cohort was population based, the obese subset contained no young subjects with obesity, and only 8% in the sample had severe obesity.
The association of ATAG with obesity phenotypes could not be explained by any single SNP. There are previous reports from the analyses of complex traits that several mutations on the same chromosome—that is, a haplotype—within a gene can interact and have a large effect on the observed phenotype that cannot be explained by the individual mutations. Examples include β2-adrenergic receptor gene haplotypes and bronchodilator response, ELAC2 and risk of prostate cancer, and the lipoprotein lipase gene and cardiovascular disease.27–29
Our results discussed so far support the notion that the GPR74 haplotype ATAG protects against obesity. This haplotype is associated with leanness (BMI <25) and a smaller waist. This latter result suggests that ATAG protects against central-fat accumulation, which is less common among women. The observation that Danish women, in contrast to Swedish women, were not protected by ATAG could be more apparent than real, because the Swedish women had, on average, much larger waists than those of the Danish women (fig. 4B).
What is the mechanism of the protective effect of ATAG? Data from samples 1 and 2 suggest that a high ability to mobilize lipids from adipose tissue is an important contributing factor. Plasma levels of the end products of lipolysis—glycerol and free fatty acids—adjusted for total body fat, were significantly elevated among subjects carrying ATAG. This finding may indicate that in vivo lipolysis activity is increased among subjects with ATAG. Our in vitro data for human fat cells from healthy nonobese subjects (in sample 4) provide evidence that ATAG improves the ability of catecholamines to stimulate lipolysis. The lipolytic sensitivity of noradrenaline was 10-fold increased among nonobese subjects carrying ATAG. In biological terms, this means that a half-maximum stimulatory effect of noradrenaline on lipolysis was obtained at ∼1 nmol/liter in the subjects carrying ATAG and at ∼10 nmol/liter in other subjects; these concentrations are within the normal physiological range. The increased noradrenaline sensitivity could be attributed to a 10-fold increase in β2-adrenoceptor sensitivity. There was no effect of ATAG on β1-adrenoceptor sensitivity or on α2-adrenoceptor sensitivity.
For the moment, molecular studies of the effect of the ATAG haplotype on GPR74 function are hampered by the fact that the haplotype contains no SNPs in the putative promoter, exons, or exon-intron borders. The lack of exon SNPs makes allele-specific mRNA quantification impossible. Intronic SNPs may also affect gene-transcript stability and mRNA levels. Unfortunately, we did not have adipose RNA from the number of subjects required to relate carriage of the ATAG haplotype to mRNA levels. Nevertheless, we provide, for the first time, evidence of a role for GPR74 in regulation of adipocyte lipolysis by use of NPFF, which is a natural ligand with high affinity for GPR74.4 Short- or long-term exposure of human fat cells in primary culture to NPFF markedly inhibited the lipolytic effect of noradrenaline. Additional data with blockade of α2-receptor–mediated effects of noradrenaline and with a cyclic AMP analogue, which stimulates lipolysis distal to the β-adrenoceptor, strongly suggest that GPR74 has inhibitory effects on catecholamine-induced lipolysis by interference with β-adrenoceptor signaling in fat cells. A more definitive proof of an inhibitory role of GPR74 on adipocyte lipolysis was obtained by siRNA experiments with primary culture of human fat cells. A decrease of GPR74 mRNA was accompanied by an increase in catecholamine-induced lipolysis.
It is an attractive hypothesis that the ATAG haplotype protects against fat accumulation by ameliorating the intrinsic inhibitory effect of GPR74 on lipolysis (seen in the siRNA experiments). Interactions between GPR74 and β2-adrenoceptors in adipocytes may be involved in the protective effect of ATAG (seen in the NPFF experiments and findings with the haplotype and pD2). Interestingly, decreased adipocyte β2-adrenoceptor function is demonstrated in human obesity,20,21 and polymorphisms in the β2-adrenoceptor gene is related to adipocyte lipolysis and body fat.30 It is quite possible, however, that the ATAG haplotype has other nonadipocyte effects on body weight that are mediated by NPFF, according to animal studies (mentioned in the introduction). Thus, effects related to cardiac regulation, stress response, reward, or food intake may influence body weight regulation among subjects with ATAG independent of the putative fat-cell effect.6–9
Obesity is accompanied by metabolic and hormonal changes. In our cohorts, ATAG was not associated with plasma levels of lipids and glucose, serum insulin, or HOMAIR as a measure of insulin resistance, which implies that ATAG has no impact on these phenotypes. However, we cannot exclude that ATAG would have a role in a sample selected for more-extreme insulin-resistance phenotypes.
