

Abstract
A pathological hallmark of Alzheimer’s disease is the senile plaque, composed of β-amyloid fibrils, microglia, astrocytes, and dystrophic neurites. We reported previously that class A scavenger receptors mediate adhesion of microglia and macrophages to β-amyloid fibrils and oxidized low-density lipoprotein (oxLDL)-coated surfaces. We also showed that CD36, a class B scavenger receptor and an oxLDL receptor, promotes H2O2 secretion by macrophages adherent to oxLDL-coated surfaces. Whether CD36 is expressed on microglia, and whether it plays a role in secretion of H2O2 by microglia interacting with fibrillar β-amyloid is not known. Using fluorescence-activated cell sorting analysis and immunohistochemistry, we found that CD36 is expressed on human fetal microglia, and N9-immortalized mouse microglia. We also found that CD36 is expressed on microglia and on vascular endothelial cells in the brains of Alzheimer’s disease patients. Bowes human melanoma cells, which normally do not express CD36, gained the ability to specifically bind to surfaces coated with fibrillar β-amyloid when transfected with a cDNA encoding human CD36, suggesting that CD36 is a receptor for fibrillar β-amyloid. Furthermore, two different monoclonal antibodies to CD36 inhibited H2O2 production by N9 microglia and human macrophages adherent to fibrillar β-amyloid by ∼50%. Our data identify a role for CD36 in fibrillar β-amyloid-induced H2O2 production by microglia, and imply that CD36 can mediate binding to fibrillar β-amyloid. We propose that similar to their role in the interaction of macrophages with oxLDL, class A scavenger receptors and CD36 play complimentary roles in the interactions of microglia with fibrillar β-amyloid.
Alzheimer’s disease (AD) is the most common neurodegenerative disease of adults. 1 It is characterized by the extracellular deposition of insoluble fibrillar β-amyloid protein (fAβ) in the brain parenchyma. 2 Intraparenchymal deposits of fAβ are composed of Aβ peptides 40 to 42 amino acids in length, surrounded by activated microglia, astrocytes, and dystrophic neurites, 1,2 all of which constitute the senile plaque. Inflammation, initiated by cellular reactions to these intraparenchymal deposits of fAβ, is thought to play an important role in the pathogenesis of AD. 3,4 Microglia, the brain’s mononuclear phagocytes, are thought to be the principal cells that mediate this inflammation. 3-5
The interaction of neonatal microglia with fAβ in vitro stimulates these cells to produce proinflammatory and potentially neurotoxic substances such as nitric oxide, tumor necrosis factor-α, 5 and reactive oxygen species (ROS). 6,7 Removal of microglia from cultures containing mixed brain cells and fAβ almost totally eliminates the toxic effects of fAβ on primary neurons, 8 suggesting that microglia, and/or substances they produce, mediate the neurotoxic effects of fAβ.
Microglia express class A scavenger receptors (SR-A). 9 In neonatal microglia these receptors promote endocytosis of fAβ in suspension, 10 and adhesion of microglia to fAβ-containing surfaces. 6 SR-A expression is enhanced in microglia in brains of AD patients compared to brains of individuals of similar age who do not have AD, 11,12 and in the brains of transgenic mice expressing a mutated form of the human amyloid precursor protein (APP23), which develop AD-like pathology. 13 It is not known whether microglial expression of other scavenger receptors is affected in AD.
Like fAβ, oxidized low-density lipoprotein (oxLDL) is a ligand for SR-A. OxLDL is also a ligand for CD36. 14 We have shown that SR-A and CD36 play complementary roles in mediating adhesion of human monocyte-derived macrophages to surfaces coated with oxLDL and in secretion of ROS. 15 SR-A participates in adhesion of macrophages to oxLDL-coated surfaces, whereas CD36 signals ROS production but is not required for adhesion to these surfaces. The similarities between interactions of microglia with fAβ and of macrophages with oxLDL led us to test expression of CD36 on microglia in vitro and in brains of AD patients and to determine whether it plays a role in fAβ-induced secretion of ROS by microglia. We report here that CD36 is expressed on microglia in vitro and in AD brains and that it cooperates with adhesion-promoting receptors in signaling secretion of ROS by microglia and macrophages in vitro.
Materials and Methods
Antibodies
We used several anti-CD36 monoclonal antibodies (mAbs) obtained as follows: SMφ (Sigma Chemical Co., St. Louis, MO), NL07 (Pharmingen, San Diego, CA), and FA6-152 (Biodesign International, Kennebunk, ME). Monoclonal antibody anti-CD11b (M1/70) was from Caltag (Burlingame, CA). Rabbit anti-human glial fibrillary acidic protein (GFAP) was a generous gift from Dr. James Goldman, Columbia University, New York, NY. 16 Biotin-SP-conjugated Affinipure F(ab)2 fragments of rabbit anti-mouse IgG, donkey anti-mouse IgG, and goat anti-human IgG were from Jackson ImmunoResearch (West Grove, PA). Control rat IgG2b was from Zymed (San Francisco, CA), control rabbit anti-serum and monoclonal mouse IgG1 were from Sigma Chemical Co. Control antibody, MOPC 104E was from Organon Teknika Corp. (Cappel Research Products, Durham, NC). Fluorescein isothiocyanate-labeled goat anti-mouse IgG for fluorescence-activated cell sorting (FACS) analysis was from BD Pharmingen (San Diego, CA).
Fluorescent Dyes and Reagents
Streptavidin-Alexa 568, Amplex Red hydrogen peroxide assay kit, and CyQuant cell proliferation assay kit were from Molecular Probes Inc. (Eugene, OR). Tyramide signal amplification kit (TSA) for immunohistochemistry was from New England Nuclear (Boston, MA). Amyloid-β peptide 25-35 was from Sigma Chemical Co. or from Bachem Bioscience Inc. (King of Prussia, PA). Amyloid-β peptides 1-42, 42-1, and 35-25 (synthetic peptides with the reverse sequences of Aβ 1-42 and 25-35, respectively) were from Bachem Bioscience Inc. or American Peptide Company (Sunnyvale, CA). Thrombospondin-1 (TSP-1) and phorbol-12-myristate acetate were from Sigma Chemical Co..
