

Abstract
Interleukin (IL)-12 and interferon (IFN)-γ are implicated in the pathogenesis of immune disorders of the central nervous system (CNS). To define the basis for the actions of these cytokines in the CNS, we examined the temporal and spatial regulation of key signal transducers and activators of transcription (STATs) and suppressors of cytokine signaling (SOCS) in the brain of transgenic mice with astrocyte production of IL-12 or in mice with experimental autoimmune encephalomyelitis (EAE). In healthy mice, with the exception of STAT4 and STAT6, the expression of a number of STAT and SOCS genes was detectable. However, in symptomatic transgenic mice and in EAE significant up-regulation of STAT1, STAT2, STAT3, STAT4, IRF9, and SOCS1 and SOCS3 RNA transcripts was observed. Although the increased expression of STAT1 RNA was widely distributed and included neurons, astrocytes, and microglia, STAT4 and STAT3 and SOCS1 and SOCS3 RNA was primarily restricted to the infiltrating mononuclear cell population. The level and location of the STAT1, STAT3, and STAT4 proteins overlapped with their corresponding RNA and additionally showed nuclear localization indicative of activation of these molecules. Thus, in both the glial fibrillary acidic protein-IL-12 mice and in EAE the CNS expression of key STAT and SOCS genes that regulate IL-12 (STAT4) and IFN-γ (STAT1, SOCS1, and SOCS3) receptor signaling is highly regulated and compartmentalized. We conclude the interaction between these positive and negative signaling circuits and their distinct cellular locations likely play a defining role in coordinating the actions of IL-12 and IFN-γ during the pathogenesis of type 1 immune responses in the CNS.
The heterodimeric cytokine interleukin-12 (IL-12) is a dominant regulator of cell-mediated immunity necessary for host defense against microbial infection and is implicated in the pathogenesis of certain autoimmune diseases. 1,2 In the central nervous system (CNS), the pathogenesis of multiple sclerosis and the animal model of experimental autoimmune encephalomyelitis (EAE), are closely linked to the influence of IL-12 and the development of a type 1 cellular immune response. 3-5 IL-12 is produced by a number of cells including microglia and astrocytes that are intrinsic to the CNS. 6-8 Principal among its actions is the ability of IL-12 to stimulate natural killer (NK) cell activity, to drive the expansion of CD4+ Th1 cells, and induce interferon (IFN)-γ production from both cells types. 1,2 IFN-γ itself is an important downstream effector of most, but not all, of the responses evoked by IL-12. 9
The many and distinct actions of cytokines such as IL-12 and IFN-γ result from their binding to specific cell surface receptors that are coupled to the activation of unique signal transduction pathways. For a significant number (>30) of cytokines (eg, IL-2, IL-3, IL-4, IL-6, IL-10, IL-12, IFN-α, IFN-β, IFN-γ, and granulocyte macrophage-colony stimulating factor), growth factors, and hormones (eg, growth hormone, prolactin, leptin, platelet-derived growth factor, and epidermal growth factor), the molecular participants in these pathways have been identified as belonging to two families of cytoplasmic proteins known as the Janus kinases (JAKs) and the signal transducers and activators of transcription (STAT). 10-12 Currently, four JAK (JAK1 and JAK2 and TYK1 and TYK2) and seven STAT (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) proteins have been identified. On binding of ligand, JAKs associate with the receptor chain and are activated by tyrosine phosphorylation. These kinases then activate the cytoplasmic tails of the receptor by phosphorylating target tyrosine residues. Depending on the receptor involved and the tyrosine site phosphorylated, docking then occurs of a specific STAT molecule via its SH2 domain leading to phosphorylation of the STAT protein by the receptor-associated JAK. This process then results in the recruitment of a further STAT molecule that also undergoes tyrosine phosphorylation. These activated STAT molecules dissociate from the receptor and form dimers with each other that then translocate to the nucleus and bind to specific target DNA sequences involved in modulating gene transcription.
IL-12 induces tyrosine phosphorylation and homodimerization of STAT4 and STAT3 in T cells and NK cells. 13,14 However, STAT4 signaling seems to be primarily involved in mediating the actions of IL-12 because the loss of STAT4 function in mice is associated with severely impaired Th1 and NK cell function, reduced production of IFN-γ, and increased susceptibility to infectious disease. 15,16 In contrast to IL-12, IFN-γ promotes tyrosine phosphorylation and homodimerization of STAT1 that translocates to the nucleus and binds to a conserved sequence motif named the γ-activated sequence or GAS. The critical role of this signaling pathway for IFN-mediated responses has been clarified in mice with a targeted disruption of the STAT1 gene. 17,18 These animals exhibit increased susceptibility to viral and other infectious diseases and cultured fibroblasts derived from these mice are unresponsive to IFN-γ.
More recently it has become clear the JAK/STAT signal transduction pathway is also subject to negative regulation. Members of the recently discovered family of molecules termed the suppressors of cytokine signaling (SOCS) 19-21 ) inhibit cytokine-activated JAK/STAT signaling. The SOCS family contains at least eight members, SOCS1 to SOCS7 and cytokine-inducible Src homology 2 (SH2) domain-containing protein (CIS). Expression of the SOCS genes can be induced by a wide range of cytokines and may thus constitute a physiological negative feedback loop in the regulation of cytokine-mediated actions. 22,23 Although the signaling pathways affected by many of the SOCS molecules are unknown, studies in mice with targeted disruptions of the SOCS1, SOCS2, or SOCS3 genes reveal pivotal roles for these molecules in IFN-γ, 24,25 growth hormone/IGF-1, 26 and erythropoietin signaling, 27 respectively. In the case of IL-12, a specific suppressor of the STAT4-signaling pathway remains to be identified.
For cytokines such as IL-12 and IFN-γ that are known to be central modulators of type 1 cellular immune responses, the activity of the JAK/STAT pathway, which ultimately determines how a cell responds, depends on the balance between positive and negative regulatory inputs. Despite this, to date, very little is known about the cellular location or regulation of the expression of key STAT and SOCS family members in the CNS during immunoinflammatory disease. Therefore, to begin to address these issues, here we examined the temporal and spatial regulation of STAT and SOCS gene expression in a recently developed transgenic mouse model with IL-12 production under the transcriptional control of the glial fibrillary acidic protein (GFAP) promoter and thus targeted to astrocytes in the CNS. 28 These mice develop a spontaneous neuroimmune disease characterized by an adult-onset of progressive wasting, ataxia, ruffled fur, hunched posture, and premature death. The corresponding neuropathological alterations include neurodegeneration, demyelination, meningoencephalitis, gliosis, and severe calcification. Inflammatory lesions can be seen both in the parenchyma and perivascular locations. The infiltrating cells in these lesions are primarily activated CD4+ and CD8+ T cells and NK cells that produce IFN-γ. This transgene-encoded IL-12-driven autoimmune disease is characteristic of a type 1 cell-mediated immune disease and shares many similarities at the molecular and cellular levels with EAE. Therefore, to determine the extent to which these two models also share their molecular-signaling circuitry we also examined the temporal and spatial regulation of the STAT and SOCS gene expression in mice with myelin oligodendrocyte glycoprotein-EAE.
Materials and Methods
Animals
Hemizygous transgenic mice expressing the combination of the IL-12 p35 and p40 subunit genes or the IL-12 p40 gene alone in astrocytes were generated as recently described. 28 The stable GFAP-IL12 transgenic mouse line GF-IL12 (expressing the IL-12 p35 plus IL-12 p40 genes), produces chronic low levels of bioactive IL-12 whereas the GF-p40 (expressing the IL-12 p40 gene alone) line produces IL-12 p40. All mice were on the C57BL6 × SJL hybrid background. Nontransgenic wild-type (control) mice were obtained from the breeding of the GF-IL12 and GF-p40 lines. GF-IL12 mice studied were between 2 to 4 or 6 to 12 months of age corresponding to before or after the onset of spontaneous CNS disease, respectively. Transgenic animals with CNS disease that were used in our study all had evidence of moderate to severe ataxia, a disease stage that typically was reached from 1 to 2 months after the initial appearance of gait disturbance. It should be noted that GF-p40 mice do not develop CNS disease at any age.
C57BL6 mice used for the induction of EAE (see below) were obtained from the rodent breeding colony of the Scripps Research Institute (La Jolla, CA).
Induction of MOG-EAE
On day 0 C57BL6 mice were immunized subcutaneously into the rear flanks with an emulsion of 100 μl of MOG35–56 peptide (3 mg/ml; Research Genetics, Huntsville, AL) in 100 μl of complete Freund’s adjuvant (CFA) supplemented with 4 mg/ml Mycobacterium tuberculosis H37RA (Difco, Detroit, MI). In addition, each mouse received an intraperitoneal injection of 500 ng of pertussis toxin (Sigma Chemical Co., St. Louis, MO) on days 0 and 2. Initially, animals were observed every second day then after 6 days every day. The disease severity was scored as follows: 0, no disease; 0.5, partial loss of tail tonus; 1, complete tail atony; 2, hind limb paraparesis; 3, hind limb paralysis; 4, moribund; and 5, death.
