

Abstract
Exposure of cells to stress, particularly oxidative stress, leads to misfolding of proteins and, if they are not refolded or degraded, to cytoplasmic protein aggregates. Protein aggregates are characteristic features of a variety of chronic toxic and degenerative diseases, such as Mallory bodies (MBs) in hepatocytes in alcoholic and non-alcoholic steatohepatitis, neurofibrillary tangles in neurons in Alzheimer’s, and Lewy bodies in Parkinson’s disease. Using 2D gel electrophoresis and mass spectrometry, we identified p62 as a novel MB component. p62 and cytokeratins (CKs) are major MB constituents; HSP 70, HSP 25, and ubiquitinated CKs are also present. These proteins characterize MBs as a prototype of disease-associated cytoplasmic inclusions generated by stress-induced protein misfolding. As revealed by transfection of tissue culture cells overexpressed p62 did not induce aggregation of regular CK filaments but selectively bound to misfolded and ubiquitinated CKs. The general role of p62 in the cellular response to misfolded proteins was substantiated by detection of p62 in other cytoplasmic inclusions, such as neurofibrillary tangles, Lewy bodies, Rosenthal fibers, intracytoplasmic hyaline bodies in hepatocellular carcinoma, and α1-antitrypsin aggregates. The presence of p62 along with other stress proteins and ubiquitin in cytoplasmic inclusions indicates deposition as aggregates as a third line of defense against misfolded proteins in addition to refolding and degradation.
An eukaryotic cell has several options for dealing with misfolded proteins which arise to a certain extent physiologically in the course of protein synthesis and can be abundantly present if cells are exposed to stress situations. Misfolded proteins may interfere with essential cellular functions and thus have to be eliminated very efficiently. They can be refolded into their native conformation by chaperones of the heat shock protein (HSP) 70 class under the consumption of ATP. 1,2 Since this rescue process has a limited capacity, large amounts of misfolded proteins require additional pathways of elimination, such as degradation by the proteasome protein degradation machinery. 3 Whereas most misfolded proteins are degraded after ubiquitination by the 26 S proteasome complex, oxidatively modified and partially unfolded proteins can be cleaved by the 20 S core proteasome in an ATP- and ubiquitin-independent manner. 4 However, under certain stress conditions, and particularly if the proteasome pathway is blocked, misfolded proteins aggregate via exposed hydrophobic amino acid residues and accumulate in the cytoplasm at the microtubule organizing center as aggresomes or sequestosomes. 5,6 The biological significance of these protein aggregates is unclear. They could, on the one hand, reflect a third line of defense against harmful misfolded proteins by depositing them in a biologically inert form. On the other hand, occurrence of large protein aggregates could trigger further cell damage and thereby aggravate the deleterious effects of stress situations.
Exploration of the conditions under which cytoplasmic protein aggregates accumulate as well as elucidation of their biological significance is all the more interesting since cytoplasmic inclusions consisting of abnormal proteins are hallmark lesions of a considerable number of human diseases, also designated as protein aggregation diseases, such as neurofibrillary tangles in neurons of patients with Alzheimer’s disease, Lewy bodies in neurons of patients with Parkinson’s disease, Lewy body-like or skein-like inclusions in amyotrophic lateral sclerosis, inclusions in skeletal muscle fibers in patients with inclusion body myopathies, aggregates of mutated α1-anti-trypsin in hepatocytes of patients with α1-antitrypsin deficiency, Mallory bodies (MBs) in hepatocytes of patients with alcoholic steatohepatitis, and a variety of non-alcoholic chronic toxic and degenerative liver disorders as well as in hepatocellular neoplasms . 7-9
To obtain new insight into the pathogenesis of such inclusions we analyzed the protein components of MBs and studied their role in MB formation. MBs are irregularly shaped cytoplasmic inclusions typically present in hepatocytes. They consist of abnormally phosphorylated, ubiquitinated, and cross-linked cytokeratins (CKs) and non-CK components. 9-15 MBs can be reproduced in mice by chronic intoxication with 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC). 16 Using mass spectrometry, we identified p62 as a novel and major component of MBs isolated from DDC intoxicated mouse livers. p62 (also designated STAP, A170, and ZIP) was originally identified as a ligand for the SH2 domain of p56lck 17-20 and was shown to contain several functional domains, such as a zinc finger motif, proline rich regions, and a PEST sequence. It was shown that p62 interacts with PKC-ζ and is involved in the activation of NF-κB and p38 MAPK. 17,19,21-23 Besides a role in signal transduction p62 might be involved in the cellular stress response since, on the one hand, its expression is markedly increased on a variety of stress stimuli, such as exposure to oxidative stress, sodium arsenite, cadmium, and ionophore and, on the other hand, p62 binds to polyubiquitinated proteins. 18,24-26 We found that p62 binds specifically to aggregates of misfolded and ubiquitinated proteins and that it is present in cytoplasmic inclusions in diverse human diseases suggesting a general role of p62 in the cellular response to abnormal proteins in protein aggregation diseases.
Materials and Methods
Induction of Mallory Bodies and Analysis by 2D Gel Electrophoresis and Immunoblotting
For induction of MBs, Swiss Albino mice (strain Him OF1 SPF; Institute of Laboratory Animal Research, Himberg, Austria) were fed a diet containing 0.1% DDC (Aldrich, Steinheim, Germany). After 2 months of DDC feeding, mice were killed and liver tissue was immediately processed for isolation of MBs. For immunofluorescence analysis aliquots were snap-frozen in methylbutane precooled with liquid nitrogen.