Cells and Transfection
The murine microglial cell line N9, was a generous gift from Dr. P. Ricciardi-Castagnoli (University of Milano, Bicocca, Italy) 17 and was cultured as previously described 13 in RPMI 1640 medium (Life Technologies, Inc., Long Island, NY) supplemented with 10% heat-inactivated fetal bovine serum and penicillin (100 U/ml) and streptomycin (100 μg/ml). Microglia were isolated from human fetal tissue provided by the Fetal Tissue Bank at Albert Einstein College of Medicine as described. 18 The cells were maintained in Dulbecco’s modified Eagle’s medium with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and 25 mmol/L Hepes. Human monocyte-derived macrophages were prepared as described. 19 Bowes human melanoma cells were obtained from American Type Culture Collection (Rockville, MD), and were maintained in Dulbecco’s modified Eagle’s medium and 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). The mammalian expression vector for human CD36 (CDM8-CD36) was a generous gift from Dr. Brian Seed (Massachusetts General Hospital, Boston, MA). 20 A stable Bowes melanoma cell line expressing human CD36 was generated by co-transfecting CDM8-CD36 with vector pcDNA-neo (Invitrogen, Carlsbad, CA) using the calcium phosphate co-precipitation method and selection in G418 sulfate (Invitrogen) as described previously for SR-A. 19 Mock-transfected cells were generated by co-transfecting CDM8 (not containing CD36 cDNA) and pcDNA-neo in Bowes cells as described above.
Preparation of Fibrillar β-Amyloid
Aβ 25-35 and Aβ 35-25 were dissolved in phosphate-buffered saline (PBS) at 1 mg/ml and incubated at 37°C for 1 day. 6 Aβ 1-42 and 42-1 were dissolved in either double-distilled H2O (ddH2O) alone at 1 mg/ml, or ddH2O followed by 10× PBS to a final concentration of 1 mg/ml and incubated at 37°C for 3 to 4 days as described. 6 Fibril formation was confirmed by several methods (see Results and Figure 1 ▶ ). Fibrils formed by Aβ 1-42 were visualized by transmission electron microscopy as described. 21 Briefly, Formvar-coated nickel grids were incubated on drops of Aβ 1-42 (1 mg/ml) in PBS for 1 minute and then on drops of 2% phosphotungstic acid for 1 minute. The samples were photographed in a Philips CM 10 transmission electron microscope at 80kV.
Figure 1.

Characterization of Aβ peptides. A and B: Solutions containing Aβ 42-1 and 1-42 were prepared as described in Materials and Methods and used to coat multispot slides. A: Spots coated with Aβ 42-1 and incubated with Thioflavin S for 1 minute remained unstained implying no fibril formation. B: Spots coated with Aβ 1-42 and incubated with Thioflavin S (Sigma) for 1 minute stained intensely. C: A transmission electron micrograph of negatively stained Aβ 1-42 fibrils (original magnification, ×65,000). D: Nonreducing SDS-PAGE of fAβ 1-42 and 42-1 peptides stained with Coomassie blue (Sigma). The lane containing fAβ 1-42 peptides shows two bands, one at ≅4.2 kd and one at ≅21 kd, whereas the lane containing 42-1 contains a single band at ≅4.2 kd.
Measurement of ROS and H2O2
Because of limitations in availability of primary human microglia, we used nitroblue tetrazolium (NBT) reduction 22 to measure ROS production by these cells. Fifty μl of RPMI containing 25 × 10 3 microglia and 1 mg/ml NBT (Molecular Probes) and 1 mg/ml bovine serum albumin (BSA) was added to each spot of Multispot slides (Shandon-Lipshaw, Philadelphia, PA) coated with the indicated amount and type of Aβ peptide and incubated for 30 minutes at 37°C. The medium on each spot was aspirated and the slides were washed three times in methanol and air-dried. (At this stage, cells can be visualized by light microscopy and the slides can be stored for additional analysis without risk of quenching or loss of signal.) To extract the reduced and precipitated formazan, 30 μl of 2 mol/L KOH was added to each spot followed by 35 μl of dimethyl sulfoxide. The entire KOH-dimethyl sulfoxide mixture was transferred to a 96-well plate and the optical density of each sample was read in a multiwell plate reader at 650 nm.
H2O2 secretion by macrophages was assayed using 10-acetyl-3,7-dihydroxy-phenoxazine (Amplex Red) and horseradish peroxidase according to the manufacturer’s protocol (Molecular Probes), using a Cytofluor II fluorescence plate reader at excitation 530 nm and emission 590 nm. Where indicated, cells were incubated with 20 μg/ml of anti-CD36-mAb (SMφ or NLO7), control antibody (MOPC 104E), or 5 μg/ml of TSP-1 in KRBG-A for 30 minutes at room temperature before plating.
Immunofluorescence
Human fetal microglia or N9 cells were plated on coverslips and allowed to adhere at 37°C for 0.5 to 12 hours. The coverslips were then rinsed in Hepes-buffered saline (HBS) (125 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L KH2PO4, 10 mmol/L NaHCO3, 20 mmol/L Hepes, 5 mmol/L glucose, 1 mmol/L CaCl2, 1 mmol/L MgCl2), incubated in HBS containing 1% BSA for 30 minutes at 4°C to block nonspecific binding of antibodies. The coverslips were further incubated without fixation at 4°C for 30 minutes with HBS containing 1% BSA and 8 μg/ml mouse monoclonal anti-CD36 antibody FA6-152, or 8 μg/ml of MOPC-31c, an IgG1 isotype-matched control. The cells were washed with HBS on ice, incubated with a 1:200 dilution of Biotin-SP-conjugated Affinipure F(ab)2 fragments of rabbit anti-mouse IgG for 30 minutes, fixed in 3.7% formaldehyde for 10 minutes on ice, washed again with HBS, and incubated at room temperature for 30 minutes with a 1:1000 dilution of Streptavidin-Alexa 568 in HBS. Alexa 568 staining was imaged using a Zeiss LSM 410 confocal microscope at the confocal imaging core facility, Department of Anatomy and Cell Biology and the Herbert Irving Comprehensive Cancer Center at Columbia University.