Antibody and Lectin Reagents
Rabbit polyclonal antibodies against cow GFAP (diluted 1:2000; DAKO, Carpinteria, CA), and human CD3 (diluted 1:500, DAKO) were used in the immunohistochemical identification of astrocytes and T cells, respectively. Monoclonal antibody against human phosphorylated neurofilament (SMI33, diluted 1:1000; Sternberger, Lutherville, MD) and lectin from Lycopersicon esculentum (tomato biotin-labeled, diluted 1:100; Sigma Chemical Co.) were used to identify neurons and macrophage/microglia, respectively. Rabbit polyclonal antibodies were used against STAT1 (Santa Cruz Biotechnology, Santa Cruz, CA), STAT2 (kindly provided by Dr. Christian Schindler, Columbia University, New York, NY), STAT3 (Zymed Laboratories, South San Francisco, CA), and STAT4 (Upstate Biotechnology, Lake Placid, NY, and Chemicon International, Temecula, CA). A nonimmune rabbit serum used as a negative control was obtained from Pharmingen, San Diego, CA.
RNA Preparation
Animals were euthanized and the brain removed and dissected into the forebrain (cerebrum and olfactory bulb) and hindbrain (cerebellum and brain stem). For EAE experiments (see below) the spinal cord was also collected. All tissues were then immediately snap-frozen in liquid nitrogen and stored at −80°C pending RNA extraction. Poly(A)+ RNA was isolated according to a previously published method. 29
RNase Protection Assays
RNase protection assays (RPAs) for the detection of cytokine RNAs were performed as described previously. 30 The RNA samples were hybridized with labeled cytokine probe sets ML11 31 and ML26 (kindly provided by Dr. Monte Hobbs, The Scripps Research Institute) as described previously. 28 For the detection of gene expression for members of the STAT and SOCS families, new multiprobe sets were constructed. The targeted genes comprising these probe sets and their specific sequence locations are given in Table 1 ▶ . The targeted sequences for each gene probe were synthesized by reverse transcriptase-polymerase chain reaction from liver or spleen of lipopolysaccharide-treated mice using specific oligonucleotide primers flanked with HindIII (antisense primer) and EcoRI (sense primer) sites. The primers were designed to generate fragments of desired length that could be separated on a standard polyacrylamide sequencing gel. After polymerase chain reaction, the amplified fragments were incubated with polynucleotide kinase (Promega, Madison, WI) and ligated with T4 ligase (Promega) and subsequently digested with HindIII/EcoRI (Promega), and then ligated into pGEM4 (Promega). The specific identity of each cDNA clone was verified by sequencing analysis. The genomic clone RPL32-4A, 32 kindly provided by Dr. Monte Hobbs (The Scripps Research Institute) served as a probe for the ribosomal protein L32 and was included as an internal control for RNA loading. For quantitation, densitometric analysis of each band was performed on scanned autoradiographs using NIH Image 1.57 software. Expression of the individual mRNA density was normalized to that of L32 and the mean ± SE was calculated using Microsoft Excel 98 (Microsoft Corporation, Seattle, WA).
Table 1.
STAT and SOCS cDNA Target Sequences Used to Derive Multiprobe RPA Sets
| Gene family | Protected fragment (bp) | Target sequence | GenBank no. | |||
|---|---|---|---|---|---|---|
| Signal transduction and activation of transcription | ||||||
| STAT1 | 250 | 121–371 | U06924 | |||
| STAT3 | 225 | 301–526 | U06922 | |||
| STAT4 | 300 | 361–661 | U06923 | |||
| STAT5a/b | 200 | 781–991 | U21103 | |||
| STAT6 | 185 | 421–606 | L47650 | |||
| IRF9/p48 | 150 | 481–631 | U51992 | |||
| STAT2 | 135 | 520–655 | AF206162 | |||
| Suppressor of cytokine signaling | ||||||
| SOCS1 | 294 | 301–595 | U88325 | |||
| SOCS2 | 164 | 401–564 | U88327 | |||
| SOCS3 | 250 | 121–371 | U88328 | |||
| SOCS5 | 125 | 81–206 | AF033187 | |||
| PIAS1 | 225 | 371–596 | AF077950 | |||
Immunoblotting
The cerebellum was removed and immediately solubilized in lysis buffer containing 1% IGEPAL CA-630 (octylphenoxy polyethoxy-ethanol), 10 mmol/L Hepes, 10 mmol/L KCl, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.5 mmol/L dithiothreitol, 50 mmol/L NaF, 1 mmol/L Na3VO4, 10 mmol/L β-glycerophosphate, 4.5 mmol/L Na-pyrophosphate (all from Sigma Chemical Co.), and 1 EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics Corp., Indianapolis, IN)/10 ml. After solubilization samples were clarified by centrifugation at 4000 × g for 15 minutes, the supernatant was kept as cytoplasmic protein. After washing twice with lysis buffer, the pellet was resuspended in nuclear extraction buffer containing 25% glycerol, 10 mmol/L Hepes, 420 mmol/L NaCl, 0.1 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 50 mmol/L NaF, 1 mmol/L Na3VO4, 10 mmol/L β-glycerophosphate, 4.5 mmol/L Na-pyrophosphate, and 1 EDTA-free protease inhibitor cocktail tablet (Roche)/10 ml. The pellet was extracted by vortexing for 20 seconds every 5 minutes during a 40-minute incubation on ice. After centrifugation at 4000 × g for 15 minutes, the supernatant was kept as nuclear protein extract. The protein concentration of the cytoplasmic and nuclear extracts was determined using a protein quantification kit (BioRad, Hercules, CA). Before fractionation and blotting, the extracts were stored at −70°C. Gel electrophoresis, blotting, and immunostaining were performed as described previously. 28
In Situ Hybridization and Immunohistochemistry
Mice were perfused intracardially with ice-cold saline followed by either 4% buffered paraformaldehyde or Zamboni’s fixative. Brains were removed, postfixed overnight in the same fixative, dehydrated through graded alcohol solutions, and embedded in paraffin. In some cases where indicated, brains were removed after saline perfusion and placed in Bouin’s fixative for 24 hours before further processing as described above.
In situ hybridization was performed on paraformaldehyde-fixed brain sections with 33P-labeled cRNA probes transcribed from the appropriately linearized STAT or SOCS cDNA containing ribovectors that were constructed for the RPA described above and in Table 1 ▶ . Dual-label in situ hybridization and immunohistochemistry was performed as described previously. 33
For immunohistochemical detection of the STAT and SOCS proteins, sections were deparaffinized, rehydrated in graded alcohol, rinsed in phosphate-buffered saline (PBS), and blocked for 1 hour at room temperature in PBS containing 3% fetal bovine serum, 3% goat serum, and 0.1% IGEPAL CA-630. The sections were then incubated overnight at 4°C with primary antibody diluted in blocking buffer. The sections were then washed in PBS and incubated with anti-rabbit avidin-biotinylated horseradish peroxidase complex (ABC kit; Vector, Burlingame, CA) used according to the manufacturer’s instructions. After washing in PBS, staining reactions used 3,3-diaminobenzidine (Sigma Chemical Co.) as substrate. Finally, the sections were counterstained with Mayer’s hematoxylin, dehydrated through graded alcohol, and air-dried. After coverslipping, slides were examined by bright-field microscopy. Controls for specificity included incubation of the sections with a nonimmune rabbit serum as well as omission of the primary antibody.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay for STAT1 DNA-binding protein was performed with whole-cell extracts essentially as described previously. 34 Whole-cell extracts were prepared from freshly removed cerebella. After homogenization of the tissue in lysis buffer (10% glycerol, 0.5% IGEPAL CA-630, 50 mmol/L Tris, pH 8.0, 100 mmol/L NaCl, 0.5 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4, 50 mmol/L NaF, 4 mmol/L β-glycerophosphate, 4 mmol/L Na-pyrophosphate, and 1 EDTA-free protease inhibitor cocktail tablet (Roche)/10 ml) the samples were incubated on ice for 60 minutes. After centrifugation at 4000 × g for 5 minutes, the supernatant was kept as whole-cell extract. The protein concentration was determined as described above, and the extracts were stored at −80°C after snap-freezing in liquid nitrogen.