MBs were isolated by sucrose gradient centrifugation and high salt extraction as described previously. 11 For 2D gel electrophoresis, MB material was dissolved in TISO buffer (2 mol/L thiourea, 8 mol/L urea, 4% CHAPS, 20 mmol/L Trizma base, and 30 mmol/L DTT; all reagents from Sigma-Aldrich, Deisenhofen, Germany) and separated by isoelectric focusing using non-linear immobilized pH gradient strips ranging from pH 3.5 to 10 (Pharmacia, Uppsala, Sweden) followed by separation on sodium dodecyl sulfate-polyacrylamide gradient gels (4 to 16%). Gels were either stained with Coomassie blue or transferred onto nylon membranes for immunoblotting as described. 27 Reactivity of the antibodies (see below) was detected using enhanced chemiluminescence (Amersham, Buckinghamshire, UK).
Analysis of Mallory Body Components by Mass Spectrometry
To obtain specific peptide fragments MB protein spots were excised from the gel, digested with trypsin in 40 mmol/L ammonium bicarbonate at 37°C overnight, and the reaction was stopped by freezing. For matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF) the samples were prepared on thin film spots according to Jensen et al. 28 Briefly, 0.3 μl of nitrocellulose containing saturated solution of α-cyno-4-hydroxycinnamic acid (Sigma-Aldrich) in acetone were deposited onto individual spots on the target. Subsequently, 0.8 μl of 10% formic acid and 0.4 μl of digested sample were loaded on top of the thin film spots and dried at ambient temperature. To remove salts the spots were washed with 10% formic acid and H2O. MALDI mass spectra were recorded in the positive ion mode with delayed extraction on a Reflex II time-of-flight instrument (Bruker-Daltonic, Bremen, Germany) equipped with a SCOUT multiprobe inlet and a 337 nm nitrogen laser. Ion acceleration voltage was set to 20.0 kV, the reflector voltage was set to 21.5 kV, and the first extraction plate was set 15.4 kV. Mass spectra were obtained by averaging 50 to 200 individual laser shots. Calibration of spectra was performed internally by two-point linear fit using the autolysis products of trypsin at m/z 842.50 and m/z 2211.10.
Sample preparation for postsource decay (PSD) analysis was achieved by cocrystallization of matrix with ZipTip C18 (Millipore, Bedford, MA) concentrated samples. In brief, the peptides in the supernatant of in-gel digestion were absorbed to a prewashed (50% acetonitrile/water) and equilibrated (0.1% trifluoroacetic acid/water) ZipTip C18 by repeated applications. Following washing of the ZipTip C18 by equilibration buffer the peptides were eluted from the ZipTip C18 with 1 μl of matrix (α-cyano-4-hydroxycinnamic acid saturated in 50% acetonitrile/water, Sigma-Aldrich). PSD analysis was performed in the reflection mode with delayed extraction by setting an ion gate width of 40 Da around the ion of interest. Data were acquired in 14 segments by decreasing the reflector voltage in a stepwise fashion. For each segment 100 to 200 individual laser shots were accumulated. The fragment ion spectrum was obtained by pasting together all segments to a single spectrum using FAST software provided by Brucker. Fragment ion calibration was performed externally with fragment masses of the adrenocorticotropic hormone (ACTH) 18–19 clip.
Singly charged monoisotopic peptide masses were used as inputs for database searching. Searches were performed for best hits against the NCBInr database using the ProFound algorithm developed at the Rockefeller University, New York (see W. Zhang & B.T. Chait, http://prowl.rockefeller.edu/cgi-bin/ProFound) and the Protein Prospector software developed at the University of California, San Francisco (see P.R. Baker and K.R. Clauser, http://prospector.ucsf.edu). Isoelectric points were allowed to range from 0 to 14, and the oxidation of methionine was included as a side reaction. Up to one missed tryptic cleavage was considered, and a mass tolerance for monoisotopic peptide masses was set to ± 0.1 Da. Searches for fragment masses from PSD experiments were performed against NCBIncr database using the MS-Tag search algorithm provided by the Protein Prospector software package. Parent mass tolerance was set to ±50 ppm and fragment ion tolerance was set to ±1500 ppm.