FACS Analysis
N9 microglia were suspended at 10 6 cells/ml in PBS containing 1% BSA and 20 μl/ml phycoerythrin-labeled SMφ (PE-SMφ) or MOPC (PE-MOPC 104E) antibody, and incubated for 30 minutes at 4°C. The cells were then washed three times in PBS to remove unbound antibodies, and suspended in PBS containing 1% BSA at 106/cells ml. To stain human monocyte-derived macrophages and Bowes melanoma cells transfected with human CD36 cDNA, the cells were incubated with the anti-CD36 antibody FA6-152 (10 μg/ml) for 30 minutes on ice in PBS containing 1% BSA, washed three times, followed by another 30-minute incubation with fluorescein isothiocyanate-labeled rabbit anti-mouse IgG. FACS analysis was performed using a Becton Dickinson FACSCalibur as previously described. 19
Immunohistochemistry
Frontal brain samples from patients with AD, Parkinson’s disease, amytropic lateral sclerosis, and other diseases were obtained from the Brain Bank, Department of Pathology, Columbia University; The Alzheimer’s Disease and Schizophrenia Brain Bank; Department of Psychiatry, Mount Sinai School of Medicine; and The Kathleen Price Bryan Brain Bank, Duke University Medical Center. Eight-μm-thick brain sections were cut using a Minotome cryostat (International Equipment Co., Needham Heights, MA), collected on Fisherbrand Superfrost/Plus precleaned slides, mounted, dried for 2 hours at room temperature, permeabilized by immersion for 10 minutes in −20°C acetone, dried for 2 hours at room temperature, and immediately frozen at −80°C until needed. Staining of CD36 was performed using monoclonal anti-CD36 antibody FA6-152 and direct tyramide signal amplification using New England Nuclear’s TSA direct kit (NEL701) to enhance the intensity of the signal achieved by traditional streptavidin-biotin staining. Tissues were stained according to manufacturer’s instructions. Briefly, the tissues were blocked for 30 minutes, incubated for 1 hour with 20 μg/ml of FA6-152, or control IgG1. Slides were then gently washed in PBS, and incubated 30 minutes with a 1:200 dilution of biotin-conjugated donkey or rabbit anti-mouse IgG. Tissues were then washed again in PBS, and incubated with a 1:100 dilution of streptavidin-horseradish peroxidase for 30 minutes then washed with PBS again. The incubation time with tyramide-fluorescein was ∼6 minutes. To visualize astrocytes, some sections were co-stained with a 1:500 dilution of rabbit polyclonal antibody against human GFAP in HBS for 30 minutes at room temperature as described. 16 To visualize microglia, some sections were co-stained with 1 μg/ml of rat monoclonal antibody M1/70 against mouse and human Mac1 (CD11b/CD18) 23 in HBS for 30 minutes at room temperature. Because microglia also are known to express IgG on their cell surface, 24 identification of microglia in some sections was also verified by staining with goat anti-human IgG (data not shown) as described. 24 All co-staining antibodies were visualized using a 1:3000 dilution of the appropriate Alexa 594-labeled secondary antibody. Identification of endothelial cells was performed using staining with a 1:500 dilution of fluorescein isothiocyanate-labeled Ricinus communis lectin (an established endothelial cell marker) 25 obtained from Sigma Chemical Co. as described. 26 Slides were mounted in Gel/Mount (Biomeda Corp., Foster City, CA) and coverslips sealed with nail polish before viewing with a Nikon Eclipse E800 microscope with a ×60 objective lens and digital camera (Scientific Instruments, MI) using advanced spot software. Images were assembled in Adobe Photoshop.
Cell Adhesion Assays
Cell adhesion assays were performed as described previously using multispot slides. 6,19 Where indicated, the Cyquan cell proliferation assay kit (Molecular Probes) was used to measure the number of adherent cells according to the manufacturer’s instructions.
Results
Characterization of Aβ Peptides Used in These Experiments
We confirmed that Aβ 1-42 peptides prepared as described in Materials and Methods form fibrils in vitro by four different methods. First, we used established methods 27 to confirm that fAβ 1-42 was birefringent under polarized light whereas Aβ 42-1 was not. Second, we coated glass slides with fAβ 1-42 or Aβ 42-1, incubated them with Thioflavin S and examined them by fluorescence microscopy as described. 28 Glass surfaces coated with Aβ 42-1 did not fluoresce after staining with Thioflavin S (Figure 1A) ▶ , whereas those coated with fAβ 1-42 did (Figure 1B) ▶ . Third, using transmission electron microscopy, we found that Aβ 1-42 formed thin elongated fibrils (Figure 1C) ▶ similar to those described by others. 21 Fourth, analysis of fAβ 1-42 by nonreducing SDS-PAGE and Coomassie-blue staining showed two bands, an ≅4.2-kd band (the monomeric form of the peptide), and a second band of ≅21 kd (likely to be pentamers of Aβ 1-42) (Figure 1D) ▶ . As expected, Aβ 42-1 peptides ran at ≅4.2 kd indicating that they did not oligomerize (Figure 1D) ▶ .
Using more sensitive Western blotting and silver staining analyses other investigators have described up to four different bands under denaturing nonreducing conditions. 21 The data presented in Figure 1D ▶ suggests that the ≅21-kd species is the dominant oligomer after denaturation of fAβ 1-42 under nonreducing conditions. Taken together, the data presented in Figure 1 ▶ confirm that Aβ 1-42 used in the experiments reported here was fibrillar, whereas Aβ 42-1 was not.
N9 Immortalized Murine Microglia and Human Fetal Microglia Express CD36 on Their Surfaces
Murine microglia express scavenger receptor activity as evidenced by their ability to endocytose DiI-AcLDL 24 and adhere to fAβ-containing surfaces. 6 SR-A is expressed by microglia in normal brains 11 and in increased amounts by microglia in the brains of patients with AD, 11,12 and in the brains of transgenic APP23 mice with AD-like pathology. 13 To test whether CD36 is expressed on microglia, we incubated N9 microglia with anti-CD36 monoclonal antibody PE-SMφ or with an isotype-matched PE-labeled control IgM (PE-MOPC 104E) and analyzed the cells by FACS. N9 cells incubated with PE-SMφ anti-CD36 were approximately four times more fluorescent than N9 cells incubated with the control antibody indicating that N9 cells express CD36 (Figure 2A) ▶ . Similarly, human fetal microglia in culture expressed significant scavenger receptor activity as evidenced by their capacity to endocytose DiI-AcLDL (Figure 2B) ▶ . They also stained with the anti-CD36 antibody FA6-152 (Figure 2, C and D) ▶ , showing they express CD36 and that the protein is present on their surfaces. Human fetal microglia in culture did not stain with control isotype-matched antibodies (Figure 2, E and F) ▶ .
Figure 2.
