

Abstract
Differential gene expression can be investigated effectively by cDNA arrays. Because tissue homogenates result inevitably in an average expression of a bulk of different cells, we aimed to combine mRNA profiling with cell-type-specific microdissection. Using a polymerase chain reaction (PCR)-based preamplification technique, the expression profile was shown to be preserved. We modified the existing protocol enabling to apply the total amount of extracted RNA from microdissected cells. A mean amplification factor of nearly 1000 allowed to reduce the demand of initial RNA to ∼10 ng. This technique was used to investigate intrapulmonary arteries from mouse lungs (∼500 cell equivalents). Using filters with 1176 spots, three independent experiments showed a high consistency of expression for the preamplified cDNAs. These profiles differed primarily from those of total lung homogenates. Additionally, in experimental hypoxia-induced pulmonary hypertension, amplified cDNA from intrapulmonary vessels of these lungs was compared to cDNA from vessels dissected from normoxic lungs. Validation by an alternative method was obtained by linking microdissection with real-time polymerase chain reaction (PCR). As suggested by the array data, nine selected genes with different factors of up-regulation were fully confirmed by the PCR technique. Thus, a rapid protocol is presented combining microdissection and array profiling that demands low quantities of initial RNA to assess reliably cell-type-specific gene regulation even within nonneoplastic complex tissues.
Oligonucleotide and cDNA arrays allow an effective investigation of functional genomics. 1 They determine differential expression of a multitude of genes, using a systematic and comprehensive approach, and give an open-minded look at the field of research. This enables rapid elucidation of regulation modalities associated with special conditions and developmental states, revealing important candidate genes or gene clusters with impact on further basic research as well as clinical applications. 2-4 Moreover, understanding of the intercellular cross-talk within complex tissues is highly challenging because it may substantially differ from culture models. However, the application of tissue homogenates results inevitably in averaging of the expression of different cell types, and the expression profile of a specific cell type may be primarily masked or even lost because of the bulk of surrounding cells.
To select cell types from complex tissues, microdissection techniques have been successfully used. Within a few years these techniques have become an accepted tool aiming to investigate complex tissues in a more detailed manner. They have been shown to isolate precisely single cells or cell clusters under optical control for use in several downstream applications for DNA, RNA, and protein analysis. 5-7 In particular, the fragile mRNA can be obtained in a quality that is even suitable for construction of cDNA libraries or array hybridization.
As the amount of RNA from microdissected material is often limited and not sufficient for hybridization, a preamplification technique has to be incorporated. This ideally results in an accurate amplification of all RNAs, thus representing the original mRNA pool and preserving the expression profile. The T7-based linear amplification 8,9 was the first approach to be shown to generate a representative mRNA profile that allows to combine microdissection with array technology. 10,11 As the linear amplification is very time consuming and susceptible to various disturbances and even failures leading to RNA loss and degradation, a PCR-based technique was suggested. 12 This approach uses the ability of reverse transcriptase to add nucleotides to the 3′ end of the cDNA strand and allows a second primer to add. Thus, cDNAs with defined 5′ and 3′ ends for PCR amplification are obtained (commercially available as SMART PCR; Clontech, Palo Alto, CA). 13 It was shown that high-, medium-, and low-abundance transcripts are amplified in a representative manner 14 and that full-length cDNAs are provided. 13,15 Moreover, application of SMART-amplified cDNA to arrays resulted in a very high homology of the expression profile of several hundred genes when compared to unamplified cDNA 16,17 indicating that the relative proportions of the mRNAs were maintained.
In intact mouse lungs, originating from control animals and those with pulmonary hypertension because of chronic hypoxia, we aimed to establish a rapid and reproducible protocol that allows a reliable mRNA profiling of intrapulmonary arteries containing a limited amount of microdissected cell profiles. Hereby, the application to nonneoplastic tissue required a special effort in precision because message regulation often varies in a smaller extent compared to tumors. Laser-assisted microdissection was used for isolation of the vessels that represent only a minor portion of the lung tissue. Using SMART PCR for preamplification, we modified the protocol to introduce the entire cDNA to PCR amplification and subsequent array hybridization. To validate differential gene expression measured by cDNA arrays, the results were verified by a second independent technique based on microdissection in combination with relative mRNA quantitation by real-time PCR. 18
Materials and Methods
Lung Preparation of Mice under Hypoxia/Normoxia
All animal experiments were approved by the local authorities (Regierungspräsidium Giessen, no. II25.3-19c20-15 1 GI20/10-Nr.22/2000). Male BALB/cAnNCrlBR mice (20 to 22 g; Charles River, Sulzfeld, Germany) were exposed to normobaric hypoxia [inspiratory O2 fraction (FiO2) 0.10] in a ventilated chamber. The level of hypoxia was held constant by an autoregulatory control unit (O2 controller model 4010; Labotect, Göttingen, Germany), supplying either nitrogen or oxygen. Excess humidity in the recirculating system was prevented by condensation in a cooling system. CO2 was continuously removed by soda lime. Mice exposed to normobaric normoxia were kept in a similar chamber at an FiO2 of 0.21. After 21 days, the animals were intraperitoneally anesthetized with 180 mg of sodium pentobarbital/kg body weight. A cannula was inserted into the trachea by tracheostomy, a midline sternotomy was performed, and the lungs were flushed via a catheter in the pulmonary artery with Krebs Henseleit buffer (125.0 mmol/L NaCl, 4.3 mmol/L KCl, 1.1 mmol/L KH2PO4, 2.4 mmol/L CaCl2, 1.3 mmol/L MgCl2, and 13.32 mmol/L glucose) at a pressure of 20 cm H2O at room temperature. It was equilibrated with a gas mixture of 1% O2, 5.3% CO2, balanced N2. NaHCO3 was adjusted to result in a constant pH range of 7.37 to 7.40. During perfusion of the lungs the buffer was allowed to drain freely from a catheter in the left ventricle. Once the effluent was clear of blood, 800 μl of prewarmed TissueTek (Sakura Finetek, Zoeterwoude, The Netherlands) were instilled into the airways via the tracheal cannula. After ligation of the trachea, the lungs were excised and immediately frozen in liquid nitrogen. Preparation of the hypoxic animals was continuously performed in the hypoxic environment. The right ventricular wall was trimmed from the left ventricle plus septum to calculate the ratio of right ventricle wall/(left ventricle plus septum) of the dried heart tissue. Right heart hypertrophy after 21 days of hypoxia was additionally ascertained by separate experiments including normoxic and hypoxic animals. Data were compared by a paired t-test.