Antibodies, Immunofluorescence Microscopy, and Immunohistochemistry
Antibodies to p62 were raised in guinea pigs against the two peptides (peptide p62NT, MASLTVKAYLLGKEDAAREIC, and p62CT, CNYDIGAALDTIQYSKHPPPL) conjugated to keyhole limpet hemocyanin as described. 29 Additional antibodies used were: 1) monoclonal antibodies: Ks 8.7 and Ks 18.04 (Progen, Heidelberg, Germany) to CK 8 and CK 18, respectively, SMI 31 that recognized phosphorylated epitopes present on tau and neurofilament proteins (Sternberger Monoclonal Inc., Baltimore, MD), anti-HSP/C 70 (Novocastra Laboratories, Newcastle-on-Tyne, UK), and anti-his tag (Roche Diagnostics, Mannheim, Germany); 2) polyclonal antibodies: anti-ubiquitin (StressGene, Victoria, BC, Canada), anti-HSP 25/27 (StressGene); anti CK 8 and CK 18 (produced in our laboratory); 3) secondary antibodies: FITC-conjugated goat anti-mouse Ig (Zymed, San Francisco, CA); TRITC-conjugated swine anti-rabbit Ig (Dako, Glostrup, Denmark); goat anti-guinea pig (Jackson Immuno Research Laboratory, West Grove, PA); horseradish peroxidase-conjugated rabbit anti-mouse Ig (Dako); and horseradish peroxidase-conjugated rabbit anti-guinea pig Ig (Dako). For negative control, primary antibodies were replaced by serum, isotype-matched immunoglobulins, and antibody binding was inhibited by addition of the corresponding peptides (160 μg/ml), respectively. Immunofluorescence specimens were analyzed with a MRC600 (BioRad, Richmond, CA) laser-scanning confocal device attached to a Zeiss Axiophot. Immunohistochemistry on paraffin-embedded human tissues using avidin-biotin complex detection method was performed as described. 27
Cloning of the p62 cDNA and Transfection Experiments
Full-length human p62 cDNA was isolated from human liver tissue by reverse transcription-PCR using the primers corresponding to positions 1425–1402 and 53–70 of the published p62 sequence. 19 PCR products were cloned into the pCRII vector using the TA cloning kit (Invitrogen, Groningen, The Netherlands). For transfection the p62 cDNA was subcloned into NSI I site of a multiple cloning site which has been introduced in the expression vector pHβ April-1 harboring the human β-actin promoter. 30 Furthermore the following cDNA expression constructs were used for transfection: LK440-H8 (human CK 8), 31 LK440-H18 (human CK 18), 31 and pRBG4-ubiquitin-his-myc (kindly provided by R. Kopito). HepG2 cells and CHO-K1 cells were seeded 18 hours before transfection onto glass coverslips. Transfection was performed using the adenovirus-augmented transferrin receptor-mediated gene delivery system essentially as described by Wagner et al. 32 Immunofluorescence analysis was performed 17 to 24 hours after transfection.
Human Tissues
Formaldehyde-fixed and paraffin-embedded human tissue samples were obtained from the Institutes of Pathology of the Universities at Graz and Zurich, respectively. Diagnoses and number of cases studied are listed in below.
Results
To investigate the protein composition of MBs, MBs were isolated from DDC-intoxicated mice livers by sucrose gradient centrifugation followed by high salt extraction. 11 MB proteins separated on 2D gels consisted of two major protein groups with an apparent molecular mass of 48 kd and 55 kd, respectively, which had previously been identified as CK A (corresponding to human CK 8) and CK D (corresponding to human CK 18) 10,11 (Figure 1a) ▶ . Besides CKs there was another major MB component with an apparent molecular mass corresponding to 65 to 68 kd and an isoelectric pH value in the range of pH 4.5. This component had been shown to be recognized by the antibody SMI 31, which binds to phosphoepitopes present on tau protein as well as on neurofilaments in neurofibrillary tangles and Lewy bodies. 14,33
Figure 1.

Analysis of MB components by MALDI-TOF mass spectrometry. a: Coomassie blue-stained 2D gel of MB proteins isolated from DDC-intoxicated mouse liver. Numbers indicate protein spots used for MALDI-TOF analysis. b: Summary of gel spot identifications using tryptic protein digests and MALDI-TOF mass spectrometry.
The MB proteins were characterized after tryptic digestion of excised protein spots by MALDI-TOF mass spectrometry (Figure 1) ▶ . According to the NCBInr database the best hit for peptide masses obtained from tryptic digest of the SMI-31-reactive protein (Figure 1a ▶ , spot 4) was the oxidative stress protein A 170 from mouse, which is a homologue to the human p62 ubiquitin-binding protein. An additional finding of MALDI-TOF analysis was that CK 8 was covalently modified by polyubiquitin (Figure 1) ▶ . Using the NCBInr database and searching with the masses of tryptic peptides obtained from spot 5 in the “single protein only” mode, by far the best hit was CK 8 from mouse; CK 8 from other species ranked next with lower probabilities and scores. Database search with the residual masses of spot 5 indicated the presence of an additional protein with ubiquitin as the best hit. CK 8 and ubiquitin were also specifically detected in spot 5 by PSD fragment ion analysis. Therefore the higher molecular weight of CK 8 in spots 5 and 6, compared to the authentic molecular weight position of CK 8 in spot 7, is due to ubiquitination (see also hits for spot 1 in Figure 1b ▶ ).
As estimated on the basis of Coomassie blue staining intensity, p62 was as abundantly present in MBs as CKs and, therefore, CKs and p62 have to be considered as the major components of MBs. In addition to p62, HSP 70 and HSP 25 were also identified as MB components in the 2D gels. However, the concentrations of the latter two components were much lower than that of p62 and CK.
To show that the proteins identified by mass spectrometry were indeed integral MB components, polyclonal antibodies to N- and C-terminal amino acid sequences of p62 were raised in guinea pigs. Antibodies to the other MB components were commercially available. 2D immunoblots showed that the antibodies to p62 were specific and recognized the same protein as the SMI 31 antibody (Figure 2) ▶ .
Figure 2.

P62 associated with MBs contains the epitope recognized by SMI 31. Coomassie blue-stained 2D gel of MB proteins isolated from DDC-intoxicated mouse liver (a) and the corresponding immunoblots with antibodies to p62 (c) and the SMI 31 (b) antibody.