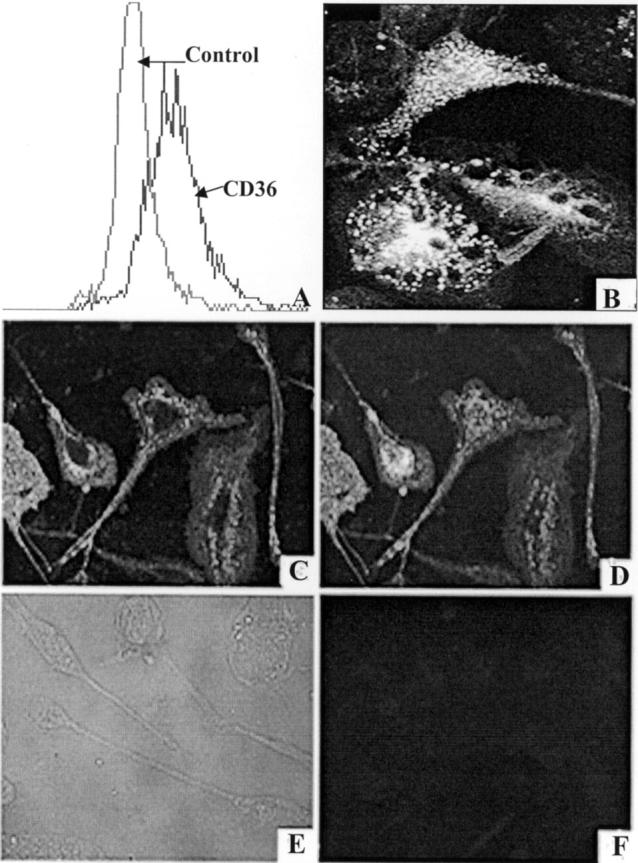
N9 mouse microglia and cultured human fetal microglia express CD36. A: N9 microglia were incubated with phycoerythrin (PE)-labeled monoclonal anti-CD36 antibody clone SMφ (PE-SMφ) or control antibody (PE-MOPC-104E). Cell-associated fluorescence was measured using FACS analysis as described in Materials and Methods. Mean fluorescence was 197.2 for PE-SMφ-labeled cells and 53.9 for PE-MOPC-labeled cells. B: Adherent human fetal microglia were incubated with PBS containing 1 mg/ml albumin and 5 μg/ml DiI-AcLDL for 4 hours at 37°C, washed, fixed in 3.7% formalin, and visualized using a Zeiss fluorescence microscope. C and D: Adherent human fetal microglia stained with the anti-CD36 monoclonal antibody FA6-152 as described in Materials and Methods. Surface staining was visualized by confocal microscopy of cells for a single optical section (C) and for the entire projection series of 16 sections (D). Cells did not stain with an isotype-matched control antibody (E and F).
Microglia and Macrophages Are Stimulated by fAβ to Produce ROS
Primary rat microglia, N9 murine microglia, and human monocyte-derived macrophages produce ROS when incubated with fAβ. 5-7 To determine whether human microglia also produce ROS in response to fAβ, we plated these cells on fAβ 1-42-containing surfaces and measured their capacity to reduce NBT. Human microglia adherent to surfaces containing fAβ 1-42 reduced significantly more NBT than human microglia adherent to surfaces coated with reverse Aβ 42-1 (control peptide) (Figure 3) ▶ .
Figure 3.

Fibrillar Aβ 1-42 stimulates cultured human fetal microglia to produce ROS. Human fetal microglia were incubated on multispot slides containing 1, 2.5, or 5 μg of fibrillar Aβ 1-42 (filled squares) or 5 μg of reverse (42-1) nonfibrillar amyloid (filled circle). The amount of ROS produced was measured by NBT reduction as described in Materials and Methods.
Capacity of fAβ-Containing Surfaces to Stimulate Mononuclear Phagocytes to Secrete H2O2 Is Correlated with the Cell’s Expression of CD36
Fresh human monocytes express low levels of CD36 29 and secrete little or no H2O2 when plated on oxLDL-containing surfaces, 15 whereas monocytes allowed to differentiate in culture for 3 to 5 days both express CD36 29 and secrete H2O2 when plated on oxLDL-containing surfaces. 15 To determine whether the capacity of fAβ to stimulate H2O2 secretion by monocytes and macrophages is correlated with the pattern of CD36 expression, we plated monocytes and monocyte-derived macrophages, at various stages of maturity, on surfaces coated with collagen IV and fAβ 1-42, fAβ 25-35, or Aβ reverse peptide 35-25. Fresh monocytes secreted very little H2O2 in response to fAβ (Figure 4A) ▶ . As expected, 30 fresh monocytes secreted substantial amounts of H2O2 in response to zymosan (10 mg/ml) (5.6 ± 1.9 nmol H2O2/2 × 10 5 cells/hour) or phorbol-12-myristate acetate (100 ng/ml) (11.42 ± 2.1 nmol H2O2/2 × 105 cells/hour), confirming that they were capable of H2O2 secretion when suitably stimulated.
Figure 4.

The capacity of monocyte-derived macrophages to produce H2O2 parallels their expression of CD36. A: Human monocytes/macrophages, cultured for 1 to 14 days in Teflon beakers, were added to wells coated with 2.5 μg of collagen IV overlayed with 2 μg of fibrillar Aβ 1-42 (open triangle), fibrillar Aβ 25-35 (filled square), or Aβ 35-25 (open circle) and incubated for 3 hours. H2O2 production was determined using the Molecular Probes Hydrogen Peroxide Assay Kit as described in Materials and Methods. Data presented are mean values ± SEM above background (collagen IV alone) for six experiments, each done in triplicate. B: H2O2 production by human monocyte-derived macrophages cultured for 5 days in Teflon beakers. Cells were incubated as in A above in wells containing collagen IV alone or collagen IV and 2 μg of fAβ 1-42 or 2 μg of Aβ 42-1. Data presented are mean values ± SEM above background (collagen IV alone) for three experiments each done in triplicate. *, Indicates values significantly different (P < 0.05) from control (open circles), using Student’s test. C: Summary of FACS analyses of CD36 expression on the surfaces of human monocytes/macrophages maintained in Teflon beakers and assayed at the times indicated as described in Materials and Methods. H2O2 production and CD36 expression were both maximal in cells cultured for 5 days.
Monocytes matured in Teflon beakers for 3 to 6 days and then plated on surfaces coated with collagen IV and fAβ 1-42 or fAβ 25-35 secreted significantly more H2O2 than fresh monocytes (Figure 4, A and B) ▶ . In contrast, monocytes matured in Teflon beakers for 3 to 6 days and then plated on surfaces coated with collagen IV and reverse peptides 42-1 or 35-25 secreted little H2O2 above background (Figure 4, A and B) ▶ . Monocytes maintained in Teflon beakers for 7 days or more before plating on surfaces coated with collagen IV and fAβ 1-42 or fAβ 25-35 secreted less and less H2O2, according to the length of time beyond 7 days they were maintained in culture before plating (Figure 4A) ▶ . The capacity of fAβ to promote H2O2 secretion by monocyte-derived macrophages paralleled their expression of CD36 (Figure 4C) ▶ .
Antibodies to CD36 Block ROS Production by Microglia and H2O2 Secretion by Macrophages Adherent to fAβ-Coated Surfaces
SR-A and CD36 play complementary roles in mediating macrophage adhesion to oxLDL-coated surfaces and in promoting ROS secretion by macrophages adherent to them. 15 We postulated that SR-A and CD36 play similar roles in ROS production by microglia. To test this hypothesis we incubated N9 cells and human monocyte-derived macrophages with monoclonal anti-CD36 antibodies SMφ or NLO7, plated them on fAβ-containing surfaces, and measured their production of oxidants. SMφ and NL07 inhibited ROS production by N9 microglia plated on fAβ by 58% and 52%, respectively (Figure 5A) ▶ . Similarly, SMφ and NL07 inhibited H2O2 secretion by macrophages plated on fAβ 1-42-containing surfaces by 58% and 44%, respectively, and on fAβ 25-35-coated surfaces by 43% and 37%, respectively (Figure 5B) ▶ . Addition of TSP-1, a physiological ligand of CD36 31 to the medium during plating inhibited H2O2 production by 4- to 6-day-old cultured macrophages on fAβ 1-42 or fAβ 25-35-coated surfaces by ∼90% (Figure 5B) ▶ .