Isolated Lung Perfusion and Measurement of Pulmonary Artery Pressure
In separate experiments pulmonary artery pressure was measured in an in situ isolated lung preparation. Mice were deeply anesthetized intraperitoneally with sodium pentobarbital and anti-coagulated with heparin (1000 U/kg) by intravenous injection. After placing on a heated table (37°C), animals were intubated via tracheostoma and ventilated with room air (300 μl tidal volume, 90 breaths/min, and 3 cmH2O positive end-expiratory pressure). Midsternal thoracotomy was followed by insertion of catheters into the pulmonary artery and left atrium. Using a peristaltic pump (ISM834A V2.10; Ismatec, Glattbrugg, Switzerland), buffer perfusion via the pulmonary artery was started at 4°C and a flow of 0.2 ml/min. The buffer contained 120 mmol/L NaCl, 4.3 mmol/L KCl, 1.1 mmol/L KH2PO4, 2.4 mmol/L CaCl2, 1.3 mmol/L MgCl2, and 2.4 g/L of glucose as well as 5% (w/v) hydroxyethylamylopectin (molecular weight, 200,000). NaHCO3 was adjusted to result in a constant pH range of 7.37 to 7.40. In parallel with the onset of artificial perfusion, ventilation was changed from room air to a mixture of 5.3% CO2, 21.0% O2, balanced N2. After rinsing the lungs with ≥20 ml of buffer, the perfusion circuit was closed for recirculation (total system volume, 13 ml) and left atrial pressure was set at 2.0 mm Hg. Meanwhile, the flow was slowly increased from 0.2 to 2 ml/min and the entire system heated to 37°C. Pressures in pulmonary artery and left atrium were registered via transducers. The given pulmonary artery pressure values were taken after an initial steady state period of 20 minutes. Data were recorded from normoxic mice and from mice after 21 days of chronic hypoxia and analyzed by a paired t-test.
Laser-Assisted Microdissection
Microdissection was performed as described in detail previously. 19,20 In brief, cryosections (6 μm) from lung tissue were mounted on glass slides. After hemalaun staining for 45 seconds, the sections were subsequently immersed in 70% and 96% ethanol and stored in 100% ethanol until use. Not more than 10 sections were prepared at once to restrict the storage time. Intrapulmonary arteries with a diameter of 250 to 500 μm were selected and microdissected under optical control using the Laser Microbeam System (P.A.L.M., Bernried, Germany). Afterward, the vessel profiles were isolated by a sterile 30-gauge needle. Needles with adherent vessels were transferred into a reaction tube containing 200 μl of RNA lysis buffer (Figure 1) ▶ .
Figure 1.

Laser-assisted microdissection of small intrapulmonary arteries. a: A vessel profile is selected. b: The laser has cut along the outer side of the tunica media. c: A sterile needle is approximated to isolate the vessel. d: Needle with adherent vessel is lifted and afterward transferred to a reaction tube. Original magnifications, ×20.
mRNA Extraction
Lysis buffer for mRNA extraction consisted of 4 mol/L of guauidine thiocyanate (GTC), 25 mmol/L of Na3 citrate, 0.5% sarcosyl, and 0.72% β-mercaptoethanol. After incubation for 10 minutes at room temperature, 20 μl of 2 mol/L NaAc, 220 μl of phenol (pH 4.3), and 60 μl of chloroform/isoamylalcohol (24:1) were added. The samples were vortexed and centrifuged for 15 minutes at 4°C. The aqueous layer was collected, 1 μl of glycogen (1 mg/ml) added, and afterward precipitated with 200 μl of isopropanol. Samples were frozen for 1 hour at −20°C and centrifuged for 15 minutes. The pellets were washed with 75% ethanol and air-dried. After resuspension in 10 μl of H20, DNase digestion (1 U, 30 minutes, 37°C; Ambion, Austin, TX) was performed. Afterward, extraction was repeated and RNA was diluted in 4 μl of H2O.
cDNA Synthesis and Amplification
Total RNA was reverse-transcribed using the SMART PCR cDNA Synthesis Kit (Clontech) with slight modifications. Four μl of total RNA, 1 μl of cDNA Synthesis (CDS) Primer (diluted to a concentration of 5 μmol/L), and 1 μl of SMART II oligonucleotide (diluted to a concentration of 5 μmol/L) were mixed and incubated at 70°C for 8 minutes. After short spinning, 2 minutes on ice and 2 minutes at 42°C, a master mix containing 2 μl of 5× buffer, 1 μl dithiothreitol (20 mmol/L), 1 μl dNTP (10 mmol/L) and 0.5 μl RNase H− Moloney murine leukemia virus reverse transcriptase (PowerScript, Clontech) was added and incubated at 42°C for 1 hour. Afterward, cDNA was mixed with 38.5 μl of TE buffer (10 mmol/L Tris, pH 7.6, 1 mmol/L ethylenediaminetetraacetic acid) and purified by the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). Therefore, 250 μl of buffer PB were added to the cDNA to load a column. According to the manufacturer‘s protocol, the columns were washed once. For elution, 45 μl of elution buffer (10 mmol/L Tris, pH 8.5) were applied to the center of the column, incubated for 2 minutes, and centrifuged. To improve the recovery, this step was repeated using the first eluate again.