Binding reactions consisted of 5 μg of whole-cell extract in DNA-binding buffer (20 mmol/L Hepes, pH 7.9, 40 mmol/L KCl, 0.5 mmol/L dithiothreitol, 0.1 mmol/L EGTA, 4% Ficoll, 2 μg/ml poly-dI/dC, 1 mg/ml bovine serum albumin) and 1 × 10 5 cpm Klenow-labeled human IRF-1 GAS probe in a 20-μl reaction volume. Reactions were performed at room temperature for 20 minutes. Supershifting polyclonal antibody (2 μg) was added to some samples and incubated for an additional 30 minutes at room temperature. DNA-binding complexes were resolved by nondenaturing 4 to 12% gradient polyacrylamide gel electrophoresis. The IRF GAS DNA probe used in this assay was as follows: gatcATTTCCCCGAAAT.
Results
Cytokine Gene Expression in the Brain of Symptomatic GF-IL12 Transgenic Mice
Adult GF-IL12 but not GF-p40 (expressing only the p40 subunit of IL-12 in astrocytes) mice develop a spontaneous neurological disorder characterized by weight loss, hunched posture, ruffled fur, ataxia, and muscle atrophy. 28 Multiprobe RPAs were used to determine cytokine gene expression in forebrain and hindbrain from wild-type, GF-p40, presymptomatic, and symptomatic GF-IL12 mice (Figure 1, A and B) ▶ . Levels of IL-12 p40 mRNA were high in GF-p40 mice, very low in presymptomatic GF-IL12 mice, and not detectable in wild-type controls (Figure 1A) ▶ . The level of IL-12 p40 mRNA was, however, increased markedly in the hindbrain of symptomatic GF-IL12 animals. Much lower levels of IL-12 p40 were also present in the forebrain of these animals. Similar levels of IL-12 p35 mRNA were observed in both wild-type and transgenic mice with the exception of the hindbrain and forebrain of symptomatic GF-IL12 animals in which levels of this cytokine transcript were elevated. The expression of the type 1-cytokine genes, IFN-γ, tumor necrosis factor, and IL-1α was induced in symptomatic GF-IL12 mice only, with high levels in the hindbrain and lower levels in the forebrain (Figure 1B) ▶ . Transforming growth factor-β mRNA was detectable in both brain regions from all animals, but increased up to fivefold in hindbrain from symptomatic GF-IL12 mice (Figure 1A) ▶ . In all, these findings confirm and extend our previous observations 28 and indicated that significant induction of the type 1 cytokines IL-1, IFN-γ, and tumor necrosis factor and the counter-regulatory cytokine-transforming growth factor-β, occurred in symptomatic GF-IL12 animals only and overlaps with the expression of the IL-12 p40 and IL-12 p35 genes.
Figure 1.

Expression of a number of cytokine genes was altered in the brain of symptomatic GF-IL12 transgenic mice. In this experiment, poly (A+) RNA was isolated from forebrain and hindbrain of wild-type or GF-p40 and presymptomatic (pres) or symptomatic (sym) GF-IL12 transgenic mice and 10 μg analyzed by RPA as outlined in Materials and Methods.
Constitutive and Regulated Expression of STAT Genes in the Brain of GF-IL12 Transgenic Mice
The STATs are pivotal components of the signaling pathway for a number of type 1-cytokines including IL-12 and IFN-γ, however, little is known about the expression of these genes in the CNS or their role and regulation during chronic inflammatory disease. To begin to address this issue, a multiprobe RPA with probes to all of the known murine STAT genes (STAT1 to STAT6) was developed to analyze STAT gene expression in the brain (Figure 2A) ▶ . With the exception of STAT4 and STAT6 whose expression was very low to undetectable, expression of the remaining STAT genes, as well as IRF9, was readily detectable in the brain from wild-type and GF-p40 mice. In presymptomatic GF-IL12 mice, with the exception of a small increase in STAT1 and IRF9 mRNAs in the hindbrain, little or no detectable alterations were observed in the expression of the other STAT genes. By contrast, in the hindbrain from symptomatic GF-IL12 mice, increased expression was noted for all of the STAT genes and for the IRF9 gene. This was most prominent in the hindbrain for STAT1 (increased 18-fold), STAT4 (increased 35-fold), and IRF9 (increased fivefold). In the forebrain, STAT1 and STAT4 were increased sevenfold and 2.5-fold, respectively (not shown). These findings establish that with the possible exception of STAT4 and STAT6, there is constitutive expression of all of the STAT genes as well as the IRF9 gene in the normal mouse brain. Furthermore, in addition to STAT4 and STAT1 that are central signaling molecules for IL-12 and IFN-γ, respectively, expression of STAT2, STAT3, and IRF9 was also markedly up-regulated in the brain after the induction of the cellular immune response in the brain of the GF-IL12 mice. Finally, the location of the increased STAT gene expression in the GF-IL12 brain overlapped with the levels and sites of cytokine gene expression with highest expression in the hindbrain regions.
Figure 2.

Regulation and anatomy of STAT and SOCS gene expression in the brain of GF-IL12 mice. RNase protection assays for STAT (A) and SOCS (B) RNA used poly (A+) RNA isolated from the hindbrain of wild-type or GF-p40 and presymptomatic (pres) or symptomatic (sym) GF-IL12 transgenic mice and 5 μg were analyzed as outlined in Materials and Methods. Quantitative analysis of RNA levels in A and B was performed by densitometry of scanned autoradiographs using NIH Image 1.57 software. The density level for each RNA band was normalized to the respective level of L32 RNA and the mean ± SEM calculated. With the exception of the SOCS2, SOCS5, and PIAS-1 genes, the levels of RNA were significantly increased (P < 0.05 or less; Student’s t-test) in the hindbrain of symptomatic GF-IL12 mice compared with wild-type, GF-p40, and presymptomatic GF-IL12 mice. Anatomical localization of STAT (C) and SOCS (D) gene expression in the brain. Autoradiographic film images (5-day exposure) of sagittal sections (10 μm) from wild-type or GF-p40 and presymptomatic (pres) or symptomatic (sym) GF-IL12 transgenic mice. Sections were hybridized with 33P-labeled antisense RNA probes as outlined in the Materials and Methods section. Focal areas of probe hybridization are indicated by the arrowheads for STAT4, SOCS1, and SOCS3 RNAs.
Increased STAT-mRNA Is Paralleled by the Corresponding Protein in the Brain of GF-IL12 Mice
We asked whether the increased STAT mRNA expression in the brain of GF-IL12 mice resulted in similar changes in protein production. To address this question, Western blots were performed for STAT1, STAT2, STAT3, STAT4, and STAT5 (Figure 3) ▶ . Constitutive production of STAT1, STAT2, STAT3, and STAT5 but not STAT4 was detectable in extracts of cerebellum from all groups of mice. However, in symptomatic GF-IL12 mice, whereas STAT5a/b protein levels remained unaltered, those for STAT1 and STAT2 were markedly increased and STAT3 moderately increased. In addition, STAT4 protein was clearly present in cerebellum from symptomatic GF-IL12 mice. These findings demonstrate that there is co-ordinate up-regulation in STAT mRNA and protein production in the cerebellum of the GF-IL12 mice. Furthermore, the changes in these STAT proteins showed good overlap with the corresponding alterations in STAT RNA levels.
Figure 3.

The level of different STAT proteins was increased in the hindbrain of symptomatic GF-IL12 mice. Immunoblot analysis was performed as outlined in the Materials and Methods section on lysates of hindbrain (cerebellum plus brain stem) prepared from wild-type or GF-p40 and presymptomatic (pres) or symptomatic (sym) GF-IL12 transgenic mice.
Differential Anatomical and Cellular Localization in the Expression of the STAT4 and STAT1 Genes in the Brain of GF-IL12 Mice
To further determine the gross anatomical localization of the STAT genes in the brain, in situ hybridization was performed (Figure 2C) ▶ . No detectable STAT4 hybridization above background levels was observed in brain from wild-type, GF-p40, or presymptomatic GF-IL12 mice. However, in brain from symptomatic GF-IL12 mice strong hybridization of the STAT4 cRNA probe was observed in focal areas of the cerebellum (Figure 2C ▶ ; arrowheads). Adjacent sections hybridized with a STAT1 cRNA probe revealed near background levels of signal in brain from wild-type and GF-p40 mice. In brain from presymptomatic GF-IL12 mice, hybridization of a highly focal nature was sometimes observed in the cerebellum. In contrast to STAT4, STAT1 hybridization in brain from symptomatic GF-IL12 mice showed a more widespread and diffuse pattern being localized predominantly to the cerebellum, brain stem, basal ganglia, cortex, and hippocampus (Figure 2C) ▶ . Hybridization with a STAT3 cRNA probe revealed a similar pattern and anatomical distribution as seen for STAT4 being detectable in brain from symptomatic GF-IL12 mice only (not shown).