In conclusion, this study suggests that a common GPR74 haplotype regulates body weight. Carriers of the ATAG haplotype, ∼8% of the population in Scandinavian countries, are protected against excessive fat accumulation. This may be the result of an increased ability to mobilize lipids from adipose tissue because of a weakened inhibitory effect of GPR74 on β2-adrenoceptor signaling in fat cells. Because of the relationship between GPR74 polymorphisms, lipolysis, and body fat, it is tempting to suggest that GPR74 blockers might be useful in the treatment of obesity.
Acknowledgments
We thank Britt-Marie Leijonhufvud, Katarina Hertel, Elisabeth Dungner, Eva Sjölin, Kerstin Wåhlén, and Gaby Åström for excellent technical assistance. The study was supported by grants from AFA Life Insurance Health Foundation, Swedish Research Council, Swedish Heart and Lung Foundation, Novo Nordic Foundation, Swedish Diabetes Association, Swedish Society of Medicine, King Gustav V and Queen Victoria Foundation, The Danish Medical Research Council, The Danish Diabetes Association, the European Union (EUGENE2 LSHM-CT-2004-512013 and HEPADIP LSHM-CT-2005-018734), Åke Wiberg Foundation, Jeanssons Foundation, and Tore Nilsson Foundation.
Web Resources
The URLs for data presented herein are as follows:
- Haploview, http://www.broad.mit.edu/mpg/haploview/
- Hardy-Weinberg equilibrium, http://ihg.gsf.de/cgi-bin/hw/hwa1.pl (for Finetti software)
- International HapMap Project, http://www.hapmap.org/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for GPR74) [PubMed]
- SNPHAP, http://www-gene.cimr.cam.ac.uk/clayton/software/snphap.txt
References
- 1.Bulik CM, Allison DB (2001) The genetic epidemiology of thinness. Obes Rev 2:107–115 10.1046/j.1467-789x.2001.00030.x [DOI] [PubMed] [Google Scholar]
- 2.Parker RM, Copeland NG, Eyre HJ, Liu M, Gilbert DJ, Crawford J, Couzens M, Sutherland GR, Jenkins NA, Herzog H (2000) Molecular cloning and characterisation of GPR74 a novel G-protein coupled receptor closest related to the Y-receptor family. Brain Res Mol Brain Res 77:199–208 10.1016/S0169-328X(00)00052-8 [DOI] [PubMed] [Google Scholar]
- 3.Elshourbagy NA, Ames RS, Fitzgerald LR, Foley JJ, Chambers JK, Szekeres PG, Evans NA, Schmidt DB, Buckley PT, Dytko GM, et al (2000) Receptor for the pain modulatory neuropeptides FF and AF is an orphan G protein-coupled receptor. J Biol Chem 275:25965–25971 10.1074/jbc.M004515200 [DOI] [PubMed] [Google Scholar]
- 4.Vyas N, Mollereau C, Cheve G, McCurdy CR (2006) Structure-activity relationships of neuropeptide FF and related peptidic and non-peptidic derivatives. Peptides 27:990–996 10.1016/j.peptides.2005.07.024 [DOI] [PubMed] [Google Scholar]
- 5.Mollereau C, Roumy M, Zajac JM (2005) Opioid-modulating peptides: mechanisms of action. Curr Top Med Chem 5:341–355 10.2174/1568026053544515 [DOI] [PubMed] [Google Scholar]
- 6.Jhamandas JH, Harris KH, Petrov T, Yang HY, Jhamandas KH (1998) Activation of neuropeptide FF neurons in the brainstem nucleus tractus solitarius following cardiovascular challenge and opiate withdrawal. J Comp Neurol 402:210–221 [DOI] [PubMed] [Google Scholar]
- 7.Cador M, Marco N, Stinus L, Simonnet G (2002) Interaction between neuropeptide FF and opioids in the ventral tegmental area in the behavioral response to novelty. Neuroscience 110:309–318 10.1016/S0306-4522(01)00587-5 [DOI] [PubMed] [Google Scholar]
- 8.Huang EY, Li JY, Wong CH, Tan PP, Chen JC (2002) Dansyl-PQRamide, a possible neuropeptide FF receptor antagonist, induces conditioned place preference. Peptides 23:489–496 10.1016/S0196-9781(01)00632-5 [DOI] [PubMed] [Google Scholar]
- 9.Nicklous DM, Simansky KJ (2003) Neuropeptide FF exerts pro- and anti-opioid actions in the parabrachial nucleus to modulate food intake. Am J Physiol Regul Integr Comp Physiol 285:R1046–R1054 [DOI] [PubMed] [Google Scholar]
- 10.Lefrere I, De Coppet P, Camelin JC, Le Lay S, Mercier N, Elshourbagy N, Bril A, Berrebi-Bertrand I, Feve B, Krief S (2002) Neuropeptide AF and FF modulation of adipocyte metabolism: primary insights from functional genomics and effects on β-adrenergic responsiveness. J Biol Chem 277:39169–39178 10.1074/jbc.M205084200 [DOI] [PubMed] [Google Scholar]