From the eluted cDNA (∼44 μl), 2 μl were separated for further determination of the amplification factor. For the PCR-based amplification, the remaining 42 μl of cDNA were mixed with 5 μl of 10× buffer, 1 μl PCR primer (10 μmol/L), 1 μl dNTP (10 mmol/L), and 1 μl Advantage 2 polymerase mix. PCR conditions were 95°C for 1 minute, followed by 19 cycles with 95°C for 15 seconds, 65°C for 30 seconds, and 68°C for 3 minutes. The resulting PCR product was purified using the QIAquick columns as described above. Forty-four μl of elution buffer were applied twice for elution and 2 μl were separated for determination of the amplification factor. All incubations were performed with a GeneAmp 2400 PCR cycler (PE Applied Biosystems, Foster City, CA).
Probe Labeling and Array Hybridization
For array hybridization, we used nylon filters with 1176 spotted cDNA (Mouse 1.2 II Atlas cDNA Arrays, Clontech). The purified PCR product was labeled with α-32P dATP using the Atlas SMART Probe Amplification Kit (Clontech). Forty-two μl of PCR product and 1 μl of CDS primer were heated at 95°C for 8 minutes. After 2 minutes at 50°C, a master mix containing 6 μl of 10× labeling buffer, 5 μl of dNTP (without dATP), 4 μl of α-32P dATP (Amersham Pharmacia Biotech, Freiburg, Germany), and 1 μl of Klenow enzyme were added, mixed, and incubated for 30 minutes. Reaction was stopped by applying 2 μl of 0.5-mol/L ethylenediaminetetraacetic acid. Labeled DNA was purified by QIAquick columns as described above, eluted twice with 100 μl of elution buffer, and resulted in ∼5 to 8 × 10 6 cpm. Afterward, array hybridization was performed according to the protocol. Filters with 32P-labeled PCR product were incubated at 68°C overnight. They were washed three times in 500 ml of 2× standard saline citrate and 1% sodium dodecyl sulfate at 68°C for 30 minutes. Finally, they were wrapped in plastic and exposed to an imaging plate (Fuji Photo Film, Tokyo, Japan) in lead sheathing. The film was read with a phosphorimaging system (BAS RPI 1000, Fuji Photo Film).
Array Analysis
Analysis was performed using the AtlasImage 1.5/2.0 software (Clontech). Global normalization was calculated by the sum method. For both arrays, differences of signal intensity minus background were added for all values over background. Afterward, normalization coefficient was determined by calculating the ratio of array 1 sum and array 2 sum. After normalization, background was substracted, ratio threshold was set at 2, and difference threshold was set at 550.
Determination of the Amplification Factor
Based on the following equation, we used comparative quantitation (ΔCT). Real-time PCR was performed by the Sequence Detection System 7700 (PE Applied Biosystems).
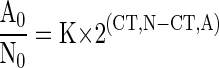 |
where Ao is initial number of cDNA copies after amplification; No is the initial number of nonamplified cDNA copies; CT,A is the threshold cycle of amplified PCR product; CT,N is the threshold cycle of nonamplified cDNA; and K is constant.
Applying either 2 μl of nonamplified cDNA or 2 μl of amplified PCR product, 25 μl Universal Master Mix (Applied Biosystems), porphobilinogen deaminase (PBGD) forward primer and reverse primer (Table 1) ▶ in a final concentration of 900 nmol/L and hybridization probe (Table 1) ▶ in a final concentration of 200 nmol/L were mixed in an end volume of 50 μl. Cycling conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 61°C for 60 seconds.
Table 1.
Sequences, Amplicon Sizes, and Position of Primers (and Probe)
| Primer name | Sequence | Position | ||
|---|---|---|---|---|
| PBGD (M28663, M28664); amplicon size: 135 bp | ||||
| FP (M28663) | 5′GGTACAAGGCTTTCAGCATCGC3′ | 103-82 | ||
| RP (M28664) | 5′ATGTCCGGTAACGGCGGC3′ | 505-522 | ||
| Probe (M28664; M28663) | 5′CCAGCTGGCTCTTACGGGTGCCCAC3′ | 2510-2490; 55-52 | ||
| Procollagen 1 α1 subunit (U08020); amplicon size: 124 bp | ||||
| FP | 5′CCAAGGGTAACAGCGGTGAA3′ | 1253-1272 | ||
| RP | 5′CCTCGTTTTCCTTCTTCTCCG3′ | 1376-1356 | ||
| Procollagen 1 α2 subunit (X58251); amplicon size: 113 bp | ||||
| FP | 5′TGTTGGCCCATCTGGTAAAGA3′ | 1412-1432 | ||
| RP | 5′CAGGGAATCCGATGTTGCC3′ | 1524-1506 | ||
| Procollagen 3 α1 subunit (X52046); amplicon size: 92 bp | ||||
| FP | 5′TCAAGTCTGGAGTGGGAGG3′ | 17600-17621 | ||
| RP | 5′TCCAGGATGTCCAGAAGAACCA3′ | 19252-19231 | ||
| Carbonic anhydrase 3 (M27796); amplicon size: 101 bp | ||||
| FP | 5′GACGGGAGAAAGGCGAGTTC3′ | 532-551 | ||
| RP | 5′CAGGCATGATGGGTCAAAGTG3′ | 632-612 | ||
| Matrix gamma-carboxyglutamate protein (D00613); amplicon size: 101 bp | ||||
| FP | 5′GTGGCGAGCTAAAGCCCAA3′ | 165-183 | ||
| RP | 5′CGTAGCGCTCACACAGCTTG3′ | 265-246 | ||
| Non-muscle myosin light chain 3 (U04443); amplicon size: 101 bp | ||||
| FP | 5′CTTTGAGCACTTCCTGCCCA3′ | 168-187 | ||
| RP | 5′CCTTCCTTGTCAAACACACGAA3′ | 269-248 | ||
| Serine proteinase inhibitor 3 (U25844); amplicon size: 82 bp | ||||
| FP | 5′TCCTGCCTCAAGTTCTATGAAGC3′ | 398-420 | ||
| RP | 5′TGTTGATGTGCTGTCGGGAC3′ | 479-460 | ||
| Cytochrome b-245 α polypeptide (M31775); amplicon size: 101 bp | ||||
| FP | 5′TTTCGGCGCCTACTCTATCG3′ | 48-67 | ||
| RP | 5′TCTGTCCACATCGCTCCATG3′ | 148-129 | ||
| AXL receptor tyrosine kinase (X63535); amplicon size: 101 bp | ||||
| FP | 5′AGTCACAGGACACAGCTCC3′ | 473-493 | ||
| RP | 5′AGGTGGTGACTCCCTTGGC3′ | 573-555 | ||
The primer/(probe) sets work under identical PCR-cycling conditions to obtain simultaneous amplification in the same run. Sequences were taken from GeneBank, Accession numbers are given.
