

Abstract
Androgen receptor expression was analyzed in the CWR22 human prostate cancer xenograft model to better understand its role in prostate cancer recurrence after castration. In androgen-dependent tumors, 98.5% of tumor cell nuclei expressed androgen receptor with a mean optical density of 0.26 ± 0.01. On day 2 after castration androgen deprivation decreased immunostained cells to 2% that stained weakly (mean optical density, 0.16 ± 0.08). Cellular proliferation measured using Ki-67 revealed <1% immunostained cells on day 6. Androgen receptor immunostained cells increased to 63% on day 6 and 84% on day 32 although immunostaining remained weak. Cellular proliferation was undetectable beyond day 6 after castration until multiple foci of 5 to 20 proliferating cells became apparent on day 120. These foci expressed increased levels of prostate-specific antigen, an androgen receptor-regulated gene product. In tumors recurrent 150 days after castration androgen receptor-immunostaining intensity was similar to CWR22 tumors from intact mice although the percentage of cells immunostained was more variable. The appearance of proliferating tumor cells that expressed androgen receptor and prostate-specific antigen 120 days after castration suggests that these cells represent the origin of recurrent tumors.
High-affinity binding of dihydrotestosterone to androgen receptor (AR) causes AR to function as a transcription factor 1,2 that regulates a network of androgen response genes. 3,4 Prostate cancer (CaP) is androgen-dependent and its growth is mediated by this AR-regulated gene network. Androgen deprivation causes reduced AR expression, 5 apoptosis, decreased cell volume, 6 and decline of serum prostate-specific antigen (PSA). However, most CaPs eventually develop the capacity for recurrent growth in the absence of testicular androgen. All of 22 specimens of testicular androgen-independent metastatic CaP showed positive immunohistochemical staining for AR protein. 7 Transurethral resection of prostate specimens from 10 untreated CaP patients and 20 patients with CaP recurrent after androgen deprivation were compared and no significant difference in the percentage of AR-positive cells was found. 8,9 Because AR expression is similar in androgen-dependent and recurrent CaP, we sought to understand how AR expression changes in relation to cellular proliferation in the interval between androgen deprivation and tumor recurrence.
CWR22 is an androgen-dependent human CaP xenograft propagated subcutaneously in nude mice. CWR22 resembles the majority of human CaPs; CWR22 secretes PSA, undergoes tumor regression after androgen deprivation, and recurs as a palpable, growing and ultimately lethal tumor after several months in the absence of testicular androgen. 10-13 We demonstrated that recurrence of CWR22 tumor after androgen deprivation was associated with re-expression of a network of androgen-regulated genes including PSA, human kallikrein-2, Nkx 3.1, AR co-activator ARA-70, cell cycle genes Cdk1 and Cdk2, 3 and insulin-like growth factor binding protein-5. 14 Recently, Amler and associates 15 have reported incomplete reactivation of the androgen response pathway despite androgen absence in recurrent CWR22 using microarray analysis. Similar expression of AR and these androgen-regulated genes in androgen-dependent and recurrent CWR22 tumors suggested a role for AR regulation of gene expression in the development of recurrent CWR22 despite the absence of testicular androgen.
Video image analysis has been used to quantitate AR expression more precisely than visual scoring. 16-19 We developed an automated method for measuring AR expression in individual cells that was used to demonstrate the dependence of AR protein levels on serum androgen levels in the CWR22 model. 20 In CWR22 tumor-bearing mice castrated for 6 days, AR mean optical density (MOD) decreased to 57% of levels in tumors from intact mice. After 72 hours of exogenous testosterone administration to 6-day castrated mice, AR MOD in CWR22 returned to the level found in tumors from intact mice. Cellular proliferation of testosterone-treated tumors reached ∼50% of the original androgen-stimulated CWR22 tumors from intact mice. 14 These data suggested that the majority of CWR22 cells on day 6 after castration had functional AR. In archived radical prostatectomy specimens, AR protein content was higher in androgen-dependent, clinically localized CaP and lower in prostate intraepithelial neoplasia than benign prostatic hyperplasia (BPH). 19,20 AR immunostaining intensity was similar in androgen-stimulated and recurrent tumors from the CWR22 xenograft and transurethral resection of the prostate specimens of BPH; all tissues were small volume and fixed immediately after procurement. 20 Finally, 12 specimens of recurrent CaP and 16 specimens of BPH, all acquired by transurethral resection of the prostate and fixed immediately, exhibited similar AR immunostaining (unpublished data). Taken together, these findings suggest that AR is expressed in androgen-stimulated CaP, diminished but recoverable after castration, and re-expressed despite androgen absence on CaP recurrence.
We sought to test the hypothesis that re-expression of AR coincided with the onset of androgen-independent cellular proliferation in CaP. To test this hypothesis, the temporal relationship between AR protein expression and cellular proliferation was determined using the CWR22 xenograft model during tumor regression and recurrence after castration. Quantitative immunohistochemistry and color video image analysis were used to measure precisely the proportion of cells expressing AR and Ki-67 and the intensity of expression of AR associated with response to androgen deprivation and emergence of the recurrent phenotype.
Materials and Methods
Research Specimens
Nude/nude athymic mice were purchased from Harlan Sprague-Dawley, Inc., Indianapolis, IN. The CWR22 tumor model has been maintained by continuous passage since December of 1995 from CWR22 cells that were a gift from Thomas A. Pretlow, MD, PhD, Case Western Reserve University). CWR22 tumors were transplanted as 1 million dissociated cells suspended in Matrigel (Collaborative Research Inc., Bedford, MA) injected subcutaneously into nude mice 4 to 5 weeks of age. 11,12 A 12.5-mg sustained-release testosterone pellet (Innovative Research of America, Sarasota, FL) was placed subcutaneously in each animal 2 days before tumor injection and every 3 months thereafter to maintain consistent serum levels of testosterone of ∼4 ng/ml. After tumors reached a volume of 1 cm 3 , animals were anesthetized with methoxyflurane, castrated, and the testosterone pellets removed. Intact mice bearing tumors and castrated animals with either regressed or recurrent CWR22 tumors were exposed to methoxyflurane and sacrificed by cervical dislocation. Tumor height, width, and depth were measured using calipers and tumor volume was calculated by multiplying these three measurements and 0.5234. Tumors were excised and cut into several pieces (∼125 mm3); half was frozen in liquid nitrogen and half was fixed in 10% buffered formalin for 24 to 48 hours, washed in phosphate-buffered saline (PBS) for 24 hours, and paraffin-embedded. Specimens of BPH prepared identically were used as positive controls. Blood was obtained on sacrifice of all tumor-bearing mice for measurement of serum PSA.