Figure 5.

CD36 mediates ROS production by microglia and H2O2 production by macrophages plated on fAβ-coated surfaces. A: N9 microglia (106/ml) were preincubated for 30 minutes with anti-CD36 monoclonal antibody SMφ or NL07 (20 μg/ml) or isotype-matched control antibody MOPC-104E (20 μg/ml). Cells (50,000) were added to each spot of multispot slides coated with fibrillar Aβ 1-42 (black bars). ROS production was measured using the NBT assay as described in Materials and Methods. B: Human monocyte-derived macrophages (4 to 6 days in culture) were preincubated with anti-CD36 mAbs (SMφ or NL07, 20 μg/ml) or TSP-1 (5 μg/ml), plated on surfaces coated with 2.5 μg of collagen IV and 2 μg of fibrillar Aβ 1-42 (black bars) or fibrillar Aβ 25-35 (gray bars) and assayed for H2O2 secretion as described in Materials and Methods. Data are mean values ± SEM of three experiments done in triplicate. ROS production in response to Aβ 42-1 was similar to background (see Figure 4B ▶ , and data not shown).
Maximal inhibition of H2O2 secretion by monoclonal antibodies SMφ and NL07 occurred at 20 μg/ml. At 1 or 5 μg/ml SMφ inhibited H2O2 secretion by 5% and 27%, respectively. Maximal inhibition of H2O2 secretion by TSP-1 was obtained at 5 μg/ml, whereas 1 and 0.1 μg/ml gave 60% and 17% inhibition, respectively.
Control experiments showed that an isotype-matched control antibody (MOPC 104E) did not inhibit NBT reduction by N9 cells plated on fAβ-coated surfaces, and that SMφ and NL07 did not inhibit zymosan-induced ROS production by these cells (data not shown). Similarly, TSP had no effect on zymosan (10 mg/ml)- or phorbol-12-myristate acetate (100 ng/ml)-induced ROS production by macrophages cultured for 4 to 6 days in vitro (data not shown). These results suggest that anti-CD36 antibodies block ROS production by microglia and macrophages by inhibiting CD36’s interaction with fAβ, and are consistent with a similar role for TSP-1.
Bowes Melanoma Cells Transfected with CD36 cDNA Adhere to fAβ-Coated Surfaces
To test whether CD36 binds to fAβ 1-42, we transfected Bowes human melanoma cells with a mammalian expression vector for human CD36 and generated a cell line expressing CD36 (Bowes-CD36) (see Materials and Methods). We confirmed expression of CD36 in these cells by FACS analysis (Figure 6A) ▶ . Bowes-CD36 cells gained the ability to bind to surfaces coated with fAβ 1-42 (Figure 6B) ▶ , confirming that CD36 promotes binding to fAβ 1-42. Furthermore, binding of Bowes-CD36 to fAβ 1-42 was inhibited by monoclonal antibody FA6-152 but not by control IgG1 (Figure 6C) ▶ , confirming that Bowes-CD36 binding to surfaces containing 100 to 500 ng of fAβ 1-42 is CD36 mediated.
Figure 6.

Bowes cells expressing CD36 gain the ability to bind to fAβ 1-42-coated surfaces. A: FACS analysis of Bowes melanoma cells transfected with a mammalian expression vector for human CD36 and stained with a monoclonal antibody FA6-152 to CD36. B: Adhesion Bowes cells (2.5 × 10 5 cells/ml), transfected with a mammalian expression vector for CD36 (CDM8-CD36) or with a control vector (PcDNAneo), and suspended in phosphate buffer containing 5 mmol/L EDTA, to multispot slides coated with 2 μg of collagen IV and the indicated amount of Aβ 1-42. C: Bowes melanoma cells expressing CD36 were incubated with the indicated concentrations of anti-CD36 monoclonal antibody FA6-152 or an isotype-matched IgG1 control and plated on multispot slides coated with 2 μg of collagen IV and 500 ng of Aβ 1-42.
Effects of Antibodies to CD36 and of TSP-1 on Adhesion of Microglia and Macrophages to fAβ-Coated Surfaces
We have shown previously that adhesion of microglia to fAβ, 6 and of macrophages to oxLDL-containing surfaces, 15 is required for ROS/H2O2 production. We have also shown that anti-CD36 antibodies block ROS production by macrophages adherent to surfaces coated with oxLDL but have no significant effect on adhesion to these surfaces, suggesting that CD36 is not required for macrophage adhesion. In parallel studies, we tested whether anti-CD36 antibodies SMφ or NL07, control antibody MOPC 104E, or the CD36 ligand TSP-1 31 affected adhesion of microglia and macrophages to surfaces coated with collagen IV and fAβ 1-42 or fAβ 25-35. None of the antibodies (data not shown) inhibited significantly adhesion of N9 microglia or of macrophages to surfaces containing fAβ 1-42 or 25-35. TSP-1 reduced adhesion of macrophages to surfaces coated with fAβ 1-42 or fAβ 25-35 by ∼28% and ∼21% respectively. Although this reduction in macrophage adhesion is significant, it is far less than the 90% reduction in H2O2 secretion effected by TSP-1 (Figure 5B) ▶ . These results imply that CD36 functions are blocked more effectively by ligand-receptor interactions than antibody-receptor interactions and/or that receptors for TSP-1 and/or fAβ other than CD36 32 may contribute to adhesion of macrophages to fAβ-containing matrices and to signaling ROS secretion.
CD36 Is Expressed on Microglia in Brains of Patients with Alzheimer’s Disease
To confirm that microglia express CD36 in vivo we stained frozen sections of unfixed brains from 11 patients using the anti-CD36 antibody FA6-152. These included five patients with AD, one patient with Parkinson’s disease, three patients with amyotrophic lateral sclerosis, and two patients without clinical signs of dementia. The immunostained sections shown in Figure 7 ▶ are derived from the brain of a patient with AD. The staining is representative of stained frozen sections from all 11 brains studied. In all sections, cells with the appearance of microglia (Figure 7, a and d) ▶ reacted with the anti-CD36 antibody FA6-152. CD36-expressing cells stained with antibodies against Mac-1 (CD11b/CD18) (Figure 7b) ▶ , a microglial marker, 33,34 but not with antibodies against GFAP (Figure 7e) ▶ , an astrocyte marker. 16,35 An overlay showing co-localization of anti-CD36 and anti-Mac-1 staining is shown in Figure 7c ▶ , and an overlay showing non-co-localization of anti-CD36 and anti-GFAP staining in Figure 7f ▶ . CD36 was expressed to variable extents on cells with microglial morphology in all 11 brains tested (data not shown). Because of the limited number of specimens examined, and the difficulty of detecting modest differences in protein expression by immunohistochemistry, we cannot determine whether CD36 expression is enhanced in microglia in brains of patients with AD compared to microglia in brains from normal aged people or from people with other neurological diseases. In addition to microglia, structures resembling vessels (Figure 7g) ▶ also stained with anti-CD36 antibodies. To confirm that these structures are blood vessels we co-stained with fluorescein isothiocyanate-labeled Ricinus communis lectin, an endothelial cell marker. As seen in Figure 7, j to l ▶ , staining with CD36 correlated with staining for endothelial cells, indicating that in addition to microglia, brain endothelial cells also express CD36.