To identify which cells in the brain of the symptomatic GF-IL12 mice were expressing the STAT4 and STAT1 genes, combined in situ hybridization and immunohistochemistry for various cell types was performed. Compared with wild-type (Figure 4A) ▶ , in symptomatic GF-IL12 mice, expression of STAT4 RNA was highly restricted being localized to infiltrating CD3+ (Figure 4B ▶ ; arrows) and CD3− mononuclear cells only. STAT4 RNA was neither detectable in neurons (not shown), nor in astrocytes (not shown). On the whole lectin-positive microglial cells were also negative for STAT4 RNA (Figure 4D ▶ ; arrowheads). Compared with wild-type brain and STAT4, STAT1 RNA was widely expressed in symptomatic GF-IL12 mice being detectable in astrocytes (Figure 5B ▶ ; arrows), various neuronal populations including cerebellar granule and molecular layer neurons, motor neurons in the brain stem (Figure 5D ▶ ; arrows), and neurons of the dentate gyrus and at high levels in infiltrating CD3+ (Figure 5H ▶ ; arrows) and CD3− mononuclear cells. Lower levels of STAT1 RNA were also found in scattered microglia (Figure 5F ▶ ; arrows) and particularly in the cerebellum and brain stem.
Figure 4.

Cellular localization of STAT4 RNA expression in the brain of wild-type (A and C) or symptomatic GF-IL12 (B and D) mice. Double-labeling experiments using in situ hybridization with 33P-labeled antisense STAT4 RNA probe and immunostaining for CD3 (A and B) or binding of tomato lectin (C and D). In B, the arrows indicate some representative cells positive for CD3 and STAT4. In D, STAT4 RNA-positive cells were negative (arrows) for lectin binding. In D, lectin-positive cells are denoted by the arrowheads. Original magnifications, ×400.
Figure 5.
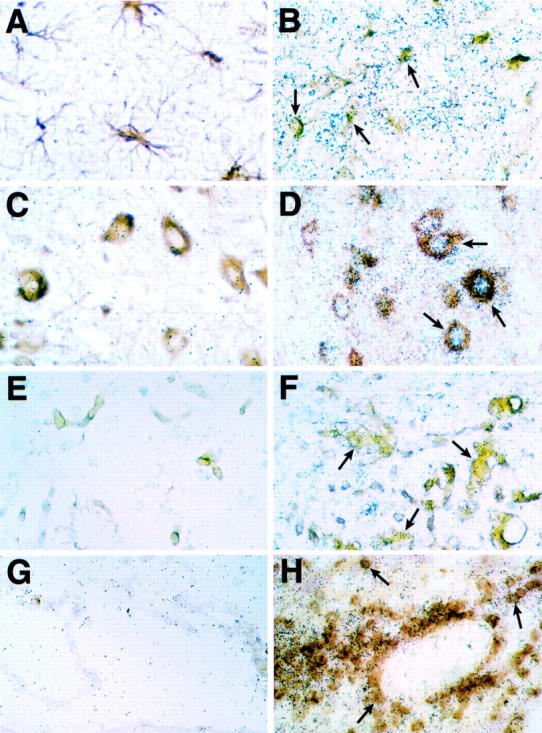
Cellular localization of STAT1 RNA expression in the brain of wild-type (A, C, E, and G) or symptomatic GF-IL12 (B, D, F, and H) mice. Double-labeling experiments using in situ hybridization with 33P-labeled antisense STAT1 RNA probe plus immunostaining for GFAP (A and B), neurofilament (C and D), and CD3 (G and H) or binding of tomato lectin (E and F) were performed as described in Materials and Methods. The arrows in each panel indicate double-positive cells. Original magnifications: ×600 (A–F); ×400 (G and H).
These experiments highlighted cellular compartmentalization in the spatial expression of the STAT4 and STAT1 genes in the CNS with the former being highly restricted to the infiltrating leukocyte population whereas the latter is widespread and in addition to the infiltrating leukocytes included neurons, astrocytes, and macrophage/microglia.
In Situ Protein Localization and Evidence for Functional Activation of STAT4, STAT1, and STAT3
Functional activation of the STAT proteins is associated with their dimerization and translocation into the nucleus. Immunostaining for STAT4, STAT1, and STAT3 was performed to visualize the cellular and subcellular localization of these proteins (Figure 6) ▶ . Similar to the expression of STAT4 RNA, STAT4 protein was not detectable in brain from wild-type, GF-p40, or presymptomatic GF-IL12 (Figure 6C) ▶ mice. However, in symptomatic GF-IL12 mice, expression of STAT4 protein was readily detected and localized entirely to the infiltrating mononuclear cells (Figure 6D) ▶ . Staining for STAT4 protein in the majority of positive cells was seen in both the cytoplasm and nucleus (Figure 6D ▶ ; arrows). Staining for STAT3 protein was also only detectable in infiltrating mononuclear cells in brain from symptomatic GF-IL12 mice (Figure 6B) ▶ . However, compared with STAT4, the number of cells positive for STAT3 protein was markedly less and these tended to be larger cells located at the boundaries of the inflammatory infiltrates. Nonetheless, STAT3-positive cells also displayed cytoplasmic and nuclear staining (Figure 6B ▶ ; arrows).
Figure 6.

Restricted cellular distribution and nuclear localization of the STAT3 and STAT4 proteins in the brain of symptomatic GF-IL12 mice. Sections from Bouin’s-fixed brain were immunostained with polyclonal antibody against either murine STAT3 (A and B) or STAT4 (C and D) protein as described in Materials and Methods. Although no staining was detectable in brain from wild-type mice (A and C), numerous immunostained cells are evident in mononuclear cell infiltrates in cerebellum from a symptomatic GF-IL12 mouse (B and D). Both STAT3-positive (B) and STAT4-positive (D) cells exhibit nuclear staining (arrows). Original magnifications, ×600.
In contrast to STAT4 and STAT3, STAT1 protein was expressed more widely throughout the brain of symptomatic GF-IL12 mice (Figure 7) ▶ . Compared with similarly stained brain from wild-type mice, in symptomatic GF-IL12 mice dense staining of the neuropil was seen particularly in the cerebellum where numerous cells present also displayed nuclear staining (Figure 7B ▶ ; arrow). Surprisingly however, and despite expressing high levels of RNA (see above), infiltrating mononuclear cells exhibited little STAT1 protein (Figure 7B ▶ ; asterisk). In addition to neurons, numerous cells in periventricular subcortical white matter tracts corresponding morphologically to oligodendrocytes exhibited nuclear staining for STAT1 (Figure 7C ▶ ; arrows). In symptomatic GF-IL12 mice, astrocytes (Figure 7D ▶ ; arrows) and microglia (Figure 7D ▶ ; arrowheads) were also found to have increased STAT1 protein levels.
Figure 7.
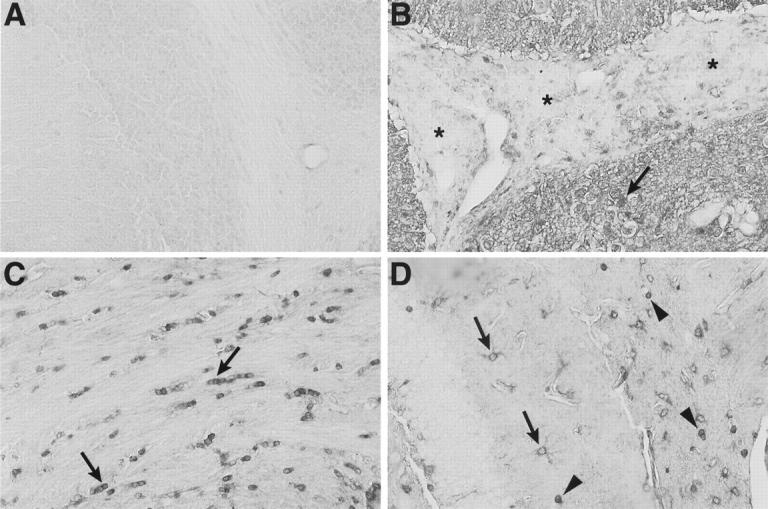
Wide cellular distribution and nuclear localization of STAT1 protein in the brain of symptomatic GF-IL12 mice. Sections from Bouin’s-fixed brain were immunostained with polyclonal antibody against murine STAT1 protein as described in Materials and Methods. Little staining was detectable in brain from wild-type mice (A). By contrast, in GF-IL12 brain (B, C, and D), strong, diffuse staining of neuropil was evident in the molecular layer of the cerebellum (B) with many nuclei exhibiting nuclear staining (arrow). However, in the same section, the majority of infiltrating mononuclear cells (shown here in the subarachnoid space) had only low staining (B, asterisks). In subcortical white matter adjacent to the lateral ventricle (C, arrows), nuclei with the morphological and spatial characteristics corresponding to oligodendrocytes, showed strong nuclear staining for STAT1. Cytoplasmic staining of cells with the morphological characteristics of astrocytes (D, arrows) and surrounding cells with strong nuclear staining (D, arrowheads) is shown in the hippocampal region. Original magnifications, ×400.