- 11.Arner P (2005) Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab 19:471–482 10.1016/j.beem.2005.07.004 [DOI] [PubMed] [Google Scholar]
- 12.Bell CG, Walley AJ, Froguel P (2005) The genetics of human obesity. Nat Rev Genet 6:221–234 10.1038/nrg1556 [DOI] [PubMed] [Google Scholar]
- 13.Kolaczynski JW, Morales LM, Moore JH Jr, Considine RV, Pietrzkowski Z, Noto PF, Colberg J, Caro JF (1994) A new technique for biopsy of human abdominal fat under local anaesthesia with Lidocaine. Int J Obes Relat Metab Disord 18:161–166 [PubMed] [Google Scholar]
- 14.Jorgensen T, Borch-Johnsen K, Thomsen TF, Ibsen H, Glumer C, Pisinger C (2003) A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99. Eur J Cardiovasc Prev Rehabil 10:377–386 10.1097/01.hjr.0000096541.30533.82 [DOI] [PubMed] [Google Scholar]
- 15.Andersen G, Wegner L, Yanagisawa K, Rose CS, Lin J, Glumer C, Drivsholm T, Borch-Johnsen K, Jorgensen T, Hansen T, et al (2005) Evidence of an association between genetic variation of the coactivator PGC-1β and obesity. J Med Genet 42:402–407 10.1136/jmg.2004.026278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter D, Hu FB, et al (2006) A common genetic variant is associated with adult and childhood obesity. Science 312:279–283 10.1126/science.1124779 [DOI] [PubMed] [Google Scholar]
- 17.Masud S, Ye S (2003) Effect of the peroxisome proliferator activated receptor-γ gene Pro12Ala variant on body mass index: a meta-analysis. J Med Genet 40:773–780 10.1136/jmg.40.10.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonora E, Targher G, Alberiche M, Bonadonna RC, Saggiani F, Zenere MB, Monauni T, Muggeo M (2000) Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 23:57–63 10.2337/diacare.23.1.57 [DOI] [PubMed] [Google Scholar]
- 19.Gallagher D, Visser M, Sepulveda D, Pierson RN, Harris T, Heymsfield SB (1996) How useful is body mass index for comparison of body fatness across age, sex, and ethnic groups? Am J Epidemiol 143:228–239 [DOI] [PubMed] [Google Scholar]
- 20.Reynisdottir S, Ellerfeldt K, Wahrenberg H, Lithell H, Arner P (1994) Multiple lipolysis defects in the insulin resistance (metabolic) syndrome. J Clin Invest 93:2590–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynisdottir S, Wahrenberg H, Carlstrom K, Rossner S, Arner P (1994) Catecholamine resistance in fat cells of women with upper-body obesity due to decreased expression of β2-adrenoceptors. Diabetologia 37:428–435 [DOI] [PubMed] [Google Scholar]
- 22.Dicker A, Ryden M, Naslund E, Muehlen IE, Wiren M, Lafontan M, Arner P (2004) Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia 47:420–428 10.1007/s00125-003-1324-0 [DOI] [PubMed] [Google Scholar]
- 23.Nordstrom EA, Ryden M, Backlund EC, Dahlman I, Kaaman M, Blomqvist L, Cannon B, Nedergaard J, Arner P (2005) A human-specific role of cell death-inducing DFFA (DNA fragmentation factor-α)-like effector A (CIDEA) in adipocyte lipolysis and obesity. Diabetes 54:1726–1734 10.2337/diabetes.54.6.1726 [DOI] [PubMed] [Google Scholar]
- 24.Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, et al (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78 10.1101/sqb.2003.68.69 [DOI] [PubMed] [Google Scholar]
- 25.Dahlman I, Eriksson P, Kaaman M, Jiao H, Lindgren CM, Kere J, Arner P (2004) α2-Heremans-Schmid glycoprotein gene polymorphisms are associated with adipocyte insulin action. Diabetologia 47:1974–1979 10.1007/s00125-004-1556-7 [DOI] [PubMed] [Google Scholar]
- 26.Qin ZS, Niu T, Liu JS (2002) Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet 71:1242–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–10488 10.1073/pnas.97.19.10483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, et al (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27:172–180 10.1038/84808 [DOI] [PubMed] [Google Scholar]
- 29.Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, et al (1998) Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Large V, Hellstrom L, Reynisdottir S, Lonnqvist F, Eriksson P, Lannfelt L, Arner P (1997) Human beta-2 adrenoceptor gene polymorphisms are highly frequent in obesity and associate with altered adipocyte beta-2 adrenoceptor function. J Clin Invest 100:3005–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]