Double-label immunofluorescence microscopy performed on DDC-intoxicated mouse livers showed that p62 was present in all MBs and that p62 reactivity colocalized with CK reactivity in MBs but not in CK intermediate filaments (Figure 3a) ▶ . Since p62 was already detectable in the smallest MBs, which, at least in part, reflect newly formed MBs, p62 is suggested to have a central role in MB formation. HSP 25 and HSP 70 were also present in MBs (Figure 3, c and d) ▶ . These two proteins, however, were detected in most but not all MBs. The same was noted for ubiquitin in accordance with previous reports (Figure 3b) ▶ . 7,12
Figure 3.
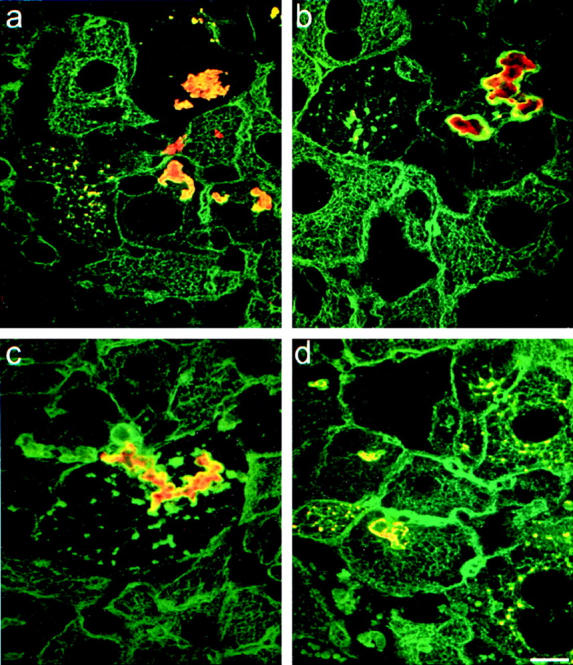
p62 and heat shock proteins are present in murine MBs. Double-label immunofluorescence microscopy using antibodies to CK (green) and the following antibodies to non-CK MB components (red): to p62 (a), to ubiquitin (b), to HSP 25 (c), and to HSP 70 (d). Scale bar, 10 μm.
To elucidate the mechanisms leading to accumulation of p62 in MBs, we cloned human p62 cDNA and studied the interaction of p62 with CKs in a series of transfection experiments. Overexpression of p62 in HepG2 human hepatoma cells led to granular accumulation of p62 in the cytoplasm. In this situation p62 neither associated with endogenous CK intermediate filaments, nor disturbed the intermediate filament architecture, nor caused aggregation of CK proteins (Figure 4a) ▶ . This means that p62 by itself is not able to induce MB formation. Based on the fact that in MBs CKs 8 and 18 are not present in an equimolar ratio and are not assembled as regular intermediate filaments, 9 we tested whether p62 is able to bind to improperly assembled CK. For this purpose, we transfected CHO-K1 cells, which do not endogenously express CK, with human CK 8. When overexpressed in the absence of its partner CK18, CK 8 was unable to form intermediate filaments and accumulated as cytoplasmic aggregates (for review on intermediate filament assembly see reference 34). With double-label immunofluorescence microscopy using antibodies to CK 8 and p62, we observed that endogenous p62 was associated with some CK aggregates (not shown). This indicates that the occurrence of improperly assembled CKs provides a stimulus for the cell to induce endogenous p62. Furthermore, p62 apparently selectively associates with misfolded but not with regularly assembled CKs. Since p62 is known to bind polyubiquitin in a non-covalent manner and ubiquitination of CKs has been reported previously, 35 we analyzed whether the association of p62 with CK 8 is mediated via ubiquitin by cotransfecting CK 8 with a tagged ubiquitin expression construct (Figure 4, b and e) ▶ . Double-label immunofluorescence microscopy using either antibodies to p62 and CK 8 or antibodies to ubiquitin and CK 8 demonstrated that endogenous p62 associated with CK 8 as described above, indicating that transfection of ubiquitin in addition to CK 8 neither influenced the distribution of CK 8 nor had any effect on the association of the endogenous p62 with CK. Furthermore, the distribution of ubiquitin matched that of CK 8, which demonstrates that improperly assembled CK becomes heavily ubiquitinated and binding of p62 to CK is most likely mediated by ubiquitin (Figure 4e) ▶ . The next question to be answered was whether the amount of endogenous p62 was a limiting factor for its association with the overexpressed CK. This was addressed in a triple transfection experiment where CK 8, tagged ubiquitin, and p62 were cotransfected into CHO-K1 cells. In this situation in which both CK 8 and p62 were overexpressed all three components colocalized and the amounts of accumulated CK 8 and p62 were comparable. The resulting cytoplasmic inclusions, therefore, closely resembled MBs (Figure 4, c and d) ▶ . To further demonstrate that the association of p62 with CK was indeed a consequence of the accumulation of abnormally folded proteins, we cotransfected CK 8 and its partner, CK 18, together with the tagged ubiquitin. 35 This experiment led to formation of CK intermediate filament bundles, which were negative both for ubiquitin and p62 (Figure 4f) ▶ .
Figure 4.