Immunohistochemistry
The avidin-biotin-immunoperoxidase technique 21 was modified for use in paraffin-embedded tissues that were immunostained using capillary action with a MicroProbe staining station (Fisher Scientific, Pittsburgh, PA). 22 Monoclonal antibody (mAb) F39.4.1 (BioGenex, San Ramon, CA) recognizes an epitope in the N-terminal region of human AR. 23 mAb MIB-1 (Oncogene, Cambridge, MA) and polyclonal antibody MIB-5 (DAKO Corp., Carpinteria, CA) react with the cell cycle-associated antigen Ki-67 expressed during the proliferative phases (G1, S, G2, and M) but absent in the resting phase (G0) of the cell cycle. 24 mAb A67-B/E3 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) corresponds to amino acids 1 to 261 representing full length PSA p30 of human origin. 25
Paraffin-embedded CWR22 tumor specimens were cut into 6-μm-thick histological sections. After deparaffinization and rehydration, tissue sections were heated to 100°C for 30 minutes in a vegetable steamer in the presence of antigen retrieval solution (CITRA, pH 6.0; BioGenex) and cooled for 10 minutes. Slides were preincubated with 2% normal horse serum for 5 minutes at 37°C and washed with automation buffer (Fisher Scientific).
AR mAb was diluted 1:300 (0.13 μg/ml in PBS containing 0.1% bovine serum albumin, pH 7.4) and sections were stained for 120 minutes at 37°C. Slides were incubated in biotinylated anti-mouse immunoglobulin (IgG) (Vector Laboratories, Inc., Burlingame, CA) for 15 minutes at 37°C (1:200 in PBS, pH 7.4) and in horseradish peroxidase-conjugated avidin-biotin complex (Vector Laboratories, Inc.) for 15 minutes at 37°C (1:100 in PBS, pH 7.4). The immunoperoxidase complexes were visualized using diaminobenzidine tetrahydrochloride (Vector Laboratories, Inc) for 8 minutes at 37°C (0.75 mg/ml in Tris buffer containing 0.03% hydrogen peroxide, pH 7.6). Slides were dehydrated through graded alcohol solutions and cleaned by Hemo-De xylene substitute (Fisher Scientific). Counterstaining was performed with hematoxylin (Gill’s formula, 1:6 dilution; Fisher Scientific) for 12 seconds. Slides were mounted with Permount and coverslips. Two representative slides were selected from each time point and stained with the polyclonal AR antibodies, AR52 and PG-21, following protocols reported previously. 5,26 AR52 was provided by Dr. Elizabeth M. Wilson (University of North Carolina at Chapel Hill) and PG-21 was provided by Dr. Gail S. Prins (University of Illinois at Chicago). Slides prepared from a CWR22 tumor on day 6 after castration and human BPH were included as external controls to avoid variation of immunostaining intensity caused during staining procedures. Nonimmune mouse IgG (Vector Laboratories, Inc.) was used instead of AR mAb at the same IgG concentration for negative control slides prepared from the same tissue blocks as specimens; negative control slides were nonreactive.
MIB-1 mAb staining was performed at an IgG concentration of 0.5 μg/ml (1:50). All other steps were as described for AR immunostaining. Serial sections adjacent to the sections stained for AR were obtained from tumors on day 120 after castration and stained with MIB-1 mAb. Colon cancer tissue served as positive controls and 0.5 μg/ml of nonimmune mouse IgG was used instead of MIB-1 mAb at the same IgG concentration for negative control slides prepared from the same tissue blocks as specimens; negative control slides were nonreactive.
PSA mAb (1:50, 4 μg/ml) was biotinylated and blocked in vitro using the Iso-IHC kit (InnoGenex, San Ramon, CA) to avoid background staining caused by infiltrated murine cells in CWR22 tumors harvested from castrated animals. Sections were digested in Proteinase-K (20 μg/ml, DAKO Corp.) for 6 minutes at room temperature. Sections were incubated in the blocking solution and labeled with PSA mAb for 1 hour at 37°C and in streptavidin-peroxidase (InnoGenex) for 5 minutes at 37°C. Immunoreaction was visualized by diaminobenzidine tetrahydrochloride for 8 minutes at 37°C. Double immunohistochemistry was performed on additional CWR22 slides to co-localize PSA expression among Ki-67-positive tumor cells. Sections were eluted by glycine buffer (pH 2.3) for 5 minutes three times at room temperature and antigen-retrieved as described previously. A mixture of normal goat serum (2%) and avidin (1:50 in PBS, Vector Laboratories, Inc.) was used for blocking for 5 minutes at 37°C. Sections were reacted with MIB-5 (1:50, 20 μg/ml) mixed with biotin (1:50 in PBS, Vector Laboratories, Inc.) for 2 hours at 37°C. The same avidin-biotin-peroxidase complex technique used for MIB-1 was performed. Immunoreaction was visualized by 3-amino-9-ethylcarbazole (AEC) (Vector Laboratories, Inc.) for 10 minutes at 37°C. BPH and CaP specimens were used as positive controls. For the negative control slide, nonimmune rabbit IgG (Vector Laboratories, Inc.) was used instead of PSA mAb at the same IgG concentration; appropriate biotinylated IgGs were replaced with PBS in PSA and MIB-5 steps to check against cross-reactions. Negative control slides showed neither nonspecific reaction nor cross-reactions.
Automated Digital Image Analysis
Automated digital image analysis was performed as described previously. 20 Briefly, imaging hardware consisted of a Zeiss Axioskop microscope, a 3-chip charge-coupled device camera (C5810; Hamamatsu Photonics Inc., Hamamatsu, Japan), a camera control unit (Hamamatsu Photonics Inc.), and a Pentium-based personal computer. Each field of view for AR-stained slides was digitized at total magnification ×1200 using a ×40 objective (numerical aperture, 1.3). For MIB-1- and PSA-stained slides, a ×20 objective (numerical aperture, 0.6) was used for total magnification at ×600. Twenty images that contained ∼200 to 250 nuclei at ×1200 and 400 to 500 nuclei at ×600 provided an adequate sample size for each tumor because the deviation of average intensity values of randomly chosen immunopositive areas became stable (within ±5%).
Immunopositivity and immunonegativity were determined using a linear discriminant analysis based on hue, saturation, and intensity of 100 immunostained cells of an intact CWR22 specimen and 100 cells of a negative control slide, respectively. The positivity for AR, Ki-67, and PSA was defined as the total number of pixels from immunopositive areas divided by the total number of pixels from all nuclear areas detected in a given specimen.