These immunohistochemical studies illustrated further the compartmentalized expression of STAT3 and STAT4 versus widespread expression of STAT1 in the brain of the GF-IL12 mice. The nuclear localization observed for STAT4, STAT3, and STAT1 is consistent with the activation of these transcriptional factors. With the exception of STAT1 in the infiltrating mononuclear cell population, STAT protein localization also showed good concordance with the corresponding STAT RNA.
Because STAT1 was by far the most abundant of all of the STAT proteins examined in the brain of the symptomatic GF-IL12 mice, further evidence for its activation was sought by electrophoretic mobility shift assay. Total protein extracts prepared from the cerebellum of wild-type or symptomatic GF-IL12 mice were examined for the presence of STAT1-GAS-binding complexes (Figure 8) ▶ . Compared with wild-type controls, extracts from the GF-IL12 mice contained a unique binding complex (Figure 8 ▶ ; arrow) that had a similar size to a positive control STAT1-binding complex present in whole-cell extract prepared from IFN-γ-stimulated HeLa cells. The binding of the unique complex to the GAS probe present in the GF-IL12 samples could be prevented by the addition of a STAT1 antibody but not by an antibody to nuclear factor-κB. The apparent reduced level of binding in the presence of the nuclear factor-κB antibody evident in Figure 8 ▶ was not found in repeated experiments. Finally, addition of unlabeled GAS probe to the binding reaction resulted in a dose-dependent reduction in binding activity. The presence of STAT1 protein-GAS DNA-binding complexes in the cerebellum of the GF-IL12 mice further confirms the functional activation of this signaling pathway.
Figure 8.

STAT1-dependent, IRF-1γ-activated sequence (GAS) DNA-binding activity in protein extracts of brain from GF-IL12 mice. Whole protein extracts were prepared from freshly removed cerebellum from wild-type or symptomatic GF-IL12 mice. Extracted protein was analyzed by electrophoretic mobility shift assay using 32P-labeled IRF-1 GAS-containing oligonucleotide probe and DNA-binding complexes were resolved by nondenaturing 4 to 12% gradient polyacrylamide gel electrophoresis and visualized by autoradiography as described in Materials and Methods. Specificity of binding was assessed either by the addition to the protein extracts before the binding assay of anti-STAT1 or nuclear factor-κB antibody or of a 10-fold (GAS 1) or 100-fold (GAS 10) excess of unlabeled GAS oligonucleotide. The positive (+ve) control was whole-cell extract prepared from IFN-γ-stimulated HeLa cells kindly provided by Dr. Christian Schindler (Columbia University, New York, NY).
SOCS1 and SOCS3 Gene Expression Is Up-Regulated and Localized Mainly to Infiltrating Mononuclear Cells in Symptomatic GF-IL12 Mice
Cytokine signaling via the JAK/STAT pathway is subject to negative feedback control by a variety of molecules that either prevent activation of the STAT proteins or prevent their binding to target DNA sequences. Little, if anything, is known about the expression of these genes in the CNS or their role in regulating cytokine signaling during inflammatory disease. As a first step toward examining this important regulatory pathway, a multiprobe RPA was developed that permitted the detection of SOCS1, SOCS2, SOCS3, SOCS5, and PIAS-1 gene expression in the brain (Figure 2B) ▶ . In brain from wild-type, GF-p40, presymptomatic, and symptomatic GF-IL12 mice high constitutive expression of PIAS1 and SOCS2 and low constitutive expression of SOCS5 was detectable. Low constitutive expression of the SOCS1 and SOCS3 genes was also found in the forebrain (not shown), and in the case of SOCS1 but not SOCS3, the hindbrain from wild-type, GF-p40, and presymptomatic GF-IL12 mice (Figure 2B) ▶ . However, in symptomatic GF-IL12 mice there was a marked increase in the expression of the SOCS1 and SOCS3 mRNA that was primarily restricted to the hindbrain (Figure 2B) ▶ .
To determine the gross anatomical localization of the SOCS1 and SOCS3 genes, in situ hybridization was performed (Figure 2D) ▶ . No detectable SOCS1 or SOCS3 hybridization above background levels was observed in brain from wild-type, GF-p40, or presymptomatic GF-IL12 mice. However, in brain from symptomatic GF-IL12 mice strong hybridization of the SOCS1 and SOCS3 cRNA probes was seen in focal areas of the cerebellum with a similar pattern and anatomical distribution as shown for STAT4 gene expression. Further analysis was performed using dual-label in situ hybridization and immunohistochemistry to identify which cells in the brain expressed the SOCS1 (Figure 9 ▶ ; A, B, and C) and SOCS3 (Figure 9 ▶ ; D, E, and F) genes. No hybridization signal above background levels was detectable in brain from wild-type mice for either SOCS1 (Figure 9A) ▶ or SOCS3 (Figure 9D) ▶ . In contrast, strong hybridization signal was detectable for these genes in brain from the symptomatic GF-IL12 mice. SOCS1 RNA was expressed predominantly by CD3+ (Figure 9B ▶ ; arrows) and CD3− (Figure 9B ▶ ; arrowhead) cells and rarely by lectin-positive cells (Figure 9C ▶ ; arrow). Similarly, SOCS3 RNA was also expressed predominantly by CD3+ (Figure 9E ▶ ; arrows) and CD3− (Figure 9E ▶ ; arrowhead) cells. Increased numbers of lesion-associated (Figure 9F ▶ ; arrows) but not parenchymal (Figure 9F ▶ ; arrowhead) lectin-positive cells also expressed SOCS3 RNA. These experiments revealed that expression of the SOCS1 or SOCS3 RNA was primarily restricted to the infiltrating CD3+ and CD3− mononuclear cells and to a small number of lesion-associated macrophage/microglia.
Figure 9.
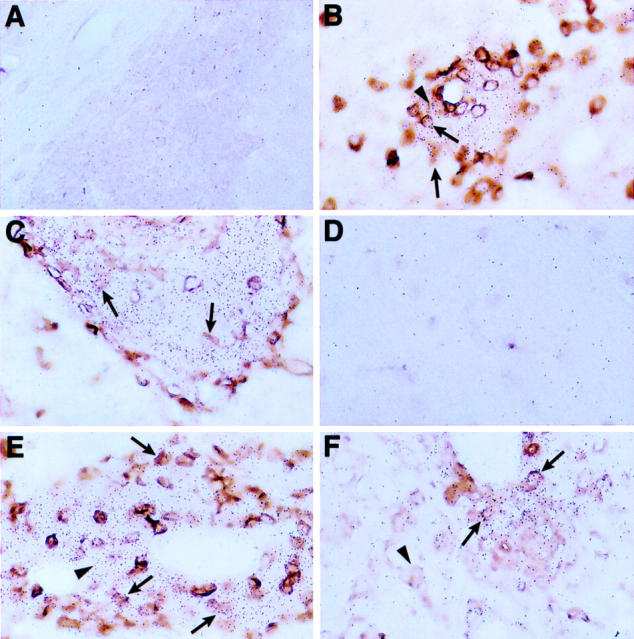
Restricted cellular distribution of SOCS1 and SOCS3 RNA in the brain of symptomatic GF-IL12 mice. Double-labeling experiments using in situ hybridization with 33P-labeled antisense SOCS1 (A, B, and C) or SOCS3 (D, E, and F) RNA probe plus immunostaining for CD3 (A, B, D, and E) or binding of tomato lectin (C and F) were performed as described in Materials and Methods. Background levels of hybridization were detectable to brain sections from the wild-type animal (A and D). In contrast, areas of high hybridization were observed in brain sections from the GF-IL12 mice (B, C, E, and F) and corresponded to mainly infiltrating CD3+ (arrows) and CD3− (arrowheads) mononuclear cells and a small number of lesion-associated lectin+ cells (arrows). Original magnifications, ×400.