Interaction of p62 with cytokeratin is ubiquitin dependent. The interaction of p62 was studied with cell lines transiently cotransfected with different combinations of CK 8, ubiquitin, and p62 expression constructs. a: Double-label immunofluorescence microscopy (IF) with antibodies to CK 8 and 18 (green) and to p62 (red) of HepG2 cells transfected (TF) with p62. b: IF with antibodies to CK 8 (green) and p62 (red) of CHO-K1 cells transfected with CK 8 and ubiquitin. c: IF with antibodies to CK 8 (green) and to p62 (red) of CHO-K1 cells transfected with CK 8, ubiquitin, and p62. d: IF with antibodies to ubiquitin (green) and to p62 (red) of CHO-K1 cells transfected with CK 8, ubiquitin, and p62. e: IF with antibodies to CK 8 (green) and to ubiquitin (red) of CHO-K1 cell transfected with CK 8 and ubiquitin. f: IF with antibodies to CK 8 (green) and to ubiquitin (red) of CHO-K1 cells transfected with CK 8, CK 18, and ubiquitin. Scale bar, 10 μm.
To see whether the association of p62 with aggregates of misfolded proteins is a more general phenomenon, a series of human diseases known to involve cytoplasmic inclusions of misfolded proteins were investigated. As shown in Figure 5 ▶ , p62 is a common denominator of cytoplasmic inclusions in a variety of chronic degenerative diseases, some genetic diseases, and tumors of the liver and brain. All 10 human liver biopsies with alcoholic hepatitis investigated showed strong reactivity of all MBs with the polyclonal p62 antibody (Figure 5a) ▶ . Furthermore, in five cases of α1-antitrypsin deficiency, in which accumulation of misfolded proteins is caused by mutation of the α1-antitrypsin gene, we observed p62 reactivity of some larger irregularly shaped α1-antitrypsin deposits (Figure 5c) ▶ . In these liver specimens, however, most of the α1-antitrypsin deposits were negative for p62. This can be explained by the fact that α1-antitrypsin is a secretory protein and mutated α1-antitrypsin accumulates in the endoplasmic reticulum. 36 Under certain circumstances, α1-antitrypsin deposits leak from the endoplasmic reticulum into the cytoplasm, where they are exposed to p62. Hepatocellular carcinoma is another liver disease which may feature different types of cytoplasmic inclusions. 27 Figure 5e ▶ shows a hepatocellular carcinoma with intracytoplasmic hyaline bodies, which are strongly stained with the p62 antibodies. These findings of occurrence of p62 in cytoplasmic inclusions in several liver diseases are also pertinent to a variety of diseases of the brain. In all 10 cases of Alzheimer’s disease, neurofibrillary tangles stained for p62 (Figure 5g) ▶ . Some tangles, however, were not detected by the antibodies. In all five cases of Parkinson’s disease, Lewy bodies constantly reacted with the p62 antibodies (Figure 5i) ▶ . Furthermore, in all five cases of pilocytic astrocytoma most of the Rosenthal fibers, which are cytoplasmic inclusions consisting of glial fibrillary acidic protein and ubiquitin, reacted with the p62 antibodies (Figure 5k) ▶ .
Figure 5.
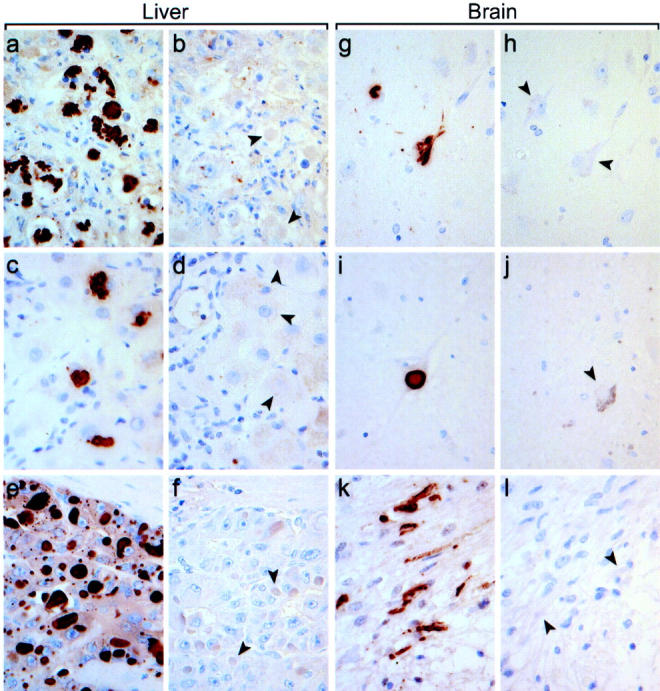
p62 is a common denominator of cytoplasmic inclusions in a variety of human diseases. Immunohistochemical detection of p62 in MBs in human alcoholic hepatitis (a), inclusions in α1-antitrypsin deficiency (c), intracytoplasmic hyaline bodies in hepatocellular carcinoma (e), neurofibrillary tangles in Alzheimer’s disease (g), Lewy bodies in Parkinson’s disease (i), and Rosenthal fibers in astrocytoma (k). For control of antibody specificity, immunoreactions were performed in parallel in the presence of the p62 peptide used for immunization resulting in complete inhibition of antibody binding (b, d, f, h, j, l). Arrowheads indicate examples of cytoplasmic inclusions in the corresponding tissues. Magnification, ×300.