Differences in MOD and percentage of AR-, Ki-67-, and PSA-positive cells from all images from all tumors at various time points were evaluated using Wilcoxon rank sum tests. Correlations between features were examined using the Pearson’s product moment correlation test. F-tests were performed to compare the variances among samples. Statistical significance was achieved if P < 0.05.
Western Immunoblot Analysis of AR
Lysates were prepared from frozen CWR22 tumors. Tumor tissue (100 mg) was pulverized in liquid nitrogen, thawed on ice, and mixed with 1.0 ml of RIPA buffer with protease inhibitors (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.5 mmol/L phenylmethylsulfonyl fluoride, 10 μmol/L pepstatin, 4 μmol/L aprotinin, 80 μmol/L leupeptin, and 5 mmol/L benzamidine). Tissue was homogenized on ice for 30 seconds using a Biohomogenizer (Biospec Products, Inc., Bartlesville, OK). Two μl of 0.2 mol/L phenylmethylsulfonyl fluoride were added and homogenates incubated 30 minutes on ice. Homogenates were centrifuged at 10,000 × g for 20 minutes; supernatants were collected and centrifuged to prepare final lysates. Supernatant protein (100 μg) from each sample was electrophoresed in 12% sodium dodecyl sulfate-polyacrylamide gels and electroblotting to Immobilon-P membrane (Millipore Corp., Bedford, MA). Immunodetection used AR mAb F39.4.1 at 1:10,000 dilution. Secondary antibody (goat anti-mouse IgG conjugated to horseradish peroxidase; Amersham Corp., Arlington Heights, IL) was used for detection by enhanced chemiluminescence (DuPont-NEN Research Products, Boston, MA).
Results
Average MOD and percentage of cells expressing AR (percent AR positivity) were determined in tumors from CWR22-bearing mice before and after castration (Table 1) ▶ . The majority of nuclei in CWR22 tumors from intact nude mice (98.5 ± 0.2%) showed intense staining of AR (MOD was 0.26 ± 0.01) (Figure 1) ▶ . On day 1 after castration, AR MOD decreased and remained low on day 4 after castration whereas AR percent positivity declined to a minimum on day 2 (2%) and remained low on day 4 (10%). On day 6 after castration, AR-positive nuclei increased sixfold to 63% and were distributed evenly throughout all tumor sections. AR positivity increased further to 71% on day 12. AR MOD decreased to a low of 0.11 on day 4 and remained low at 0.15 to 0.17 on days 12 through 120 after castration. Recurrent CWR22 tumors obtained ∼150 days after castration exhibited lower percent AR positivity (72.1 ± 7.6%) than CWR22 tumors from intact, androgen-stimulated mice (P = 0.03). However, among malignant nuclei expressing AR, MOD was similar (P = 0.99) in the original CWR22 under androgen stimulation and recurrent CWR22 in the absence of testicular androgens. Immunostaining of CWR22 tumors before and after castration yielded similar results when the polyclonal antibodies AR-52 and PG-21 were used instead of AR mAb F39.4.1 (data not shown). Western blotting (Figure 2) ▶ of CWR22 tumor lysates revealed similar AR levels in androgen-dependent and recurrent CWR22 tumors and reduced AR levels after castration until tumor recurrence.
Table 1.
Quantitation of Tumor Volume (cm3), AR Expression (AR MOD and %AR Positivity), Tumor Cellular Proliferation (%Ki-67 Positivity), and PSA Serum Levels (ng/ml) and Tissue Expression (%PSA Positivity) Measured in Androgen-Stimulated, Androgen-Deprived and Recurrent CWR22 Tumors*
| Days after castration | No. of tumors | Tumor volume | AR MOD | %AR positivity | %Ki-67 positivity | Serum PSA | %PSA positivity |
|---|---|---|---|---|---|---|---|
| intact CWR22 | 12 | 1.11 ± 0.94† | 0.26 ± 0.01‡ | 98.5 ± 0.2 | 73.5 ± 4.4 | 246.7 ± 55.3† | 17.4 ± 3.6 |
| Day 1 | 2 | 0.15 ± 0.10 | 28.5 ± 7.4 | 56.9 ± 7.5 | 3.4 ± 1.0 | ||
| Day 2 | 4 | 0.81 ± 0.09 | 0.16 ± 0.08 | 2.3 ± 5.5 | 26.1 ± 5.6 | 169.0 ± 53.7 | 5.5 ± 2.8 |
| Day 4 | 2 | 0.11 ± 0.09 | 9.9 ± 8.6 | 9.4 ± 8.1 | 4.8 ± 1.1 | ||
| Day 6 | 6 | 0.72 ± 0.49 | 0.15 ± 0.06 | 62.7 ± 4.8§ | 0.8 ± 0.5 | 109.9 ± 76.3 | 5.6 ± 1.5 |
| Day 12 | 6 | 0.81 ± 0.27 | 0.17 ± 0.06 | 70.9 ± 4.9§ | 0.3 ± 0.3 | 18.8 ± 10.8 | 0.8 ± 0.9 |
| Day 32 | 4 | 0.64 ± 0.25 | 0.17 ± 0.11 | 70.4 ± 7.3§ | 0.1 ± 0.3 | 5.6 ± 4.1 | 0.6 ± 0.3 |
| Day 64 | 2 | 0.15 ± 0.05 | 71.7 ± 12.2§ | 0.4 ± 0.2 | 1.1 ± 0.1 | ||
| Day 90 | 4 | 0.64 ± 0.30 | 0.17 ± 0.05 | 73.4 ± 6.5§ | 0.8 ± 0.5 | 11.1 ± 1.8 | 1.5 ± 0.7 |
| Day 120 | 4 | 0.78 ± 0.37 | 0.17 ± 0.03 | 72.3 ± 8.4§ | 3.3 ± 1.2 | 21.3 ± 4.1∥ | 3.4 ± 0.1∥ |
| Recurrent CWR22 | 12 | 1.63 ± 0.38 | 0.26 ± 0.01¶ | 72.1 ± 7.6§ | 49.1 ± 7.4¶ | 261.5 ± 123.0¶ | 7.2 ± 1.3¶ |
*AR mean optical density (MOD), percent AR positivity, percent Ki-67 positivity, serum PSA level, and percent PSA positivity at all time points after castration decreased significantly (P < 0.01) compared to intact CWR22 except no significant differences were found for AR MOD in recurrent CWR22 and percent AR positivity on days 90 and 120 after castration compared to day 0 (P > 0.05).
†Tumor volume and serum PSA is described by mean ± SD and therefore the data for time points containing only two measurements were not presented.
‡The image analysis data for all nuclei for all tumors at each time point is described by mean ± SD. Each tumor is represented by 20 images containing 200 to 250 nuclei for AR and 400 to 500 nuclei for Ki-67.