Figure 7.
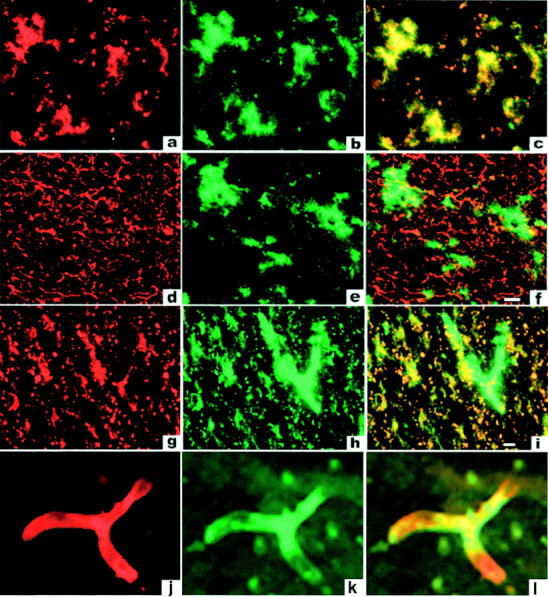
CD36 is expressed by microglia and endothelial cells in AD brains. Staining of frontal cortex from 13 patients was performed using New England Nuclear’s Tyramide Signal Amplification kit as described under Materials and Methods. Each row represents immunofluorescent staining of a single section from an AD brain and is typical for all sections examined in this study. Staining with anti-CD36 antibody is shown in a, d, g, and j, anti-MAC-1 (CD11b) antibody in b and h, anti-GFAP in e, and staining with Ricin in k. Overlays of staining in each row are shown in c, f, i, and l. Cells lining brain vasculature (likely endothelial cells) (k) express CD36 (j and l) but not the microglial marker Mac-1 (g, h, and i).
Discussion
It is now well documented that interactions between microglia and fAβ stimulate human microglia to produce ROS and tumor necrosis factor-α, and rodent microglia to produce ROS, nitric oxide, and tumor necrosis factor-α. 6-8 These and other findings have led us, and a number of other investigators to suggest that these microglial secretory products contribute to the progression of AD. The studies reported here provide further support for this concept and demonstrate for the first time that human microglia express CD36 in vitro and in vivo, and that interactions between CD36 and fAβ signal ROS production by microglia and macrophages. Consistent with this suggestion, macrophage colony-stimulating factor, a known stimulator of macrophage CD36 expression 36 is also expressed in brains of mice with AD-like pathology. 37 Macrophage colony-stimulating factor-M enhances the proinflammatory effects of fAβ on microglia, 38 possibly by enhancing their expression of CD36.
The mechanism(s) by which fAβ stimulates CD36 to signal H2O2 secretion is unknown. Anti-CD36 antibodies such as SMφ and NL07 and soluble CD36 ligands in suspension such as oxLDL, AcLDL, and TSP-1 15 do not stimulate macrophages to secrete H2O2. However, anti-CD36 antibodies stimulate H2O2 secretion by macrophages when cross-linked by a secondary antibody, 39 and surfaces containing immobilized CD36 ligands such oxLDL, acetyl LDL, and TSP-1 stimulate macrophages to adhere and secrete H2O2 (and data not shown). 15 Thus oligomerization of CD36 by fAβ, a multivalent ligand, may be required for CD36 to signal macrophage H2O2 secretion.
In addition to ROS production, CD36 can mediate adhesion to fAβ-coated surfaces. Bowes melanoma cells, which normally do not bind to surfaces coated with 100 to 500 ng of fAβ in the presence of ethylenediaminetetraacetic acid, gain the ability to adhere to such surfaces when transfected with CD36. Monoclonal anti-CD36 antibodies block this process suggesting that CD36 is a receptor for fAβ. But CD36-blocking antibodies do not affect the adhesion of microglia and macrophages to fAβ-coated surfaces. This finding suggests that microglia and macrophages express other receptors, in addition to CD36, that can mediate adhesion to fAβ. This result is also consistent with our previous finding that antibodies to SR-A block most of the adhesion of neonatal microglia to fAβ-coated surfaces 6 and with findings of Husemann and colleagues 40 and Chung and colleagues 41 that macrophages and microglia from SRA-KO mice endocytose and/or bind ∼60% less fAβ than cells from wild-type mice. We propose that multiple scavenger- and adhesion-promoting receptors cooperate in mediating adhesion of microglia to fAβ in senile plaques.
CD36 also may play a role in Alzheimer’s angiopathy. Endothelial cells of the cerebral vasculature, the site of fAβ deposition in Alzheimer’s angiopathy, 42,43 also express CD36. 44 Malaria-infected erythrocytes interact with CD36 on endothelial cells in the cerebral microvasculature and cause disruption of the blood brain barrier. 45 Fibrillar Aβ also causes disruption of the blood brain barrier. 46 We suggest that interaction of CD36 with fAβ deposited in the subendothelial space may contribute to the increase in permeability of cerebral microvessels observed in AD. 47
There are conflicting reports about whether mice genetically deficient in SR-A and maintained on an atherosclerosis-promoting diet develop larger or smaller vascular lesions than wild-type mice. 48,49 Similarly, there are discrepancies between in vitro studies done with microglia from SR-A KO mice and in vivo studies done with transgenic PD-APP mice genetically deficient in SR-A. Although neonatal microglia from SR-A-KO mice endocytose ∼60% less fAβ, 40,41 Huang and colleagues 50 reported that transgenic PD-APP mice genetically deficient in SR-A show no obvious change in their AD-like pathology compared to transgenic PD-APP mice expressing SRA. PD-APP mice may not be the best model to study the role of microglia in AD because they exhibit a weaker microglial response compared to other transgenic APP mouse models of AD and to AD patients. 13,51,52 Nonetheless, the findings reported by Huang and colleagues 50 suggest that other receptors in addition to SR-A may be involved in mediating the interactions of microglia with fAβ in mice. Mice genetically deficient in CD36 and maintained on a high- fat diet have 75% fewer atherosclerotic lesions than wild-type mice maintained on the same diet. 53 We are currently investigating whether transgenic APP mice genetically deficient in CD36 have altered AD-like pathology when compared to transgenic APP mice expressing CD36.