Discussion
The MB is a prototype of a disease-associated cytoplasmic inclusion consisting of misfolded proteins. From that point of view, better understanding of the pathogenesis of MBs and elucidation of their biological significance can also shed some light on other protein aggregation diseases. As we show here, MBs consist of CKs, ubiquitinated CKs, p62, and HSPs 70 and 25. It is interesting to note that MBs contain two major types of constituents, namely, CKs on the one hand and various stress proteins on the other. The role of CKs in MB formation has recently been investigated in mice in which either CK 8 or CK 18, the two partner CKs forming the hepatocytic intermediate filaments, have been deleted. 37,38 CK 8−/− mice revealed markedly increased DDC toxicity leading to death of a considerable number of animals. 39 In the absence of CK 8, intoxicated mice were unable to form MBs and none of the non-CK MB components accumulated as MBs or MB-like structures. In contrast, CK 18−/− mice, which like the CK 8−/− mice lack CK intermediate filaments in hepatocytes, tolerated DDC intoxication, as did wild-type mice, and formed typical MBs (unpublished observation). Furthermore, CK 18−/− mice even spontaneously formed MBs at advanced age. 38 This indicates that in the CK 18−/− mice the still expressed CK 8 cannot assemble into intermediate filaments but occurs as misfolded CK 8 and that age-associated metabolic cellular alterations are sufficient to lead to accumulation of CK 8 protein aggregates. These findings demonstrate an essential role of CKs, particularly CK 8, in MB formation but also pose the question of the significance of the stress proteins in MBs.
All stress proteins detected in MBs are known to be involved in the cellular response to misfolded proteins. HSP 70 mediates refolding whereas HSP 25 prevents formation of misfolded protein aggregates by temporarily binding misfolded proteins which are then delivered either to HSP 70 for refolding or to the proteasome complex for degradation. 40 In acute stress situations these mechanisms are apparently able to prevent deposition of misfolded proteins as aggregates. In chronic stress, as it is the case in alcoholic liver disease, age-associated degenerative diseases as well as in certain genetic disorders, however, the capacity of these rescue mechanisms might become limiting, leading to accumulation of aggregated proteins. 5 It has been reported that chronically stressed cells have reduced capacities to induce HSP expression. 41 Furthermore, chronic stress may result in impairment of the proteasome degradation machinery. 42 With regard to MB formation it is noteworthy that chronic oxidative stress seems to be involved in all human diseases associated with MB occurrence, such as alcoholic and non-alcoholic steatohepatitis as well as drug-induced liver injury. 42,43 Moreover, chronic oxidative stress might also be the common pathogenetic principle in neurodegenerative diseases featuring cytoplasmic inclusions. 44-46 It is interesting in this context, particularly with respect to chronic neurodegenerative diseases associated with ubiquitinated neuronal protein inclusions and apoptotic cell death, that p62 is up-regulated in cultured neuronal cells during initiation of apoptosis and proteasomal inhibition. 47 This suggests a protective role of p62 in pathological conditions affecting the central nervous system, eg, oxidative stress, which favor the appearance of ubiquitinated abnormal proteins and protein aggregation.
This study is one of the first to demonstrate p62 as a common component of cytoplasmic inclusions in various chronic toxic and degenerative diseases. p62 is induced in cells in response to misfolded CKs and is associated with protein aggregates. p62 per se does not lead to aggregate formation but binds to aggregated misfolded proteins and the binding is dependent on prior ubiquitination. The role of p62 in these aggregates, however, is still elusive. One possible function of p62 could be modulation of the stability and biological behavior of cytoplasmic protein aggregates. Aggregated misfolded proteins expose hydrophobic residues at their surface. These hydrophobic residues could act as a trap for other misfolded cellular proteins which are expected to occur in repeated or chronic stress situations. As mentioned above, MBs consist mostly of CKs and stress proteins. It is surprising that CKs preferentially accumulate in MBs while other cellular proteins, which would also be expected to be misfolded in stress situations, do not. An explanation for this situation could be that, on the one hand, CKs become a preferred substrate to modification by oxidative stress, and, on the other hand, the accumulation of HSPs and particularly the association of p62 prevent further binding of other misfolded proteins.
Another consequence of the accumulation of p62 in cytoplasmic aggregates could be that the redistribution of p62 within the cell has some influence on the other cellular functions of p62. p62 not only acts as a ubiquitin-binding protein but also is involved in different signaling pathways. Its role in the activation of NF-κB could be particularly important in the context of oxidative stress since the level of NF-κB is known to be a major modulator of the cellular effects of stress-induced injury. 48,49
In conclusion, we identified p62 as a novel and common component of cytoplasmic inclusions in various protein aggregation diseases. The presence of p62 along with other stress proteins, such as HSP 70, HSP 25, and ubiquitin in protein aggregates indicates that, particularly in chronic stress situations, a third line of defense against misfolded proteins, namely deposition as aggregates, gains importance in addition to refolding and degradation. The nature of the misfolded protein as well as of proteins that associate with aggregate could determine whether the resulting protein aggregate is harmful to the cell and thus aggravates tissue injury or whether it lacks deleterious effects, as it is the apparently case with MBs. 38,39
Acknowledgments
We gratefully acknowledge the excellent technical assistence of Andrea Raicht (Graz) in cloning of the p62 cDNA, Stefanie Winter-Simanowski (German Cancer Research Center, Heidelberg) for gel electrophoresis experiments, and we cordially thank Dr. Hans-Richard Rackwitz (Peptide Speciality Laboratories GmbH, Heidelberg; www.peptid.de) for peptide synthesis and KLH coupling. Furthermore we thank Ron Kopito (Stanford) for providing us with the pRBG4-ubiquitin-his-myc, and Bernhard Bader (Heidelberg) for providing the CK expression constructs LK440-H8 and LK440-H18. We also thank Eugenia Lamont (Graz) for carefully reading the manuscript.