In analogy, preservation of the expression profile was investigated by determination of the amplification factor for the four genes PBGD, tumor necrosis factor-α, cyclooxygenase 2 (COX2), and tissue factor. Preparation and PCR conditions were identical to those described before, primer/probe sets are given elsewhere. 21,22
Relative mRNA Quantitation
The above-mentioned equation was also used for relative mRNA quantitation by real-time PCR. The target gene was normalized to an internal standard gene. Therefore, PBGD mRNA was used, an ubiquitously as well as consistently expressed standard gene that is free of pseudogenes. For cDNA synthesis, reagents and incubation steps were applied as described. 18 The reactions were set up with the SYBR Green PCR Core Reagents (Applied Biosystems) according to the manufacturer‘s protocol. Using the oligonucleotide primer pairs given in Table 1 ▶ , for each gene 1 μl of the concerning primers (final concentration, 200 nmol/L) and 2 μl of cDNA were added to a final volume of 50 μl. Cycling conditions were 95°C for 6 minutes, followed by 45 cycles of 95°C for 20 seconds, 58°C for 30 seconds, and 73°C for 30 seconds. Because of the nonselective dsDNA binding of the SYBR Green, gel electrophoresis was performed to confirm the exclusive amplification of the expected PCR product.
Results
Maintenance of the Expression Profile after cDNA Preamplification
To investigate the preservation of the mRNA expression profile during the cDNA preamplification procedure, extracted mRNA from lungs treated with lipopolysaccharide and interferon-γ 20 was diluted to amounts comparable to microdissected material. Afterward, preamplification was performed using SMART PCR. The generated PCR product as well as the unamplified cDNA then underwent real-time quantitative PCR. To test the impact of preamplification on different amounts of initial mRNA copy numbers, four genes were selected representing different levels of mRNA expression. Tumor necrosis factor-α was seen to be a highly expressed gene after lipopolysaccharide/interferon-γ stimulation, PBGD and COX2 were moderately expressed, and tissue factor expression was very low. When comparing the ratios of cDNA amplified by 15 cycles to preamplified status, consistency of the amplification step for all genes was noted (Table 2) ▶ .
Table 2.
Maintenance of the Expression Profile after PCR-Based Amplification
| PBGD | TNF-α | COX2 | Tissue factor | |
|---|---|---|---|---|
| Mean amplification factor | 39.56 | 28.80 | 29.18 | 35.08 |
| SEM | 14.70 | 12.40 | 8.24 | 12.37 |
RNA from a lipopolysaccharide and interferon-γ-stimulated lung was diluted. Aliquots were amplified by SMART PCR (15 cycles). Four genes representative for the total mRNA pool were selected. Using real-time PCR, ratios of amplified PCR product to original cDNA were calculated. These mean amplification factors ± SEM from three independent experiments are given. Among one another, they varied markedly by less than a factor of 2.
Factor of Amplification
Microdissection of lung vessels was performed to isolate 30 to 40 vessel profiles corresponding to ∼500 cell equivalents (Figure 1) ▶ . RNA was extracted, transcribed to cDNA, and applied to PCR-based amplification. In preliminary experiments undertaken for the gene that first reached the plateau phase (PBGD), we noted that amplification of more than 22 cycles did not result in a further increase of PCR product. Thus, we stopped the preamplification after cycle 19 to ascertain analytic conditions within the exponential phase of PCR. After RNA extraction of the microdissected vessels, we routinely determined the factor of amplification obtained by SMART PCR. Therefore, 2 μl of cDNA were separated from the purified cDNA (=1/22 of the original cDNA) as were 2 μl of the purified DNA (1/22 of total PCR product) after 19 PCR cycles. Both were subjected to real-time PCR. The mean threshold cycle of the original purified cDNA amounted to 33.68 ± 0.37 (mean ± SEM; n = 14). Comparing the threshold cycle after amplification to the nonamplified cDNA, an amplification factor of 882 ± 144 (mean ± SEM; n = 14) was calculated for PBGD mRNA.
Real-time PCR for PBGD mRNA also allowed assessment of the full-length transcription of this mRNA and thus predicted the quality and integrity of multiplied DNA. In the used primer/probe system (Table 1) ▶ one primer was positioned in exon 1 at the start codon so that only completely transcribed mRNA is measured.