§Percent AR positivity on days 6, 12, 32, 64, 90, and 120 after castration and upon recurrence increased significantly (P < 0.001) compared to days 1, 2, and 4 after castration.
¶Recurrent CWR22 showed a significant increase in AR MOD, percent Ki-67 positivity, serum PSA level, and percent PSA positivity compared to time points after castration (P < 0.01).
∥Serum PSA level and percent PSA positivity on day 120 after castration were significantly higher than on day 90 after castration (P < 0.05).
Figure 1.
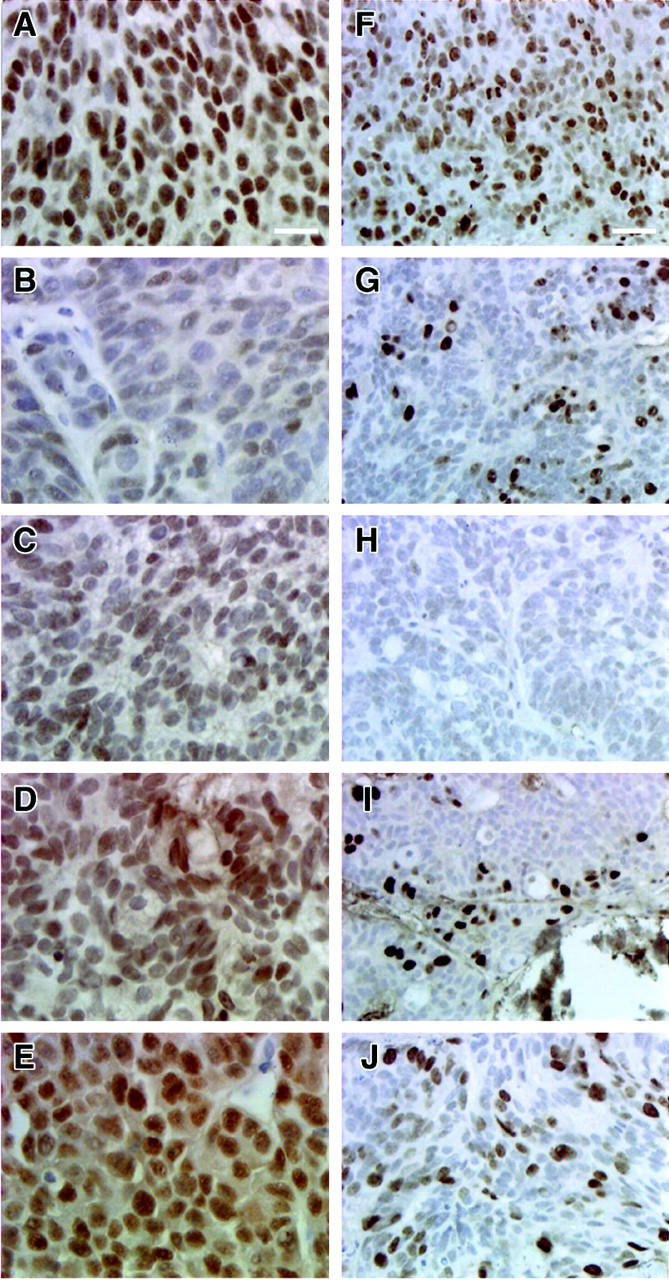
AR and Ki-67 immunohistochemistry of CWR22 tumors. AR expression was similar in androgen-dependent and recurrent tumors. AR-positive cells decreased to a minimum on day 2 after castration. Nuclear localization of AR returned on day 6 after castration but its intensity was lower than in androgen-dependent tumors. Higher percentages of AR-positive cells with lower levels of AR-staining intensity were recognized on days 32, 64, 90, and 120 after castration. Left: AR immunohistochemistry (scale bar, 10 μm). A: CWR22 before castration. B: Day 2 after castration. C: Day 6 after castration. D: Day 120 after castration. E: Recurrent CWR22 ∼150 days after castration. MIB-1 detection of the Ki-67 nuclear proliferation antigen showed similar rates of cellular proliferation in T-stimulated and recurrent tumors. Proliferation decreased on day 2 after castration and reached a level that was barely detectable on days 6 and 12 after castration. Proliferation was detectable as small nests of Ki-67-positive cells on day 120 after castration. Right: Ki-67 immunohistochemistry (scale bar, 20 μm). F: CWR22 before castration. G: Day 2 after castration. H: Day 6 after castration. I: Day 120 after castration. J: Recurrent CWR22 ∼150 days after castration.
Figure 2.

Western immunoblot analysis of AR protein in CWR22 tumors. An AR protein of 110 to 114 kd was present in lysates of androgen-dependent CWR22 tumors from intact mice. AR protein decreased after castration until tumor recurred ∼150 days after castration. AR protein levels found in recurrent tumors in the absence of androgens were similar to those found in the original androgen-dependent tumors. The position of the molecular mass marker (kd) is indicated. This experiment was performed with two to six different tumors at each time point with similar results.
The number of cells expressing Ki-67 in CWR22 tumors before and after castration were summarized in Table 1 ▶ . Ki-67 positivity was high in CWR22 tumors from intact nude mice (Figure 1) ▶ , decreased gradually on days 1 to 4 after castration, and remained at low or undetectable levels on day 6 through day 90 after castration. Although most CWR22 cells were in growth arrest, proliferating cells occurred randomly at a frequency <1% beyond day 6 until day 120 when multiple foci of 5 to 20 proliferating cells were detected throughout the tumors (Figure 1) ▶ . Recurrent CWR22 tumors, compared to the original CWR22 tumors, exhibited lower cellular proliferation (49.1 ± 7.4%, P = 0.006). Mean serum PSA levels (261.5 ± 123.0 ng/ml) of mice bearing recurrent tumors (n = 6) increased to levels similar to those of tumor bearing intact mice (246.7 ± 55.3 ng/ml) (Table 1) ▶ .
Foci of recurrent cellular proliferation, as indicated by Ki-67 staining, appeared on day 120 day after castration. When these proliferating foci were immunostained for PSA expression using double-staining immunohistochemistry, positive cytoplasmic staining for PSA was observed in the foci that contained Ki-67-positive cells (Figure 3) ▶ . Moreover, the appearance of proliferating cells on day 120 after castration was associated with increased (P = 0.01) serum PSA levels (24 ± 3 ng/ml) compared to day 90 after castration (11 ± 2 ng/ml).
Figure 3.