Footnotes
Address reprint requests to Kurt Zatloukal M.D., Institute of Pathology, Karl-Franzens University, Auenbruggerplatz 25, A-8036 Graz, Austria. E-mail: kurt.zatloukal@kfunigraz.ac.at.
Supported by grant S7401-MOB from the Austrian Science Fund to K.Z.
References
- 1.Gething MJ, Sambrook J: Protein folding in the cell. Nature 1992, 355:33-45 [DOI] [PubMed] [Google Scholar]
- 2.Hartl FU: Molecular chaperones in cellular protein folding. Nature 1996, 381:571-580 [DOI] [PubMed] [Google Scholar]
- 3.Hershko A, Ciechanover A: The ubiquitin system. Annu. Rev Biochem 1998, 67:425-479 [DOI] [PubMed] [Google Scholar]
- 4.Grune T, Reinheckel T, Davies KJA: Degradation of oxidized proteins in mammalian cells. FASEB J 1997, 11:526-534 [PubMed] [Google Scholar]
- 5.Johnston JA, Ward CL, Kopito RR: Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998, 143:1883-1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin J: p62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharmacal Res 1998, 21:629-633 [DOI] [PubMed] [Google Scholar]
- 7.Lowe J, Blanchard A, Morell K, Lennox G, Reynolds L, Billet M, Landon M, Mayer RJ: Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibers in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol 1988, 155:9-15 [DOI] [PubMed] [Google Scholar]
- 8.Carrell RW, Lomas DA: Conformational disease. Lancet 1997, 350:134-138 [DOI] [PubMed] [Google Scholar]
- 9.Denk H, Stumptner C, Zatloukal K: Mallory body revisited. J Hepatol 2000, 32:689-702 [DOI] [PubMed] [Google Scholar]
- 10.Franke WW, Denk H, Schmid E, Osborn M, Weber K: Ultrastructural, biochemical, and immunologic characterization of Mallory bodies in livers of griseofulvin-treated mice: fimbriated rods of filaments containing prekeratin-like polypeptides. Lab Invest 1979, 40:207-220 [PubMed] [Google Scholar]
- 11.Denk H, Krepler R, Lackinger E, Artlieb U, Franke WW: Immunological and biochemical characterization of the keratin-related component of Mallory bodies: a pathological pattern of hepatocytic cytokeratins. Liver 1982, 2:165-175 [DOI] [PubMed] [Google Scholar]
- 12.Ohta M, Marceau N, Perry G, Manetto V, Gambetti P, Autilio-Gambetti L, Metuzals J, Kawahara H, Cadrin M, French SW: Ubiquitin is present on the cytokeratin filaments and Mallory bodies of hepatocytes. Lab Invest 1988, 59:848-856 [PubMed] [Google Scholar]
- 13.Zatloukal K, Fesus L, Denk H, Tarcsa E, Spurej G, Böck G: High amount of ε-(γ-glutamyl)lysine cross-links in Mallory bodies. Lab Invest 1992, 66:774-777 [PubMed] [Google Scholar]
- 14.Preisegger K-H, Zatloukal K, Spurej G, Riegelnegg D, Denk H: Common epitopes of human and murine Mallory bodies and Lewy bodies as revealed by a neurofilament antibody. Lab Invest 1992, 66:193-199 [PubMed] [Google Scholar]
- 15.Stumptner C, Omary MB, Fickert P, Denk H, Zatloukal K: Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a Mallory body mouse model. Am J Pathol 2000, 156:77-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsunoo C, Harwood TR, Arak S, Yokoo H: Cytoskeletal alterations leading to Mallory body formation in livers of mice fed 3,5-diethoxycarbonyl-1,4-dihydrocollidine. J Hepatol 1987, 5:85-97 [DOI] [PubMed] [Google Scholar]
- 17.Joung I, Strominger JL, Shin J: Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci USA 1996, 93:5991-5995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii T, Yanagawa T, Kawane T, Yuki K, Seita J, Yoshida H, Bannai S: Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem Biophys Res Commun 1996, 226:456-460 [DOI] [PubMed] [Google Scholar]
- 19.Puls A, Schmidt S, Grawe F, Stabel S: Interaction of protein kinase C ζ with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci USA 1997, 94:6191-6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okazaki M, Ito S, Kawakita K, Takeshita S, Kawai S, Makishima F, Oda H, Kakinuma A: Cloning, expression profile, and genomic organization of the mouse STAP/A170 gene. Genomics 1999, 60:87-95 [DOI] [PubMed] [Google Scholar]
- 21.Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT: Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol 1998, 18:3069-3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanz L, Sanchez P, Lallena M-J, Diaz-Meco MT, Moscat J: The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. EMBO J 1999, 18:3044-3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudo T, Maruyama M, Osada H: p62 functions as a p38 MAP kinase regulator. Biochem Biophys Res Commun 2000, 269:521-525 [DOI] [PubMed] [Google Scholar]
- 24.Ishii T, Yanagawa T, Yuki K, Kawane T, Yoshida H, Bannai S: Low micromolar levels of hydrogen peroxide and proteasome inhibitors induce the 60-kDa A170 stress protein in murine peritoneal macrophages. Biochem Biophys Res Commun 1997, 232:33-37 [DOI] [PubMed] [Google Scholar]