The roles we propose for microglial scavenger receptors in AD mirror in several respects the roles reported for macrophage scavenger receptors in atherosclerosis. Oxidation of LDL in the intima of the arterial wall is one of the first events in the development of atheromata and is thought to be a major factor in the initiation and progression of atherosclerosis. SR-A mediates the adhesion of monocytes and macrophages to surfaces containing oxLDL and probably contributes to the localization of these cells at sites of oxLDL deposition 15 and basement membrane modification. 19 Adhesion of monocytes/macrophages to oxLDL-containing surfaces facilitates the interaction of CD36 with oxLDL, 15 and signals them to secrete ROS, 15 and other proinflammatory substances. We suggest that a similar inflammatory cascade is initiated when microglia interact with fAβ deposits in the brain parenchyma and perivascular spaces. Microglia adhere to fAβ and possibly other components of the senile plaque via SR-A, 6 CD36 (Figure 6) ▶ , and other adhesion promoting receptors. This signals microglia to secrete ROS (Figures 3, 5, and 8) ▶ ▶ ▶ , and possibly other neurotoxic substances. We propose that SR-A, CD36, and possibly other adhesion and/or scavenger receptors cooperate in mediating binding to Aβ fibrils in senile plaques, thereby initiating a signaling cascade that leads to production of ROS and other proinflammatory and neurotoxic substances. CD36 may therefore be a key microglial receptor contributing to fAβ-induced neuronal damage in AD.
Figure 8.

Cooperation between scavenger receptors and CD36 mediates microglial release of ROS. Diagram summarizing proposed effects of CD36 on ROS production by microglia adherent to surfaces coated with Aβ fibrils. SR-A and other scavenger receptors mediate microglial adhesion to fAβ-coated surfaces. Engagement of CD36 by substrate bound fAβ signals ROS and H2O2 production. Pre-exposure of cells to antibodies against CD36 or the CD36 ligand TSP-1 blocks interactions between CD36 and fAβ resulting in inhibition of ROS and H2O2 production (see Figures 2 and 4 ▶ ▶ ).
Acknowledgments
We thank the Brain Bank of the Department of Pathology, Columbia University and the Alzheimer’s Disease and Schizophrenia Brain Bank of the Department of Psychiatry, Mt. Sinai School of Medicine, for brain tissue that was used to stain for CD36 in the present work; Suquin Fung, Department of Physiology, Columbia University, for cutting frozen sections; Michael Cammer of the Albert Einstein College of Medicine Confocal Microscopy Facility and Theresa Swanye of the Columbia University Confocal Microscopy Facility for assistance; and Marie McKee from the renal unit at Massachusetts General Hospital for assistance with electron microscopy.
Footnotes
Address reprint requests to Joseph B. El Khoury, M.D., Center for Immunology and Inflammatory Diseases, Room 8301, Massachusetts General Hospital East, CNY Building 149, 13th Street, Charlestown MA, 02129-2000. E-mail: jelkhoury@partners.org.
Supported by Postdoctoral Fellowship HL 10196 (to I. S. C.); grants RG1-96-067 from the Alzheimer Association; grant R37-AI 20516 from the National Institute of Allergy and Infectious Disease and HL43310 from National Heart, Lung, and Blood Institute (to S. C. S.); site grants PO1-AG02219 and P50-AG05138 to the Department of Psychiatry, Mt. Sinai School of Medicine; and grant NS041330 from National Institute of Neurological Disorders and Stroke (to J. E. K.).
References
- 1.Selkoe DJ: The origins of Alzheimer disease: a is for amyloid. JAMA 2000, 283:1615-1617 [DOI] [PubMed] [Google Scholar]
- 2.Wisniewski HM, Robe A, Zigman W, Silverman W: Neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol 1989, 48:606-609 [DOI] [PubMed] [Google Scholar]
- 3.El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC: Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol Aging 1998, 19:S81-S84 [DOI] [PubMed] [Google Scholar]
- 4.Akiyama H: Inflammatory response in Alzheimer’s disease. Tohoku J Exp Med 1994, 174:295-303 [DOI] [PubMed] [Google Scholar]
- 5.Meda L, Cassatella MA, Szendrei GI, Otvos Jr L, Baron P, Villalba M, Ferrari D, Rossi F: Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995, 374:647–650 [DOI] [PubMed]
- 6.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD: Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382:716-719 [DOI] [PubMed] [Google Scholar]
- 7.Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F: Beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J Biol Chem 1999, 274:15493-15499 [DOI] [PubMed] [Google Scholar]
- 8.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE: Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 1996, 16:6021-6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell MD, Lopez-Gonzalez R, Lawson L, Hughes D, Fraser I, Gordon S, Perry VH: Upregulation of the macrophage scavenger receptor in response to different forms of injury in the CNS. J Neurocytol 1994, 23:605-613 [DOI] [PubMed] [Google Scholar]
- 10.Paresce DM, Ghosh RN, Maxfield FR: Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 1996, 17:553-565 [DOI] [PubMed] [Google Scholar]
- 11.Christie RH, Freeman M, Hyman BT: Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. Am J Pathol 1996, 148:399-403 [PMC free article] [PubMed] [Google Scholar]
- 12.Honda M, Akiyama H, Yamada Y, Kondo H, Kawabe Y, Takeya M, Takahashi K, Suzuki H, Doi T, Sakamoto A, Ookawara S, Mato M, Gough PJ, Greaves DR, Gordon S, Kodama T, Matsushita M: Immunohistochemical evidence for a macrophage scavenger receptor in Mato cells and reactive microglia of ischemia and Alzheimer’s disease. Biochem Biophys Res Commun 1998, 245:734-740 [DOI] [PubMed] [Google Scholar]
- 13.Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M: Aβ-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 2001, 158:63-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA: CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem 1993, 268:11811-11816 [PubMed] [Google Scholar]
- 15.Maxeiner H, Husemann J, Thomas CA, Loike JD, El Khoury J, Silverstein SC: Complementary roles for scavenger receptor A and CD36 of human monocyte-derived macrophages in adhesion to surfaces coated with oxidized low-density lipoproteins and in secretion of H2O2. J Exp Med 1998, 188:2257-2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldman JE, Chiu FC: Growth kinetics, cell shape, and the cytoskeleton of primary astrocyte cultures. J Neurochemistry 1984, 42:175-184 [DOI] [PubMed] [Google Scholar]
- 17.Righi M, Sassano M, Valsasnini P, Shammah S, Ricciardi-Castagnoli P: Activation of the M-CSF gene in mouse macrophages immortalized by retroviruses carrying a v-myc oncogene. Oncogene 1991, 6:103-111 [PubMed] [Google Scholar]