Array Analysis 1: Expression Profile Comparison of Homogenized Lung Tissue versus Microdissected Vessels
First, RNA from homogenized lung tissue as well as PCR-preamplified cDNA from microdissected vessels were applied to prespotted membranes. The expression profiles were found to differ completely (Figure 2) ▶ . Although the filter with vessel RNA showed spots that are weak or nondetectable on the homogenate array (ie, cardial/slow skeletal troponin C, cardiac troponin T2, non-muscle myosin light chain), several strong spots of the homogenate array were much weaker on the isolated vessel array (ie, 68-kd DNA polymerase α2, GAPDH).
Figure 2.

Comparison of a filter hybridized with lung homogenate RNA versus a filter hybridized with amplified cDNA from lung arteries. Sectors with 14 × 7 spots from block B and F of the membrane were selected and opposed. The expression profiles differ completely. Although 68-kd DNA polymerase α2 and GAPDH are prominent on the homogenate filter, the troponin, myosin, and procollagen components are markedly stronger expressed on a vessel filter.
Array Analysis 2: Comparison of Microdissected Vessels; Reproducibility
Three independent hybridization experiments were performed with microdissected vessels from two different normoxic lungs to assess the reproducibility of the technique. Normalization was calculated by the sum method, afterward the background was subtracted. On filter A, 237 spots were detectable at least twice over the background (20.2%), filter B exhibited 266 spots (22.6%), and filter C exhibited 228 spots (19.4%). Comparing filter A to B, 192 spots were common, corresponding to 81.0% of A and 72.2% of B. Comparing filter A to C, 175 spots were common, corresponding to 73.8% of A and 76.8% of C. Thus, high consistency of the expression profile among different microdissected vessels was noted.
Array Analysis 3: Comparison of Microdissected Vessels; Normoxia versus Hypoxia
Within 21 days of chronic hypoxia a significant increase in the ratio of the right ventricular wall/left ventricle plus septum from 0.31 ± 0.01 to 0.44 ± 0.02 (mean ± SEM, n = 9 each, P < 0.0002) was induced as compared to normoxic controls. Pulmonary artery pressure, measured in the isolated perfused lung preparation increased from 8.5 ± 0.2 in normoxic mice to 11.1 ± 0.2 after chronic hypoxia (mean ± SEM, n = 6 each, P < 0.006). In accordance, a remodeling process effecting a thickening of the vessel wall with proliferation of smooth muscle cells was seen (Figure 3) ▶ . To investigate differential gene expression, vessels from the two mouse lungs that were kept in normoxia were opposed to vessels from two mouse lungs that were kept in hypoxia. Three independent hybridization experiments were performed. Overall, the expression profiles were very similar between normoxic and hypoxic vascular tissue. In the example of hypoxic vessels demonstrated in Figure 4 ▶ , 207 spots (=17.6%) were detected at least two times higher than the background. One hundred seventy-five of these spots were common with those detected in the microdissected vessels of a normoxic lung analyzed in parallel. Twenty-two cDNA signals differed to an extent of the hypoxia/normoxia ratio of <0.5 or >2.0. Among those, 14 genes were up-regulated and 8 down-regulated. Corresponding results were obtained for the second hypoxic lung undergoing cDNA array hybridization.
Figure 3.

Pulmonary vessels from lungs of mice that were kept in hypoxia (10% FiO2, 21 days) or normoxia, respectively. Cryosections were stained with hemalaun. Vessels with a diameter of 250 to 500 μm were selected for further analysis. Original magnification, ×20.
Figure 4.

Expression profile comparison of vessels from hypoxic versus normoxic lungs. Sectors with 14 × 7 spots from block B and F of the membrane were selected and opposed. The expression profiles are primarily comparable. After normalization and background substraction, hypoxia-induced up-regulation was calculated for procollagen 1(subunit α1), procollagen 1(subunit α2), procollagen 3 (subunit α1), non-muscle myosin light chain 3, cytochrome b-245 (α polypeptide), and AXL receptor tyrosine kinase (Table 3) ▶ .
Performing spot-by-spot comparison, we selected nine genes that showed an up-regulation during hypoxia with different factors of induction. An increased signal in hypoxic vessels was found for procollagen 1 (subunit α1), procollagen 1 (subunit α2), procollagen 3 (subunit α1), carbonic anhydrase 3, matrix γ-carboxyglutamate protein, non-muscle myosin light chain 3, serine proteinase inhibitor 3, cytochrome b-245 (α polypeptide), and AXL receptor tyrosine kinase. The factors of up-regulation in response to hypoxia measured in the three experiments are given in Table 3 ▶ . The mean induction factor varied between 1.22 and 5.43.
Table 3.
Comparison of Array-Derived and Real-Time PCR Measured Ratios of Gene Expression in Hypoxic versus Normoxic Vessels
| Gene/accession no. | Array: signal ratio in experiment 1/2/3 | Array: mean signal ratio ± SEM | Real-time PCR (normalized to PBGD): ratio ± SEM |
|---|---|---|---|
| Procollagen 1 α1 subunit/U08020 | 7.79 /4.29/4.20 | 5.43 ± 1.18 | 2.49 ± 0.41 |
| Procollagen 1 α2 subunit/X58251 | 4.46 /3.60/3.30 | 3.79 ± 0.35 | 2.34 ± 0.20 |
| Procollagen 3 α1 subunit/X52046 | 2.81 /1.11/2.37 | 2.10 ± 0.51 | 2.26 ± 0.53 |
| Carbonic anhydrase 3/M27796 | Up /4.11/6.46 | 5.29 | 3.39 ± 0.78 |
| Matrix gamma-carboxy-glutamate (gla) protein/D00613 | 3.29 /3.34/3.96 | 3.53 ± 0.22 | 2.38 ± 0.45 |
| Non-muscle myosin light chain 3/U04443 | 2.22 /1.26/3.41 | 2.30 ± 0.62 | 1.35 ± 0.35 |
| Serine proteinase inhibitor 3/U25844 | 1.94 /1.45/2.31 | 1.90 ± 0.25 | 1.84 ± 0.26 |
| Cytochrome b-245 α polypeptide/M31775 | 2.15 /1.26/1.24 | 1.55 ± 0.30 | 1.45 ± 0.31 |
| AXL receptor tyrosine kinase/X63535 | 1.53 /1.00/1.14 | 1.22 ± 0.16 | 1.52 ± 0.36 |
In three independent experiments, array comparison was performed. Nine genes with different extent of mRNA up-regulation during hypoxia were then additionally investigated by an alternative method. RNA was extracted from isolated vessels and quantified by real-time PCR. After normalization to the standard gene PBGD, the ratios of expression in hypoxic versus normoxic vessels were calculated (n = 3 each; mean ± SEM is given).