- 25.Ishii T, Itoh K, Sato H, Bannai S: Oxidative stress-inducible proteins in macrophages. Free Radic Res 1999, 31:351-355 [DOI] [PubMed] [Google Scholar]
- 26.Vadlamudi RK, Joung I, Strominger JL, Shin J: p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 1996, 271:20235-20237 [DOI] [PubMed] [Google Scholar]
- 27.Stumptner C, Heid H, Fuchsbichler A, Hauser H, Mischinger H-J, Zatloukal K, Denk H: Analysis of intracytoplasmatic hyaline bodies in a hepatocellular carcinoma: demonstration of p62 as a major constituent. Am J Pathol 1999, 154:1701-1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen ON, Podtelejnikov A, Mann M: Delayed extraction improves specificity in database searches by matrix-assisted laser desorption/ionization peptide maps. Rapid Commun Mass Spectrom 1996, 10:1371-1378 [DOI] [PubMed] [Google Scholar]
- 29.Schnoelzer M, Alewood P, Jones A, Alewood D, Kent SBH: In situ neutralization in Boc-chemistry solid phase peptide synthesis: rapid, high yield assembly of difficult sequences. Int J Peptide Protein Res 1992, 40:180-193 [DOI] [PubMed] [Google Scholar]
- 30.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L: A human β-actin expression vector system directs high-level accumulation of antisense transcripts. Proc Natl Acad Sci USA 1987, 84:4831-4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bader BL, Magin TM, Freudenmann M, Stumpp S, Franke WW: Intermediate filaments formed de novo from tail-less cytokeratins in the cytoplasm and in the nucleus. J Cell Biol 1991, 115:1293-1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner E, Zatloukal K, Cotton M, Kirlappos H, Mechtler K, Curiel DT, Birnstiel ML: Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes. Proc Natl Acad Sci USA 1992, 89:6099-6103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nukina N, Kosik KS, Selkoe DJ: Recognition of Alzheimer paired helical filaments by monoclonal neurofilament antibodies is due to crossreaction with tau protein. Proc Natl Acad Sci USA 1987, 84:3415-3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs E, Weber K: Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem 1994, 63:345-382 [DOI] [PubMed] [Google Scholar]
- 35.Ku N-O, Omary MB: Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J Cell Biol 2000, 149:547-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teckman JH, Gilmore R, Perlmutter DH: Role of ubiquitin in proteasomal degradation of mutant α1-antitrypsin Z in the endoplasmic reticulum. Am J Physiol 2000, 278:G39-G48 [DOI] [PubMed] [Google Scholar]
- 37.Baribault H, Penner J, Iozzo RV, Wilson-Heiner M: Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev 1994, 8:2964-2974 [DOI] [PubMed] [Google Scholar]
- 38.Magin TM, Schröder R, Leitgeb S, Wanninger F, Zatloukal K, Grund C, Melton DW: Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7, and accumulation of liver-specific keratin 8-positive aggregates. J Cell Biol 1998, 140:1441-1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zatloukal K, Stumptner C, Lehner M, Denk H, Baribault H, Eshkind LG, Franke WW: Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am J Pathol 2000, 156:1263-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehrnsperger M, Gräber S, Gaestel M, Buchner J: Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 1997, 16:221-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu AY-C, Choi H-S, Lee Y-K, Chen KY: Molecular events involved in transcriptional activation of heat shock genes become progressively refractory to heat stimulation during aging of human diploid fibroblasts. J Cell Physiol 1991, 149:560-566 [DOI] [PubMed] [Google Scholar]
- 42.Fataccioli V, Andraud E, Gentil M, French SW, Rouach H: Effects of chronic ethanol administration on rat liver proteasome activities: relationship with oxidative stress. Hepatology 1998, 29:14-20 [DOI] [PubMed] [Google Scholar]
- 43.James OFW, Day CP: Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol 1998, 29:495-501 [DOI] [PubMed] [Google Scholar]
- 44.Simonian NA, Coyle JT: Oxidative stress in neurodegenerative disease. Annu Rev Pharmacol Toxicol 1996, 36:83-106 [DOI] [PubMed] [Google Scholar]
- 45.Berlett BS, Stadtman ER: Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997, 272:20313-20316 [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ: Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 1999, 154:1423-1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuusisto E, Suuronen T, Salminen A: Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun 2001, 280:223-228 [DOI] [PubMed] [Google Scholar]
- 48.Heck S, Lezoualch F, Engert S, Behl C: Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor κB. J Biol Chem 1999, 274:9828-9835 [DOI] [PubMed] [Google Scholar]
- 49.Mattson MP, Culmsee C, Yu ZF, Camandola S: Roles of nuclear factor κB in neuronal survival and plasticity. J Neurochem 2000, 74:443-456 [DOI] [PubMed] [Google Scholar]