- 18.McManus CM, Brosnan CF, Berman JW: Cytokine induction of MIP-1 alpha and MIP-1 beta in human fetal microglia. J Immunol 1998, 160:1449-1455 [PubMed] [Google Scholar]
- 19.El Khoury J, Thomas CA, Loike JD, Hickman SE, Cao L, Silverstein SC: Macrophages adhere to glucose-modified basement membrane collagen IV via their scavenger receptors. J Biol Chem 1994, 269:10197-10200 [PubMed] [Google Scholar]
- 20.Oquendo P, Hundt E, Lawler J, Seed B: CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell 1989, 58:95-101 [DOI] [PubMed] [Google Scholar]
- 21.Walsh DM, Hartley DM, Condron MM, Selkoe DJ, Teplow DB: In vitro studies of β-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692→Gly) Alzheimer’s disease. Biochem J 2001, 355:869-877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rook GA, Steele J, Umar S, Dockrell HM: A simple method for the solubilisation of reduced NBT, and its use as a calorimetric assay for activation of human macrophages by gamma-interferon. J Immunol Methods 1985, 82:161-167 [DOI] [PubMed] [Google Scholar]
- 23.Ault KA, Springer TA: Cross-reaction of a rat-anti-mouse phagocyte-specific monoclonal antibody (anti-Mac-1) with human monocytes and natural killer cells. J Immunol 1981, 126:359-364 [PubMed] [Google Scholar]
- 24.Giulian D, Vaca K: Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke 1993, 24(Suppl 12):184-190 [PubMed] [Google Scholar]
- 25.Nag S: Ultrastructural localization of monosaccharide residues on cerebral endothelium. Lab Invest 1985, 52:553-558 [PubMed] [Google Scholar]
- 26.Mannoji H, Yeger H, Becker LE: A specific histochemical marker (lectin Ricinus communis agglutinin-1) for normal human microglia, and application to routine histopathology. Acta Neuropathol 1986, 71:341-343 [DOI] [PubMed] [Google Scholar]
- 27.Lorenzo A, Yankner BA: Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA 1994, 91:12243-12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caputo CB, Fraser PE, Sobel IE, Kirschner DA: Amyloid-like properties of a synthetic peptide corresponding to the carboxy terminus of beta-amyloid protein precursor. Arch Biochem Biophys 1992, 292:199-205 [DOI] [PubMed] [Google Scholar]
- 29.Huh HY, Pearce SF, Yesner LM, Schindler JL, Silverstein RL: Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood 1996, 87:2020-2028 [PubMed] [Google Scholar]
- 30.Nakagawara A, DeSantis NM, Nogueira N, Nathan CF: Lymphokines enhance the capacity of human monocytes to secret reactive oxygen intermediates. J Clin Invest 1982, 70:1042-1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asch AS, Barnwell J, Silverstein RL, Nachman RL: Isolation of the thrombospondin membrane receptor. J Clin Invest 1987, 79:1054-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bornstein P: Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 1995, 130:503-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V: Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA 1991, 88:7438-7442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akiyama H, McGeer PL: Brain microglia constitutively express beta-2 integrins. J Neuroimmunol 1990, 30:81-93 [DOI] [PubMed] [Google Scholar]
- 35.Mancardi GL, Liwnicz BH, Mandybur TI: Fibrous astrocytes in Alzheimer’s disease and senile dementia of Alzheimer’s type. Acta Neuropathol 1983, 61:76-80 [DOI] [PubMed] [Google Scholar]
- 36.Yesner LM, Huh HY, Pearce SF, Silverstein RL: Regulation of monocyte CD36 and thrombospondin-1 expression by soluble mediators. Arterioscler Thromb Vasc Biol 1996, 16:1019-1025 [DOI] [PubMed] [Google Scholar]
- 37.Murphy GM, Zhao F, Yang L, Cordell B: Expression of macrophage colony-stimulating factor receptor is increased in the AbetaPP(V717F) transgenic mouse model of Alzheimer’s disease. Am J Pathol 2000, 157:895-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy Jr GM, Yang L, Cordell B: Macrophage colony-stimulating factor augments beta-amyloid-induced interleukin-1, interleukin-6, and nitric oxide production by microglial cells. J Biol Chem 1998, 273:20967–20971 [DOI] [PubMed]
- 39.Trezzini C, Jungi TW, Spycher MO, Maly FE, Rao P: Human monocytes CD36 and CD16 are signaling molecules. Evidence from studies using antibody-induced chemiluminescence as a tool to probe signal transduction. Immunology 1990, 71:29-37 [PMC free article] [PubMed] [Google Scholar]
- 40.Husemann J, Loike JD, Kodama T, Silverstein SC: Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol 2001, 114:142-150 [DOI] [PubMed] [Google Scholar]
- 41.Chung H, Brazil MI, Irizarry MC, Hyman BT, Maxfield FR: Uptake of fibrillar beta-amyloid by microglia isolated from MSR-A (type I and type II) knockout mice. Neuroreport 2001, 12:1151-1154 [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ: Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease. An immunoelectron microscopic study. Am J Pathol 1992, 141:249-252 [PMC free article] [PubMed] [Google Scholar]
- 43.Esiri MM, Wilcock GK: Cerebral amyloid angiopathy in dementia and old age. J Neurol Neurosurg Psychiatry 1986, 49:1221-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnwell JW, Asch AS, Nachman RL, Yamaya M, Aikawa M, Ingravallo P: A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. J Clin Invest 1989, 84:765-772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown H, Hien TT, Day N, Mai NT, Chuong LV, Chau TT, Loc PP, Phu NH, Bethell D, Farrar J, Gatter K, White N, Turner G: Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol 1999, 25:331-340 [DOI] [PubMed] [Google Scholar]
- 46.Claudio L: Ultrastructural features of the blood brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol 1996, 91:6-14 [DOI] [PubMed] [Google Scholar]
- 47.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M: Beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996, 380:168-171 [DOI] [PubMed] [Google Scholar]
- 48.De Winther MP, Gijbels MJ, van Dijk KW, van Gorp PJ, Suzuki H, Kodama T, Frants RR, Havekes LM, Hofker MH: Scavenger receptor deficiency leads to more complex atherosclerotic lesions in A-POE3 Leiden transgenic mice. Atherosclerosis 1999, 144:315-321 [DOI] [PubMed] [Google Scholar]
- 49.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Kodama T: A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 1997, 386:292-296 [DOI] [PubMed] [Google Scholar]
- 50.Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, Mucke L: Elimination of the class A scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am J Pathol 1999, 155:1741-1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998, 152:307-317 [PMC free article] [PubMed] [Google Scholar]
- 52.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D: Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996, 16:5795-5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL: Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 2000, 105:1049-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]