Confirmation by Real-Time PCR-Based Relative mRNA Quantitation
RNA was extracted from microdissected vessels of the same lungs. Applying SYBR Green and specific primers for the nine genes, mRNA was quantified in three independent experiments each. Normalized to the standard gene PBGD mRNA, we found an up-regulation of the genes that matched very well the array results (Table 3) ▶ .
Discussion
cDNA arrays were shown to be powerful tools for broad analysis of the transcriptome profile. The comparison of these profiles among different states enables the discovery of differential gene expression for hundreds or thousands of genes in parallel. Targeting cell-type-specific gene regulation within complex tissues, microdissection techniques offer a combination with array technology. To obtain a sufficient amount of RNA for array hybridization (5 to 10 μg of total RNA), several thousands of cells have to be collected. 23 This is at least in nonneoplastic tissues a tremendous effort—-if possible at all—and was tried to circumvent by a preamplification technique. T7-based linear amplification was used to this end and was shown to preserve the expression profile. 9,10,24 However, this technique is labor-intensive, long-lasting, and because of multiple steps highly susceptible to the slightest influences. Calculating an amplification factor of ∼10 to 50 per working cycle, it has to be repeated two or three times, including several incubation and cleaning steps, thereby exposing the RNA to the danger of loss and degradation. In our hands, the profile of the mRNA pool was maintained when using the T7-based technique, but the mean amplification factor ranged far below that described for the PCR-based technique in this approach, especially when introducing small amounts of original RNA.
The currently used PCR-based amplification technique (SMART, Clontech) has already been shown to result in confidential amplification of the RNA pool. 14,16,17 Using four selected genes representing different levels of RNA copy numbers, we confirmed this observation demonstrating preservation of the mRNA profile by real-time quantitative PCR. Thus, the SMART PCR is suitable for efficient amplification with sufficient accuracy.
The successful combination of PCR-based cDNA preamplification and array hybridization has been described several times. 16,17,25 Leethanakul and colleagues 26 applied the technique to microdissected material. Collecting ∼5000 cells, only a minor portion of the cDNA could be introduced to PCR amplification. This procedure corresponds to the protocol provided by the manufacturer (Probe amplification kit, Clontech), which recommends to amplify only one-tenth of the initially transcribed RNA, most probably because of an inhibitory effect of the added cDNA primers during the following PCR amplification. Indeed, the attempt to apply a larger fraction of cDNA to the PCR resulted in a loss of amplification efficiency when measured by real-time PCR (data not given). Thus, we modified the protocol by introduction of a purification step after cDNA synthesis. Without significant loss, this allows the separation of the disturbing primer fraction from the cDNA and the total amount of cDNA may then be applied to the subsequent preamplification. After SMART PCR, the product is cleaned again and completely introduced to the probe labeling process, guaranteeing that the total amount of initial RNA is applied to the hybridization reaction.
To assess the power of amplification, both the initial cDNA and the amplified PCR product were routinely measured by real-time PCR. In addition, knowledge of the absolute threshold cycle for PBGD mRNA enabled us to foresee the success of the hybridization procedure. Obtaining a threshold cycle earlier than 26 for amplified cDNA, the product was sufficient to result in a good hybridization signal. Using this modified protocol, the PCR-based preamplification could be performed much faster than the linear T7-based technique and resulted reliably in a suitable quality and quantity of amplified cDNA for hybridization.
When aiming to generate cDNA profiles representative for the input mRNA it is crucial to stop the PCR cycling before the first gene reaches its plateau phase. Because the expression profile would inevitably change, the timely termination has to be fixed in preliminary experiments. Although we noted plateauing after 22 cycles of amplification for PBGD, we stopped the PCR already after 19 cycles to be safely within the exponential phase. The amount of cycles may vary and can be adapted, potentially increasing the factor of amplification.
For SMART amplification the quality of RNA is crucial for its success. Using our microdissection protocol, the time of potential RNA degradation was minimized. To assess the quality of mRNA especially after amplification, our PBGD primer/probe system for real-time PCR is considered to be a good measure. With a length of ∼1100 bp, 27 the PBGD mRNA belongs to the medium-length RNA fraction and might be representative for the RNA pool. Because only completely transcribed and amplified PBGD cDNA copies are detected, it allows estimation of the shape of the mRNA pool.