PSA and Ki-67 double immunohistochemistry of CWR22 tumors harvested from a mouse on day 120 after castration. Tissue PSA expression was visualized with diaminobenzidine tetrahydrochloride (cytoplasmic brown staining, white arrows) and Ki-67 expression was visualized with AEC (dark red nuclear staining, black arrows). Counterstaining was done with hematoxylin. Proliferating tumor cells emerged from the same foci where PSA was expressed (scale bar, 20 μm). Ki-67 immunostaining from the double immunohistochemistry showed a pattern of numerous blobs in the nucleoplasm rather than a typical uniform nuclear staining. Protease treatment during PSA immunohistochemistry may have degraded Ki-67 protein.
Recurrent CWR22 tumors showed more heterogeneous AR positivity, AR MOD, and Ki-67 positivity (Figure 4 ▶ ; A, B, and C) than CWR22 tumors from intact mice before castration (F-tests; P < 0.001, 0.05, and 0.05, respectively). Intertumor heterogeneity of cellular proliferation in recurrent CWR22 was reflected in serum PSA levels that ranged from 25 to 740 ng/ml when measured in recurrent tumor-bearing mice. Throughout the study period, AR MOD showed a parallel trend with Ki-67 positivity (r = 0.64, P = 0.03) and PSA levels (r = 0.77, P = 0.02) (Table 1) ▶ . In addition, Ki-67 positivity was highly correlated with PSA levels (r = 0.90, P = 0.002).
Figure 4.
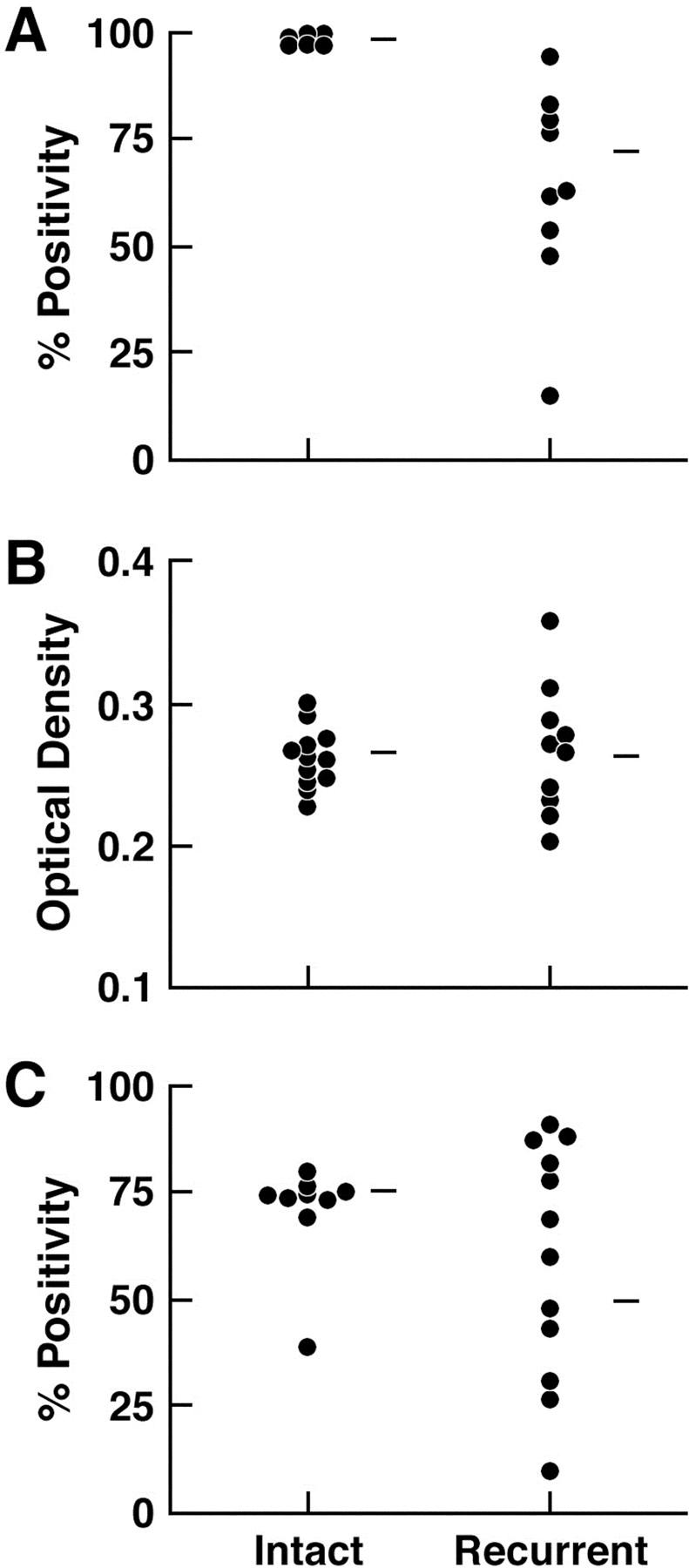
Comparison of heterogeneity of AR expression and Ki-67 expression in CWR22 specimens from intact mice and on recurrence ∼150 days after castration. A: Mean percent AR positivity. B: MOD of AR. C: Mean percent Ki-67 positivity.
Immunopositivity of PSA was highest in tumors from intact mice, decreased after castration, and remained low until sometime between 90 to 120 days after castration when an increase was noted within foci of proliferating cells. Immunopositivity of PSA was significantly correlated with AR MOD (r = 0.658, P = 0.028), Ki-67 positivity (r = 0.773, P = 0.006), and serum PSA levels (r = 0.818, P = 0.014).