Expression of the STAT and SOCS Genes Is Highly Regulated in CNS of Mice with EAE and Shows Concordance with Symptomatic GF-IL12 Mice
MOG-EAE is an inflammatory demyelinating disease perpetrated by CD4+Th1 cells reactive to components of the myelin sheath in whose pathogenesis IL-12 plays an essential role. 4 We therefore examined the regulation of the STAT and SOCS genes in mice in different CNS regions and at different phases in the development of MOG-induced EAE (Figure 10, A and B) ▶ . With the exception of STAT5 and STAT6 whose expression remained unaltered, significant increases in the expression of STAT1, STAT2, STAT3, STAT4, and IRF9 was seen at the height (day 14) of clinical disease in all CNS regions and declined during the remission phase (day 28) (Figure 10A) ▶ . Significant increases in the expression of STAT1, STAT3, and STAT4 were also found, particularly in the cerebellum, at day 6 after MOG immunization and before the development of clinical EAE. In situ hybridization analysis of brain sections from mice with EAE (day 14) showed widespread expression of STAT1 RNA throughout the brain and spinal cord, whereas expression of STAT4 RNA was limited and localized to highly focal areas in the cerebellum, brain stem, and spinal cord (Figure 10 ▶ ; arrows). Further analysis of STAT4 and STAT1 protein was performed by immunohistochemical staining of brain sections from control mice or mice with EAE (Figure 11) ▶ . Although not detectable in the control brain (Figure 11 ▶ A), high levels of STAT4 protein were present in the cytoplasm and nucleus of a subset of cells present in the mononuclear cell infiltrates (Figure 11B ▶ ; arrows). By contrast, and in comparison with the control (Figure 11 ▶ ; C, E, and G), STAT1 protein was markedly increased in various cell types (Figure 11 ▶ ; D, F, and H). Similar to the GF-IL12 mice, oligodendrocytes in periventricular white matter (Figure 11D ▶ ; arrows) and hippocampal astrocytes (Figure 11F ▶ ; arrows) were both positive for STAT1. Although not evident at these high magnifications, infiltrating mononuclear cells were prominent in the choroid plexus and blood vessels adjacent to the ventricle. In contrast with the GF-IL12 mice, infiltrating mononuclear cells in EAE displayed high levels of STAT1 protein (Figure 11H) ▶ .
Figure 10.

Regulation and anatomy of STAT and SOCS gene expression in the CNS of mice with EAE. Cerebrum (C), cerebellum (CB), and spinal cord (SC) were removed at the times shown from mice after immunization with MOG-peptide. Poly (A+) (cerebrum and cerebellum) or total (spinal cord) RNA was then extracted and 5 μg or 10 μg, respectively, analyzed by RPA for the detection of STAT (A) or SOCS (B) RNA as outlined in Materials and Methods. Densitometric analysis of each band was performed on scanned autoradiographs and quantitated using NIH Image 1.57 software. The density level for each RNA was normalized to the respective level of L32 RNA and the mean plus SEM calculated using Microsoft Excel. For statistical significance asterisks indicate P < 0.05 or less (Student’s t-test) compared with corresponding control (day 0). Anatomical localization (C) of STAT1 and STAT4 or SOCS1 and SOCS3 gene expression in the brain during EAE. Autoradiographic film images (5-day exposure) of sagittal sections (10 μm) from control mice or mice with EAE. Sections were hybridized with 33P-labeled antisense RNA probes as outlined in Materials and Methods. Focal areas of probe hybridization are indicated by the arrows for STAT4, SOCS1, and SOCS3 RNAs.
Figure 11.
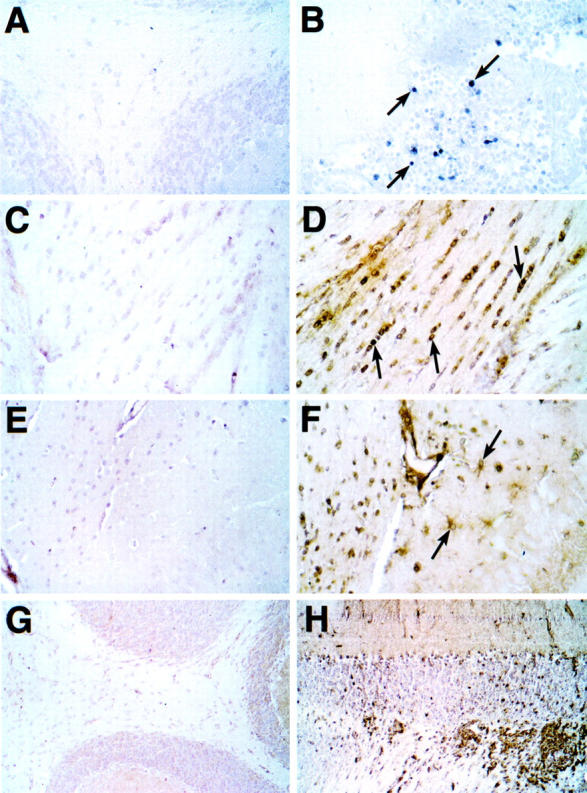
Distribution of STAT4 and STAT1 proteins in the brain during EAE. Sections from Bouin’s-fixed brain were immunostained with polyclonal antibody against either murine STAT4 (A and B) or STAT1 (C–H) protein as described in Materials and Methods. Although little staining was detectable for STAT4 in brain from control mice (A), numerous mononuclear cells were seen with strong cytoplasmic and nuclear staining in infiltrates in cerebellum (B, arrows) and spinal cord from mice with EAE. Very low staining for STAT1 was observed in brain from control mice (C, E, and G). In brains from mice with EAE, STAT1 staining was increased dramatically in most regions of the brain including subcortical periventricular white matter tracts (D), hippocampus (F), and cerebellum (H). In white matter nuclei with the morphological and spatial characteristics corresponding to oligodendrocytes showed strong nuclear staining for STAT1 (D, arrows). Cytoplasmic staining of cells with the morphological characteristics of astrocytes (F, arrows) is shown in the hippocampal region. Original magnifications: ×400 (A–F); ×100 (G and H).
Of the SOCS genes examined, only the expression of SOCS1 and SOCS3 was altered during EAE. The levels of these transcripts increased significantly in cerebellum and spinal cord at the height of disease (day 14) and then declined, but remained increased above control levels during the remission phase (Figure 10B) ▶ . Finally, in situ localization analysis for these genes revealed highly restricted and focal expression in spinal cord and cerebellum of mice with EAE (Figure 10C ▶ ; arrows).
To summarize, and in comparison with the GF-IL12 mice, we found in EAE that there is remarkable parallelism in the regulation and localization of the STAT and SOCS genes that are predominantly involved in IL-12R and IFN-γR-mediated signaling.
Discussion
Multiple sclerosis and EAE are closely linked to the influence of IL-12, IFN-γ, and the development of type 1 cellular immune responses. 4,35,36 STAT4 37,38 and STAT1 12,39 are known to be pivotal components of the molecular circuitry involved in IL-12 and IFN-γ signaling, respectively. Currently, little is known about the biology of these key molecular transducers of IL-12 and IFN-γ signaling during actual cellular immune responses in vivo. We have documented here that both STAT4 and STAT1 expression is highly regulated and exhibits cellular compartmentalization in the brain of transgenic mice undergoing a spontaneous type 1 cellular autoimmune response that is induced by the astrocyte-targeted expression of IL-12 or in mice with EAE. The cerebral expression of the STAT2- and STAT3-signaling molecules as well as IRF9 was also up-regulated in these models. Additionally, we demonstrated that the cerebral expression of two molecules known to be involved in the physiological feedback down-regulation of the JAK/STAT-signaling pathway, namely SOCS1 and SOCS3, was increased and restricted primarily to the CNS-infiltrating mononuclear cell population during the evolution of type 1 immune responses in these models.
STAT4 is expressed by NK cells, T cells, monocytes, dendritic cell, and spermatogonia. 40 It is now well established that IL-12 induces STAT4 activation in T cells and NK cells and this process is critical in the initiation and control of cellular immunity by this cytokine. 13-16 The extent to which IL-12 exerts actions on cells intrinsic to the CNS, and indeed, in other tissues, is unknown. In the GF-IL12 mice before the initiation of the immune response there are no detectable molecular and cellular alterations suggesting that IL-12 exerts little if any direct effects in the CNS. This correlates well with our observation here that there is an absence of detectable STAT4 gene expression by cells intrinsic to this tissue. Moreover, the restricted localization of activated STAT4 to the CNS-infiltrating mononuclear cell population in both the GF-IL12 mice and in EAE, argues further that these immune cells are the primary targets for the action of IL-12. In these cells, the engagement of the IL-12R with its ligand likely results in the activation of the STAT4-signaling pathway and subsequent modulation of gene transcription including the induction of IFN-γ gene expression and protein production. In ongoing studies, we are using mice with a targeted disruption of the STAT4 gene 16 to directly test the validity of this hypothesis.
IL-12 also activates STAT3 in T cells and NK cells, 13,14 however, the role of this signaling molecule in mediating the effects of IL-12 is unknown. In contrast to STAT4, STAT3 is expressed constitutively in the normal rodent brain 41,42 and is up-regulated with nuclear translocation in astrocytes and macrophage/microglia after excitotoxic injury in vivo. 43 Here we found that similar to STAT4, up-regulated STAT3 protein expression and nuclear localization was restricted primarily to the infiltrating mononuclear cell population and not detectable in resident brain cells of the adult mouse. The lower numbers and different location of the STAT3-containing cells within the infiltrates suggests they may represent a distinct IL-12 target population of leukocytes from those that express STAT4. The extent to which IL-12 actions in the CNS of the GFAP-IL12 mouse or in EAE are mediated via the STAT3-signaling pathway is unclear. By default, insights to this might come from our ongoing studies of GFAP-IL12 mice deficient for the STAT4.