To test our protocol, we selected lung vessels for microdissection. The laser was drawn along the tunica adventitia to comprise the gene expression profile of tunica media and intima in total. However, to increase the specificity, the thin endothelial layer may be separated from the tunica media by the laser beam so that both structures can be analyzed separately. Thirty to 40 vessel sections were collected with a diameter of 250 to 500 μm. Smooth muscle cells represent a cell type bearing only a minor amount of mRNA per cell. In total, we forwarded mRNA from ∼500 cell equivalents to the array analysis. Based on the calculated amplification factor of ∼1000 on the one hand and the necessity to apply ∼5 to 10 μg total RNA to the array for a good signal on the other hand, ∼10 ng of extracted RNA after microdissection are requested. Because most cell types contain 20 to 40 pg of total RNA per cell, the overall quantity of introduced cells into the analytical procedure is easily calculated. As most of the applied arrays need similar amounts of total RNA for hybridization the described approach is correspondingly applicable to the diverse available array systems. We used nylon filters to take advantage of an increased sensitivity because of the use of radioactive labeled cDNA (32P). In addition, the membranes can be stripped and reused for at least three times thus reducing costs.
As vessels represent only a minor fraction of the lung tissue we expected a different expression profile of the homogenized lung tissue compared to the isolated vessels. Indeed, the pattern of the homogenate differed primarily from that of the microdissected lung arteries. Addressing the regulation of gene expression of defined structures and/or specific cell types within complex tissues, the microdissection is an indispensable precondition to generate valid data. The consistency of the expression profile noted on analysis of microdissected vessels from independent experiments supports the validity and reproducibility of this approach.
Chronic hypoxia regularly induces an increase in pulmonary artery pressure and provokes remodeling of the vasculature such as the proliferation of smooth muscle cells and thickening of the arterial tunica media. Therefore, the chronic hypoxia represents a common model to investigate pulmonary hypertension. After 21 days of chronic hypoxia in mice, increases in the relative right ventricular weight and thickness of the vessel walls were detectable suggesting up-regulation of genes related to structural growth. Of interest, extracellular matrix components such as procollagens have already been shown to be up-regulated during the remodeling process of pulmonary arteries. Berg and colleagues 28 found elevated mRNA levels for procollagen 4 α2, 1 α1, and 3 α1 regulated in response to short-term and long-term hypoxia. In the context of primary pulmonary hypertension, Botney and colleagues 29 described the detection of procollagen 1 α1 in the tunica media and neointima of small muscular arteries whereas normal lung vessels were negative. Thereby, the increased extracellular matrix synthesis might be induced by transforming growth factor-β subtypes. 30
Apart from the new chances and possibilities opened by array technology, many voices warn against potential pitfalls. 31 These pitfalls concern correct assignment of the spotted oligonucleotides or cDNAs, mistakes during array analysis (ie, wrong evaluation of normalization parameters), as well as measurement of smear and false-positive spots. Additionally, although the SMART amplification may well preserve the representation of most of the original RNAs, exceptions may be possible. 14 To exclude these mistakes it is advisable to check the results by an independent approach. To this end, Sgroi and colleagues 23 suggested the use of real-time PCR. Following this line, we verified differential expression derived from the array analysis by our established combination of microdissection and mRNA quantitation applying real-time PCR. Comparison of the selected genes’ mRNA to the standard gene PBGD mRNA is used for normalization of the technique. The ratio of relative expression in hypoxic versus normoxic vessels is equivalent to the factor of hypoxic induction. These factors of hypoxia-induced gene up-regulation matched very well with the array data. Even if a factor less than 2 is not regarded to be significant as noted for some genes of interest, both techniques resulted in a similar ratio.
In conclusion, we present an optimized, rapid, and reproducible strategy to combine laser-assisted microdissection of ∼500 cell equivalents with array hybridization. Modification of the protocol of PCR-based SMART preamplification allows the introduction of the total amount of the extracted RNA from microdissected cells. To test the validity of the technique, real-time PCR was used as an alternative method to confirm the differential gene expression that was seen by array comparison. Apart from tumor-specific analysis, gene regulation even within nonneoplastic complex tissues and intact organs can be determined in a cell-type-specific manner. Thus, the presently described method combines the advantages of microdissection with the advantages of a broad transcriptome analysis by cDNA arrays.
Acknowledgments
We thank Professor H. Bojar and Dr. O. Modlich, Institute of Oncological Chemistry, Heinrich-Heine-University Düsseldorf, for introduction to array technique; K. Quanz for excellent technical assistance; G. Jurat for photographic arrangement; Dr. R. Snipes for critical reading of the manuscript; and Professor W. H. Gerlich, Institute of Virology, Justus-Liebig-University Giessen, for the use of the phosphorimaging system.
Footnotes
Address reprint requests to Dr. Ludger Fink, Institut für Pathologie, Universität Giessen, Langhansstr. 10, 35392 Giessen, Germany. E-mail: ludger.fink@patho.med.uni-giessen.de.
Supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 547, project Z1.