Discussion
In the present study, the temporal relationship between AR expression and cellular proliferation was investigated in CWR22 CaP xenografts during the transition from androgen-dependent to recurrent growth. Reduced AR protein immunostaining in CWR22 tumors after castration was similar to that observed in ventral prostates of castrated rats and mice. 5,26,27 However, on day 6 after castration, percent AR positivity increased several fold and remained at higher levels on days 12, 32, 64, 90, and 120. Although AR was expressed in most nuclei, nuclear AR staining (MOD) remained relatively low throughout the period of tumor regression after castration. Proliferation of tumor cells ceased by day 6 after castration and remained at low to undetectable levels through day 90. Thus, during the period of tumor remission after androgen deprivation, nuclear AR levels were low and tumors remained quiescent. Agus and associates 28 reported in their studies of cell-cycle regulators in the CWR22 xenograft that cellular proliferation assessed using Ki-67 and visual scoring fell to very low levels by day 10 after castration and remained so through day 30 after castration. The percentage of cells that stained intensely with AR mAb (F39.4.1) was low (10%) on days 3, 5, 7, and 10 after castration and reached the level of intact tumors by day 25 and thereafter. The difference between their and our reports may result from their dependence on visual scoring of AR immunostaining or their use of higher concentrations of AR mAb (2 μg/ml) that may have increased nonspecific staining. 20
In recurrent tumors 150 days after castration, AR expression and cellular proliferation were more heterogeneous than in tumors from intact mice; recurrent tumors exhibited lower percent-positive nuclei and greater variation between tumors than CWR22 tumors from intact mice. However, among nuclei immunostained for AR, there was no difference in AR MOD between intact and recurrent tumors. Previous studies have established that AR activation subsequent to binding of androgen results in increased nuclear levels of AR, 29 homodimerization of AR, 30 and binding of AR to DNA sequences that function as enhancers for AR-induced transcriptional activation. 1,2,31 The similar levels of AR expression in intact and recurrent tumors suggests that nuclear AR may be stabilized despite the absence of testicular androgen in recurrent CWR22 by a ligand-independent or synergistic mechanism. AR activation is linked closely to stabilization of AR protein; binding of androgen stabilizes AR causing it to have a slower rate of degradation. 32 AR MOD, by its definition, represents a mean nuclear staining intensity relative to cytoplasm. 20 Therefore, increased AR MOD may reflect AR activation. Another possible mechanism of increased AR expression in recurrent CWR22 is AR gene amplification. AR gene amplification was detected in 7 of 23 cases of recurrent human CaP whereas none was detected before androgen-deprivation therapy. 33 However, AR gene amplification in recurrent CWR22 tumors was not detected using Southern blot analysis and competitive reverse transcriptase-polymerase chain reaction. 34
Because AR was expressed in a lower percentage of cells and at a lower MOD in those cells expressing AR on days 1 to 4 after castration and AR re-expression occurred on gross tumor recurrence, the temporal relationship between recovery of AR expression and the onset of cellular proliferation should provide insight into the role of AR in CaP recurrence after castration. On day 120 after castration, when recurrent tumor growth was indicated by increased serum PSA but tumor sites did not yet demonstrate growth grossly, small foci of tumor cells were recognized in harvested samples that immunostained for Ki-67 and expressed PSA. These findings suggested these foci were precursors of the recurrent CWR22 tumor that appears grossly ∼150 days after castration. Two groups of investigators have reported on the relationship between serum PSA and tumor recurrence after castration in the CWR22 model. Serum PSA increase before gross tumor recurrence in the CWR22 model was reported first by Nagabhushan and associates. 12 The time course of these events cannot be compared to our results because they performed castration at higher tumor volumes. Agus and associates 28 castrated CWR22 tumor-bearing mice at tumor volumes similar to our studies. They reported increased serum PSA ∼115 days after castration that preceded tumor recurrence recognized grossly ∼20 days later. We reported previously that PSA is one of several known androgen-regulated genes whose mRNA was expressed at increased levels in recurrent CWR22 despite the absence of testicular androgen. 3 In the current study, these RNA findings were confirmed by PSA protein quantitation at the tissue level. PSA is a well known androgen-regulated gene, 35,36 however, other factors such as vitamin D 37 and transforming growth factor-β1 38 have been shown to be involved in transcriptional regulation of the PSA gene. One or more of these same factors may also be an initiator of recurrent growth. Nonetheless, coincidental increased PSA serum levels and tissue expression and the recurrence of tumor cellular proliferation might be caused by the same mechanisms, one of which is reactivation of AR.
Understanding the mechanisms driving recurrent growth is one of the most important issues in CaP research. 39-41 Because AR is a growth-stimulating transcription factor in CaP, reactivation of AR in the absence of testicular androgen could be one of the molecular events that initiates cellular proliferation and leads to tumor recurrence. Several mechanisms have been proposed for activation of AR in the absence of testicular androgen. AR mutations that alter ligand specificity may influence tumor progression subsequent to androgen deprivation by making AR more responsive to adrenal androgens. CWR22 cells express a mutant AR (His 874 to Tyr) that has normal transcriptional activity in response to testosterone and dihydrotestosterone but has altered ligand specificity making it more sensitive to activation by adrenal androgens including dehydroepiandrosterone. 13 Alternatively, a ligand-independent mechanism might cause transcriptional activation of AR. Protein kinase A and C modulators might activate AR in the absence of ligand by altering phosphorylation of AR 42-45 or AR co-activators. 46,47 Stimulation of PKA activity resulted in activation of the N-terminal domain of AR in LNCaP cells. 48 Transfected AR was reported to be activated by insulin-like growth factor I, epidermal growth factor, and keratinocyte growth factor in DU-145 cells and PSA was increased by insulin-like growth factor I in LNCaP cells. 49 Overexpression of HER-2/neu receptor tyrosine kinase was reported to increase expression of PSA and enhance growth in the androgen-dependent human CaP LAPC-4 xenograft. 50 These effects required AR expression and seemed to occur through cross-talk between the AR and HER-2/neu pathways. We reported recently that high-level expression, increased stability, and nuclear localization of AR in recurrent tumor cells were associated with increased sensitivity to the growth-promoting effects of dihydrotestosterone at concentrations as low as the femtomolar range. 34 Additionally, we have shown that high expression of transcriptional intermediary factor 2 and steroid receptor coactivator 1 in recurrent CaP increases AR transactivation in response to physiological concentrations of adrenal androgens or other steroids with affinity for AR. 51 A single event or combination of events that affect AR function may lead to recurrent tumor growth in the absence of testicular androgen.
The re-expression of AR and known androgen-regulated genes in the 150-day recurrent CWR22 3,14,15 suggests that AR reactivation has a role in stimulating recurrent tumor growth. AR MOD decreased after castration and failed to increase until sometime between 120 days after castration and gross tumor recurrence. At the 120-day time point, if proliferating cells exhibited increased AR MOD, their rarity would preclude detection because <5% of malignant cells were proliferating. Quantitation of antigens using double-staining immunohistochemistry is technically difficult; our attempts to measure AR expression in Ki-67-immunopositive versus immunonegative cells have been unsuccessful thus far. However, if the small foci of tumor cells on day 120 after castration are precursors of recurrent tumors, direct comparison of AR, androgen-regulated gene products, and other molecules in these foci of cellular proliferation versus other regions of the tumor may be useful to evaluate specific mechanisms driving recurrent growth. Further study of this transition from androgen-dependent to androgen-independent growth in CWR22 may provide valuable insights into the mechanism of androgen-deprivation treatment failure in patients.
Acknowledgments
We thank Dr. Madhabananda Sar for technical advice on immunohistochemistry; Dr. Charles Bagnell for help with image registration; Patricia Magyar, Yeqing Chen, Gail Grossman, and Natalie Edmund for technical assistance; Dominic Moore and Dr. Michael Schell for statistical assistance; and Sidney Holdrege for assistance with manuscript preparation.