The stimulation of IFN-γ production from Th1 cells and NK cells is a key action of IL-12 in the development of type 1 cellular immune responses. 9,44 Consistent with this, in a previous 28 and in the present study we have documented IFN-γ gene expression by infiltrating CD3+ and CD3− (presumed NK) mononuclear cells in the CNS of symptomatic GF-IL12 mice. In addition, IFN-γ is found in the CNS during a variety of cell-mediated immune responses including those associated with viral infection 45,46 as well as in multiple sclerosis and EAE. 35,47 IFN-γ has diverse actions in the CNS ranging from immunoregulation and inhibition of viral replication to modulation of the function and viability of many neural cell types including neurons, astrocytes, oligodendrocytes, and microglia. 35,48,49 IFN-γ binding to its receptor promotes tyrosine phosphorylation and homodimerization of STAT1 that then translocates to the nucleus and binds to GAS sites activating gene transcription. 39 Mice with targeted disruption of the STAT1 gene lack responsiveness to IFN-γ. 17,18 Studies in vitro show that IFN-γ induction of the class II transactivator and MHC class II, 50 ICAM-1, 51 and MCP-1 52 genes in astrocytes, and the CD40 53 and FAS 54 genes in microglia involves STAT1. STAT1 is known to be present in the developing and adult CNS although its cellular localization was not reported. 41 Here we confirmed that there is low constitutive expression of the STAT1 gene in the adult mouse CNS. However, this is clearly not static as indicated by the markedly increased levels seen in symptomatic GF-IL12 mice and in EAE. The increased expression of STAT1 RNA and protein and its nuclear translocation was widely disseminated both at a regional and cellular level and included neurons, astrocytes, microglia, and oligodendrocytes. Despite this, there was a relationship between the magnitude and topography of STAT1 expression with the inflammatory process. In the GF-IL12 mice, this was highest in the cerebellum and brain stem where diffuse expression of the STAT1 gene was observed by all of the neural cells whereas expression was lowest in the frontal region of the brain where a small number of more specialized cells showed increased expression. We have previously noted 28 that the expression of the transgene encoded IL-12 and the development of immune pathology exhibit a similar trend being highest and more widespread in the cerebellum and brain stem and least so in the frontal regions of the brain. Consistent with this, the cytokine RPA shown in Figure 1 ▶ showed higher levels of IL-12 p40, p35, and IFN-γ in the cerebellum/brain stem compared with the forebrain. IFN-γ in particular is a good marker for the presence of activated T cells and NK cells and therefore reflects the extent of the inflammatory process. A further point made by our findings is that there may be regional differences in the regulation of STAT1 gene expression in specific brain cells. Thus, although astrocytes located in the hippocampus showed increased levels of STAT1 protein, in periventricular white matter where oligodendrocytes were clearly STAT1-positive this was not detected in the astrocytes.
The presence of STAT1-GAS-binding complexes in cerebellar extracts from symptomatic GF-IL12 mice further confirmed the functional activation of the STAT1-signaling pathway. STAT1 RNA and protein expression can be significantly up-regulated in a variety of cells in vitro, particularly by IFN-γ 55,56 and the type 1 IFNs 55 and this may be further augmented by tumor necrosis factor. 57,58 The increased CNS expression of STAT1 in the GF-IL12 mice and in EAE is most likely mediated directly by IFN-γ alone or in combination with other cytokines such as tumor necrosis factor. Therefore, IFN-γ and STAT1 likely constitute a positive autoregulatory loop the function of which is not known, but conceivably it might amplify IFN-γR-activated STAT1-dependent responses to include cells such as those intrinsic to the CNS that under nonstimulated conditions have very low levels of STAT1. Another possibility is that STAT1 has functions independent of IFN-γR-mediated signaling. 59,60 Currently, we are using mice with a targeted disruption of the STAT1 gene to determine the precise function of STAT1 in IFN-γ-regulated gene expression and actions in the CNS of the GF-IL12 mice as well as in EAE.
Of the remaining (ie, STAT2, STAT5, and STAT6) STAT genes examined, only the expression of STAT2 was elevated significantly in the CNS of symptomatic GF-IL12 mice and in EAE. STAT2 activation is closely linked to IFNα/β-receptor binding, 61-63 however, IFN-γR binding 64 may also activate STAT2. 65 Activated STAT2 associates with STAT1 and IRF9 to form the transcriptionally active complex IFN-stimulated gene factor-3 (ISGF3) that binds to the interferon-stimulated response element sequence. 66,67 IRF9 expression was also increased in the brain of symptomatic GF-IL12 mice and in EAE. Activation of both STAT2 68 and IRF9 (which is also known as p48 and ISGF3γ) 55,69 gene transcription can be induced directly by IFN-γ. The finding that both STAT2 and IRF9 were increased in the brain of symptomatic GF-IL12 mice and in EAE raises the possibility these additional transcriptional factors might also contribute to IFN-γ- or other cytokine-receptor-mediated signaling. Indeed, IRF9 can associate with IFN-γR-activated STAT1 homodimers to form a complex that mediates secondary responses by binding to the interferon-stimulated response element. 70 However, because the affinity of this binding is somewhat lower than for ISGF3, only a restricted subset of interferon-stimulated response element-containing genes are modulated by IFN-γ. At this time the precise function of the STAT2 and IRF9 transcriptional factors in the genesis of the cellular responses to IL-12 and IFN-γ in the CNS remains unknown.
Our findings with regard to the regulation of the STAT genes in EAE somewhat agree with and extend on those of Jee and colleagues. 71 However, in contrast to our findings here, these workers found that the expression of STAT1 protein was much more restricted being localized to mainly neurons, whereas STAT4 or STAT3 proteins, in addition to being found in neurons in the brain of healthy rats showed increased expression in microglia and endothelial cells or astrocytes, endothelial cells, and meninges, respectively. We could neither confirm the constitutive expression of STAT4 nor the nature of the cell types expressing STAT4, STAT1, and STAT3 in EAE. There are clear technical differences between the two studies that might explain these dichotomies. In addition, in the studies of Jee at al 71 rats were used. This raises the possibility that there may be species differences in the regulation and localization of the STAT genes.
Negative regulation of the JAK/STAT pathway is an important process that contributes to the overall response of cells to cytokine receptor-mediated signaling. The SOCS and PIAS genes are cytokine-inducible and the molecules they encode play a central role in the down-regulation of cytokine receptor signaling. 21,72,73 Little is known concerning the temporal and spatial expression of these genes in immunoinflammatory diseases of the CNS. Polizzotto and colleagues 74 reported high levels of SOCS2 RNA expression by neurons in the developing and adult CNS of mice. Studies in SOCS2-deficient mice show this molecule is important in the negative regulation of IGF-1 signaling and may therefore play an important role in neuronal development. 26 Here we confirmed the high constitutive cerebral expression of SOCS2 but also revealed even higher constitutive expression of PIAS1. PIAS1 binds to activated STAT1 and blocks its DNA-binding activity and may be an important negative regulator of IFN-R signaling. 23 In the brain this molecule might have a general role as a negative regulator of signaling by IFN-γ and perhaps other cytokines.
In contrast to SOCS2 and PIAS1, the expression of the SOCS1 and SOCS3 genes that was very low in normal brain, increased markedly in the brain of symptomatic GF-IL12 mice and in EAE. In both models, this expression was highly restricted and limited almost entirely to the infiltrating mononuclear cell population. It should be noted that despite our best efforts using antibodies from different sources it was not possible to detect either the SOCS1 or SOCS3 proteins. The difficulty in detecting these SOCS proteins in vivo may reflect their instability and rapid turnover. 75 Structurally, SOCS1 and SOCS3 are similar to each other and not SOCS2. 20 Moreover, in vitro studies reveal that the SOCS1 and SOCS3 but not the SOCS2 molecules are functionally promiscuous inhibiting signaling by a number of the same cytokines, including IFN-γ, IL-6, LIF, and granulocyte macrophage-colony stimulating factor. 76,77 However, mice with targeted disruptions of the SOCS1 24,25 or SOCS3 27 genes have divergent phenotypes that highlight critical dominant primary roles for these molecules in IFN-γ or erythropoietin receptor signaling, respectively. Together, these observations suggest it is likely that the expression of SOCS1 and SOCS3 noted by us in symptomatic GF-IL12 mice is associated with a general down-regulation in the responses of the infiltrating mononuclear cells to IFN-γ and perhaps other cytokines. The importance of this regulatory process is dramatically illustrated by the case of SOCS1 null mice. SOCS1 deficiency causes perinatal lethality because of the uncontrolled emergence of activated T cells that produce high levels of IFN-γ. Consequently, target tissues that normally are able to down-regulate IFN-γR signaling become susceptible to the toxic actions of this cytokine.