L. F. and S. K. contributed equally to these studies.
References
- 1.Schena M, Heller RA, Theriault TP, Konrad K, Lachenmeier E, Davis RW: Microarrays: biotechnology’s discovery platform for functional genomics. Trends Biotechnol 1998, 16:301-306 [DOI] [PubMed] [Google Scholar]
- 2.Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, Mahlamaki E, Schraml P, Moch H, Willi N, Elkahloun AG, Pretlow TG, Gasser TC, Mihatsch MJ, Sauter G, Kallioniemi OP: Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst 1999, 91:1758-1764 [DOI] [PubMed] [Google Scholar]
- 3.Wang K, Gan L, Jeffery E, Gayle M, Gown AM, Skelly M, Nelson PS, Ng WV, Schummer M, Hood L, Mulligan J: Monitoring gene expression profile changes in ovarian carcinomas using cDNA microarray. Gene 1999, 229:101-108 [DOI] [PubMed] [Google Scholar]
- 4.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson Jr J, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403:503–511 [DOI] [PubMed]
- 5.Sirivatanauksorn Y, Drury R, Crnogorac-Jurcevic T, Sirivatanauksorn V, Lemoine NR: Laser-assisted microdissection: applications in molecular pathology. J Pathol 1999, 189:150-154 [DOI] [PubMed] [Google Scholar]
- 6.Walch A, Specht K, Smida J, Aubele M, Zitzelsberger H, Höfler H, Werner M: Tissue microdissection techniques in quantitative genome and gene expression analyses. Histochem Cell Biol 2001, 115:269-276 [DOI] [PubMed] [Google Scholar]
- 7.Fend F, Raffeld M: Laser capture microdissection in pathology. J Clin Pathol 2000, 53:666-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH: Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA 1990, 87:1663-1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J: Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci USA 1996, 93:14152-14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, Erlander MG: Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med 1999, 5:117-122 [DOI] [PubMed] [Google Scholar]
- 11.Ohyama H, Zhang X, Kohno Y, Alevizos I, Posner M, Wong DT, Todd R: Laser capture microdissection-generated target sample for high-density oligonucleotide array hybridization. BioTechniques 2000, 29:530-536 [DOI] [PubMed] [Google Scholar]
- 12.Chenchik A, Zhu YY, Diachenko L, Li R, Hill J, Siebert PD: Generation and use of high-quality cDNA from small amounts of total RNA by SMART PCR. Siebert PD Larrick JW eds. Gene cloning and analysis by RT-PCR. 1998, :pp 305-319 BioTechniques Books, Westborough [Google Scholar]
- 13.Franz O, Bruchhaus II, Roeder T: Verification of differential gene transcription using virtual northern blotting. Nucleic Acids Res 1999, 27:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endege WO, Steinmann KE, Boardman LA, Thibodeau SN, Schlegel R: Representative cDNA libraries and their utility in gene expression profiling. BioTechniques 1999, 26:542-550 [DOI] [PubMed] [Google Scholar]
- 15.Hung HL, Song F, Gewirtz A: A method for identifying differentially expressed genes in rare populations of primary human hematopoietic cells. Leukemia 1999, 13:295-297 [DOI] [PubMed] [Google Scholar]
- 16.Spirin KS, Ljubimov AV, Castellon R, Wiedoeft O, Marano M, Sheppard D, Kenney MC, Brown DJ: Analysis of gene expression in human bullous keratopathy corneas containing limiting amounts of RNA. Invest Ophthalmol Vis Sci 1999, 40:3108-3115 [PubMed] [Google Scholar]
- 17.Vernon SD, Unger ER, Rajeevan M, Dimulescu IM, Nisenbaum R, Campbell CE: Reproducibility of alternative probe synthesis approaches for gene expression profiling with arrays. J Mol Diagn 2000, 2:124-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fink L, Seeger W, Ermert L, Hanze J, Stahl U, Grimminger F, Kummer W, Bohle RM: Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med 1998, 4:1329-1333 [DOI] [PubMed] [Google Scholar]
- 19.Fink L, Stahl U, Ermert L, Kummer W, Seeger W, Bohle RM: Rat porphobilinogen deaminase gene: a pseudogene-free internal standard for laser-assisted cell picking. BioTechniques 1999, 26:510-516 [DOI] [PubMed] [Google Scholar]
- 20.Fink L, Kinfe T, Seeger W, Ermert L, Kummer W, Bohle RM: Immunostaining for cell picking and real-time mRNA quantitation. Am J Pathol 2000, 157:1459-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hölschermann H, Bohle RM, Schmidt H, Zeller H, Fink L, Stahl U, Grimm H, Tillmanns H, Haberbosch W: Hirudin reduces tissue factor expression and attenuates graft arteriosclerosis in rat cardiac allografts. Circulation 2000, 102:357-363 [DOI] [PubMed] [Google Scholar]
- 22.Grandel U, Fink L, Blum A, Heep M, Buerke M, Kraemer HJ, Mayer K, Bohle RM, Seeger W, Grimminger F, Sibelius U: Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation 2000, 102:2758-2764 [DOI] [PubMed] [Google Scholar]
- 23.Sgroi DC, Teng S, Robinson G, LeVangie R, Hudson Jr JR, Elkahloun AG: In vivo gene expression profile analysis of human breast cancer progression. Cancer Res 1999, 59:5656–5661 [PubMed]
- 24.Luzzi V, Holtschlag V, Watson MA: Expression profiling of ductal carcinoma in situ by laser capture microdissection and high-density oligonucleotide arrays. Am J Pathol 2001, 158:2005-2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez P, Zigler Jr JS, Epstein DL, Borras T: Identification and isolation of differentially expressed genes from very small tissue samples. BioTechniques 1999, 26:884–892 [DOI] [PubMed]
- 26.Leethanakul C, Patel V, Gillespie J, Pallente M, Ensley JF, Koontongkaew S, Liotta LA, Emmert-Buck M, Gutkind JS: Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene 2000, 19:3220-3224 [DOI] [PubMed] [Google Scholar]
- 27.Cardalda CA, Batlle A, Juknat AA: Sequence and structure of the rat housekeeping PBG-D isoform. Biochem Biophys Res Commun 1998, 249:438-443 [DOI] [PubMed] [Google Scholar]
- 28.Berg JT, Breen EC, Fu Z, Mathieu-Costello O, West JB: Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor-B in lung parenchyma. Am J Respir Crit Care Med 1998, 158:1920-1928 [DOI] [PubMed] [Google Scholar]
- 29.Botney MD, Liptay MJ, Kaiser LR, Cooper JD, Parks WC, Mecham RP: Active collagen synthesis by pulmonary arteries in human primary pulmonary hypertension. Am J Pathol 1993, 143:121-129 [PMC free article] [PubMed] [Google Scholar]
- 30.Botney MD, Bahadori L, Gold LI: Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol 1994, 144:286-295 [PMC free article] [PubMed] [Google Scholar]
- 31.Knight J: When chips are down. Nature 2001, 410:860-861 [DOI] [PubMed] [Google Scholar]