Footnotes
Address reprint requests to James L. Mohler, M.D., University of North Carolina, Division of Urology, CB# 7235, Chapel Hill, NC 27599-7235. E-mail: jmohler@med.unc.edu.
Supported by the National Institutes of Health [grants RO1-AG-11343 (to F. S. F., J. L. M.), RO1-CA-64865 (to G. J. S., J. L. M.), P01-CA-77739 (to J. L. M., F. S. F., G. J. S.) and P30-HD-18968 (DNA and Tissue Culture Cores)], the United States Army Medical Research and Materiel Command (grant 98-1-8538 to J. L. M.), and the American Foundation for Urologic Disease and Merck U.S. Human Health (to C. W. G.).
D. K. and C. W. G. contributed equally to this work.
References
- 1.Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS: Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 1995, 16:271-321 [DOI] [PubMed] [Google Scholar]
- 2.Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, Bi BY, Chatterjee B: Regulation of androgen action. Vitam Horm 1999, 55:309-352 [DOI] [PubMed] [Google Scholar]
- 3.Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, French FS: Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res 1998, 58:5718-5724 [PubMed] [Google Scholar]
- 4.Eid MA, Kumar MV, Iczkowski KA, Bostwick DG, Tindall DJ: Expression of early growth response genes in human prostate cancer. Cancer Res 1998, 11:2461-2468 [PubMed] [Google Scholar]
- 5.Prins GS, Birch L: Immunocytochemical analysis of androgen receptor along the ducts of the separate rat prostate lobes after androgen withdrawal and replacement. Endocrinology 1993, 132:169-178 [DOI] [PubMed] [Google Scholar]
- 6.Kyprianou N, Isaacs JT: Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology 1988, 122:552-562 [DOI] [PubMed] [Google Scholar]
- 7.Hobisch A, Culig Z, Radmayr C, Bartsch G, Klocker H, Hittmair A: Androgen receptor status of lymph node metastases from prostate cancer. Prostate 1996, 28:129-135 [DOI] [PubMed] [Google Scholar]
- 8.Ruizeveld de Winter JA, Janssen PJA, Sleddens HMEB, Verleun-Mooijman MCT, Trapman J, Brinkmann AO, Santerse AB, Schröder FH, van der Kwast TH: Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer Am J Pathol 1994, 144:735–746 [PMC free article] [PubMed]
- 9.DeVere White RW, Myers F, Chi S-G, Chamberlain S, Siders D, Lee F, Stewart S, Gumerlock PH: Human androgen receptor expression in prostate cancer following androgen ablation. Eur Urol 1997, 31:1-6 [DOI] [PubMed] [Google Scholar]
- 10.Pretlow TG, Wolman SR, Micale MA, Pelley RJ, Kursh ED, Resnick MI, Bodner DR, Jacobberger JW, Delmoro CM, Giaconia JM: Xenografts of primary human prostatic carcinoma. J Natl Cancer Inst 1993, 85:394-398 [DOI] [PubMed] [Google Scholar]
- 11.Wainstein MA, He F, Robinson D, Kung H-J, Schwartz S, Giaconia JM, Edgehouse NL, Pretlow TP, Bodner DR, Kursh ED, Resnick MI, Seftel A, Pretlow TG: CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res 1994, 54:6049-6052 [PubMed] [Google Scholar]
- 12.Nagabhushan M, Miller CM, Pretlow TP, Giaconia JM, Edgehouse NL, Schwartz S, Kung HJ, de Vere White RW, Gumerlock PH, Resnick MI, Amini SB, Pretlow TG: CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res 1996, 56:3042-3046 [PubMed] [Google Scholar]
- 13.Tan J-A, Sharief Y, Hamil KG, Gregory CW, Zang D-Y, Sar M, Gumerlock PH, deVere White RW, Pretlow TG, Harris SE, Wilson EM, Mohler JL, French FS: Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol 1997, 11:450-459 [DOI] [PubMed] [Google Scholar]
- 14.Gregory CW, Kim D, Ye P, D’Ercole AJ, Mohler JL, French FS: Androgen receptor up-regulates insulin-like growth factor binding protein-5 (IGFBP-5) expression in a human prostate cancer xenograft. Endocrinology 1999, 140:2372-2381 [DOI] [PubMed] [Google Scholar]
- 15.Amler LC, Agus DB, LeDue C, Sapinosa ML, Fox WD, Kern S, Lee D, Wang V, Leysens M, Higgins B, Martin J, Gerald W, Dracopoli N, Cordon-Cardo C, Scher HI, Hampton GM: Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R. Cancer Res 2000, 60:6134-6141 [PubMed] [Google Scholar]
- 16.Sadi MV, Barrack ER: Image analysis of androgen receptor immunostaining in metastatic prostate cancer. Heterogeneity as a predictor of response to hormonal therapy. Cancer 1993, 71:2574-2580 [DOI] [PubMed] [Google Scholar]
- 17.Tilley WD, Lim-Tio SS, Horsfall DJ, Aspinall JO, Marshall VR, Skinner JM: Detection of discrete androgen receptor epitopes in prostate cancer by immunostaining: measurement by color video image analysis. Cancer Res 1994, 54:4096-4102 [PubMed] [Google Scholar]
- 18.Prins GS, Sklarew RJ, Pertschuk LP: Image analysis of androgen receptor immunostaining in prostate cancer accurately predicts response to hormonal therapy. J Urol 1998, 159:641-649 [PubMed] [Google Scholar]
- 19.Magi-Galluzzi C, Xu X, Hlatky L, Hahnfeldt P, Kaplan I, Hsiao P, Chang C, Loda M: Heterogeneity of androgen receptor content in advanced prostate cancer. Mod Pathol 1997, 10:839-845 [PubMed] [Google Scholar]
- 20.Kim D, Gregory CW, Smith GJ, Mohler JL: Immunohistochemical quantitation of androgen receptor expression using color video image analysis. Cytometry 1999, 35:2-10 [DOI] [PubMed] [Google Scholar]
- 21.Sar M: Application of avidin-biotin complex technique for the localization of estradiol receptor in target tissues using monoclonal antibodies. Bullock GR Petrusz P eds. Techniques in Immunocytochemistry, 1985, vol 3.:pp 43-54 Academic Press, New York [Google Scholar]
- 22.Brigati DJ, Budgeon LR, Unger ER, Koebler D, Cuomo C, Kennedy T, Perdomo JM: Immunocytochemistry is automated: development of a robotic workstation based upon the capillary action principle. J Histotechnol 1988, 11:165-183 [Google Scholar]
- 23.Zegers ND, Claassen E, Neelen C, Mulder E, van Laar JH, Vorrhorst MM, Berrevoets CA, Brinkmann AO, van der Kwast TH, Ruizeveld de Winter JA, Trapman J, Boersma WJA: Epitope prediction and confirmation for the human androgen receptor: generation of monoclonal antibodies for multi-assay performance following the synthetic peptide strategy. Biochim Biophys Acta 1991, 1073:23-32 [DOI] [PubMed] [Google Scholar]
- 24.Gerdes J, Lemke H, Baisch H, Wacher HH, Schwab U, Stein H: Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984, 133:1710-1715 [PubMed] [Google Scholar]
- 25.Lundwall A: Characterization of the gene for prostate-specific antigen: a human glandular kallikrein. Biochem Biophys Res Commun 1989, 161:1151-1159 [DOI] [PubMed] [Google Scholar]
- 26.Sar M, Lubahn DB, French FS, Wilson EM: Immunohistochemical localization of the androgen receptor in rat and human tissues. Endocrinology 1990, 127:3180-3186 [DOI] [PubMed] [Google Scholar]
- 27.Takeda H, Nakamoto T, Kokontis J, Chodak GW, Chang C: Autoregulation of androgen receptor expression in rodent prostate: immunohistochemical and in situ hybridization analysis. Biochem Biophys Res Commun 1991, 177:488-496 [DOI] [PubMed] [Google Scholar]
- 28.Agus D, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde D, Scher H: Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst 1999, 91:1869-1876 [DOI] [PubMed] [Google Scholar]
- 29.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM: A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. J Biol Chem 1994, 269:13115-13123 [PubMed] [Google Scholar]
- 30.Wong C, Zhou ZX, Sar M, Wilson EM: Steroid requirement for androgen receptor dimerization and DNA binding. Modulation by intramolecular interactions between the NH2-terminal and steroid-binding domains. 1900, :4-19012 268: J Biol Chem 1993 [PubMed] [Google Scholar]
- 31.Avellar MCW, Gregory CW, Power SGA, French FS: Androgen-dependent protein interactions within an intron-1 regulatory region of the 20-kDa protein gene. J Biol Chem 1997, 272:17623-17631 [DOI] [PubMed] [Google Scholar]
- 32.Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM: Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol 1995, 9:208-218 [DOI] [PubMed] [Google Scholar]
- 33.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP: In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995, 9:401-406 [DOI] [PubMed] [Google Scholar]
- 34.Gregory CW, Johnson RT, Mohler JL, French FS, Wilson EM: Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res 2001, 61:2892-2898 [PubMed] [Google Scholar]
- 35.Young CY, Montgomery BT, Andrews PE, Qui SD, Bilhartz DL, Tindall DJ: Hormonal regulation of prostate-specific antigen messenger RNA in human prostatic adenocarcinoma cell line LNCaP. Cancer Res 1991, 51:3748-3752 [PubMed] [Google Scholar]
- 36.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J: Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem 1996, 271:6379-6388 [DOI] [PubMed] [Google Scholar]
- 37.Skowronski RJ, Peehl DM, Feldman D: Vitamin D and prostate cancer: 1,25 dihydroxyvitamin D3 receptors and actions in human prostate cancer cell lines. Endocrinology 1993, 132:1952-1960 [DOI] [PubMed] [Google Scholar]
- 38.Gleave ME, Hsieh JT, Wu HC, von Eschenbach AC, Chung LW: Serum prostate specific antigen levels in mice bearing human prostate LNCaP tumors are determined by tumor volume and endocrine and growth factors. Cancer Res 1992, 52:1598-1605 [PubMed] [Google Scholar]
- 39.Koivisto P, Kolmer M, Visakorpi T, Kallioniemi OP: Androgen receptor gene and hormonal therapy failure of prostate cancer. Am J Pathol 1998, 152:1-9 [PMC free article] [PubMed] [Google Scholar]
- 40.Klocker H, Culig Z, Eder IE, Nesssler-Menardi C, Hobisch A, Putz T, Bartsch G, Peterziel H, Cato ACB: Mechanism of androgen receptor activation and possible implications for chemoprevention trials. Eur Urol 1999, 35:413-419 [DOI] [PubMed] [Google Scholar]
- 41.Sadar MD: Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase: a signal transduction pathway. J Biol Chem 1999, 274:7777-7783 [DOI] [PubMed] [Google Scholar]
- 42.Ikonen T, Palvimo JJ, Kallio PJ, Reinikainen P, Janne OA: Stimulation of androgen-regulated transactivation by modulators of protein phosphorylation. Endocrinology 1994, 135:1359-1399 [DOI] [PubMed] [Google Scholar]
- 43.De Ruiter PE, Teuwen R, Trapman J, Dijkema R, Brinkmann AO: Synergism between androgens and protein kinase C on androgen-regulated gene expression. Mol Cell Endocrinol 1995, 110:51-56 [DOI] [PubMed] [Google Scholar]
- 44.Nazareth LV, Weigel NL: Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem 1996, 271:19900-19907 [DOI] [PubMed] [Google Scholar]
- 45.Culig Z, Hobisch A, Hittmair A, Cronauer MV, Radmayr C, Zhang J, Bartsch G, Klocker H: Synergistic activation of androgen receptor by androgen and leutinizing hormone-releasing hormone in prostatic carcinoma cells. Prostate 1997, 32:106-114 [DOI] [PubMed] [Google Scholar]
- 46.Rowan BG, Weigel NL, O’Malley BW: Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem 2000, 275:4475-4483 [DOI] [PubMed] [Google Scholar]
- 47.Rowan BG, Garrison N, Weigel NL, O’Malley BW: 8-bromo-cyclic AMP induces phosphorylation of two sites in SRC-1 that facilitate ligand-independent activation of the chicken progesterone receptor and are critical for functional cooperation between SRC-1 and CREB binding protein. Mol Cell Biol 2000, 20:8720-8730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadar MD, Hussain M, Bruchovsky N: Prostate cancer: molecular biology of early progression to androgen independence. Endocr-Relat Cancer 1999, 6:487–502 [DOI] [PubMed]
- 49.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H: Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor and epidermal growth factor. Eur Urol 1995, 27(Suppl 2):S45-S47 [DOI] [PubMed] [Google Scholar]
- 50.Craft N, Shostak Y, Carey M, Sawyers CL: A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signalling by the HER-2/neu tyrosine kinase. Nat Med 1999, 5:280-285 [DOI] [PubMed] [Google Scholar]
- 51.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM: A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res 2001, 61:4315-4319 [PubMed] [Google Scholar]