In conclusion, we have defined the CNS expression patterns and regulatory control of crucial components of the signaling pathways that facilitate cellular communication in vivo by cytokines during the evolution of type 1 immunity. The results indicated that the expression of key positive, ie, STAT, and negative, ie, SOCS, regulatory factors involved in IL-12 (STAT4) and IFN-γ (STAT1, SOCS1, and SOCS3) receptor-mediated signaling is highly regulated and compartmentalized during active immune responses in the CNS in both the GF-IL12 and EAE models. These findings document great similarities in the molecular and cellular pathological processes at play in the brain in these two different models. Therefore, the interaction between the positive (ie, STAT) and negative (ie, SOCS) signaling circuits and their distinct cellular locations likely play a defining role in the actions of IL-12 and IFN-γ during the pathogenesis of type 1 immune responses in the CNS.
Acknowledgments
We thank Dr. Christian Schindler (Columbia University, New York) for providing key reagents, and Heather Kemlein for administrative assistance in the preparation of this manuscript.
Footnotes
Address reprint requests to Dr. Iain L. Campbell, The Scripps Research Institute, Maildrop SP-315, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. E-mail: icamp@scripps.edu.
Supported by National Institutes of Health Grant NS 36979.
Current address of J. M.: Department of Neuropathology, University of Freiburg, Freiburg, Germany.
References
- 1.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH: The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol 1988, 16:495-521 [DOI] [PubMed] [Google Scholar]
- 2.Trinchieri G: Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol 1998, 70:83-243 [DOI] [PubMed] [Google Scholar]
- 3.Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL: Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci USA 1997, 94:599-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segal BM, Dwyer BK, Shevach EM: An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J Exp Med 1998, 187:537-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Windhagen A, Newcombe J, Dangond F, Strand C, Woodroofe MN, Cuzner ML, Hafler DA: Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med 1995, 182:1986-1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aloisi F, Penna G, Cerase J, Iglesias B, Adorini L: IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol 1997, 159:1604-1612 [PubMed] [Google Scholar]
- 7.Becher B, Dodelet V, Fedorowicz V, Antel JP: Soluble tumor necrosis factor receptor inhibits interleukin 12 production by stimulated human adult microglial cells in vitro. J Clin Invest 1996, 98:1539-1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stalder A, Pagenstecher A, Yu NC, Kincaid C, Chiang CS, Hobbs MV, Bloom FE, Campbell IL: Lipopolysaccharide induced interleukin-12 expression in the CNS and cultured astrocytes and microglia. J Immunol 1997, 159:1344-1351 [PubMed] [Google Scholar]
- 9.Trinchieri G: Interleukin-12 and interferon-γ. Do they always go together? Am J Pathol 1995, 147:1534-1538 [PMC free article] [PubMed] [Google Scholar]
- 10.Ransohoff RM: Cellular responses to interferons and other cytokines: the JAK-STAT paradigm. N Engl J Med 1998, 338:616-618 [DOI] [PubMed] [Google Scholar]
- 11.Schindler C, Strehlow I: Cytokines and STAT signaling. Adv Pharmacol 2000, 47:113-174 [DOI] [PubMed] [Google Scholar]
- 12.Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD: How cells respond to interferons. Annu Rev Biochem 1998, 67:227-264 [DOI] [PubMed] [Google Scholar]
- 13.Yu CR, Lin JX, Fink DW, Akira S, Bloom ET, Yamauchi A: Differential utilization of janus kinase-signal transducer and activator of transcription signaling pathways in the stimulation of human natural killer cells by IL-2, IL-12, and IFN-α. J Immunol 1996, 157:126-137 [PubMed] [Google Scholar]
- 14.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Murphy KM: Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med 1995, 181:1755-1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN: Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996, 382:171-174 [DOI] [PubMed] [Google Scholar]
- 16.Kaplan MH, Sun YL, Hoey T, Grusby MJ: Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 1996, 382:174-177 [DOI] [PubMed] [Google Scholar]
- 17.Meraz MA, White JM, Sheehan KCF, Bach EA, Rodig SJ, Dighe AS, Kaplan DS, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD: Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996, 84:431-442 [DOI] [PubMed] [Google Scholar]
- 18.Durbin JE, Hackenmiller R, Simon MC, Levy DE: Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996, 84:443-450 [DOI] [PubMed] [Google Scholar]
- 19.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A: A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387:921-924 [DOI] [PubMed] [Google Scholar]
- 20.Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA: Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci USA 1998, 95:114-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starr R, Willson TA, Viney EM, Murray LJL, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ: A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387:917-921 [DOI] [PubMed] [Google Scholar]
- 22.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K: Specific inhibition of Stat 3 signal transduction by PIAS3. Science 1997, 278:1803-1805 [DOI] [PubMed] [Google Scholar]
- 23.Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K: Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA 1998, 95:10626-10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TWH, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ: SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999, 98:597-608 [DOI] [PubMed] [Google Scholar]
- 25.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN: SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 1999, 98:609-616 [DOI] [PubMed] [Google Scholar]
- 26.Metcalf D, Greenhaigh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ, Alexander WS: Gigantism in mice lacking suppressor of cytokine signaling-2. Nature 2000, 405:1069-1073 [DOI] [PubMed] [Google Scholar]
- 27.Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, Pendeville H, Yasukawa H, Sasaki A, Yoshimura A, Ihle JN: SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell 1999, 98:617-627 [DOI] [PubMed] [Google Scholar]
- 28.Pagenstecher A, Lassmann S, Carson MJ, Kincaid CL, Stalder AK, Campbell IL: Astrocyte-targeted expression of IL-12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J Immunol 2000, 164:4481-4492 [DOI] [PubMed] [Google Scholar]
- 29.Badley JE, Bishop GA, St. John T, Frelinger JA: A simple, rapid method for the purification of poly A+ RNA. Biotechniques 1988, 6:114-116 [PubMed] [Google Scholar]
- 30.Stalder A, Pagenstecher A, Kincaid C, Campbell IL: Analysis of gene expression by multiprobe RNase protection assay. Harry J Tilson HA eds. Neurodegeneration Methods and Protocols. 1999, :pp 53-66 Totowa, Human Press [DOI] [PubMed] [Google Scholar]
- 31.Hobbs MV, Weigle WO, Noonan DJ, Torbett BE, McEvilly RJ, Koch RJ, Cardenas GJ, Ernst DN: Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J Immunol 1992, 150:3602-3068 [PubMed] [Google Scholar]
- 32.Dudov KP, Perry RP: The gene family encoding the mouse ribosomal protein L32 contains a uniquely expressed intron-containing gene and an unmutated processed gene. Cell 1984, 37:457-468 [DOI] [PubMed] [Google Scholar]
- 33.Asensio VC, Lassmann S, Pagenstecher A, Steffensen SC, Henriksen SJ, Campbell IL: C10 is a novel chemokine expressed in experimental inflammatory demyelinating disorders that promotes the recruitment of macrophages to the central nervous system. Am J Pathol 1999, 154:1181-1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park C, Schindler C: Protein-DNA interactions in interferon-γ signaling. Methods 1998, 15:175-188 [DOI] [PubMed] [Google Scholar]
- 35.Owens T, Renno T, Taupin V, Krakowski M: Inflammatory cytokines in the brain: does the CNS shape immune responses? Immunol Today 1994, 15:566-571 [DOI] [PubMed] [Google Scholar]
- 36.Segal BM, Shevach EM: IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med 1996, 184:771-775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplan MH, Grusby MJ: Regulation of T helper cell differentiation by STAT molecules. J Leukoc Biol 1998, 64:2-5 [DOI] [PubMed] [Google Scholar]
- 38.Murphy KM, Ouyang W, Szabo SJ, Jacobson NG, Guler ML, Gorham JD, Gubler U, Murphy TL: T helper differentiation proceeds through STAT-1 dependent, STAT4-dependent and STAT4-independent phases. Curr Top Microbiol Immunol 1999, 238:13-26 [DOI] [PubMed] [Google Scholar]
- 39.Darnell JE, Kerr IM, Stark GR: Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264:1415-1421 [DOI] [PubMed] [Google Scholar]
- 40.Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, Centola M, Wu CY, Yamada N, Gabalawy HE, O’Shea JJ: Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J Immunol 2000, 164:4659-4664 [DOI] [PubMed] [Google Scholar]
- 41.De-Fraja C, Conti L, Magrassi L, Govoni S, Cattaneo E: Members of the JAK/STAT proteins are expressed and regulated during development in the mammalian forebrain. J Neurosci Res 1998, 54:320-330 [DOI] [PubMed] [Google Scholar]
- 42.Cattaneo E, Conti L, De-Fraja C: Signalling through the JAK-STAT pathway in the developing brain. Trends Neurosci 1999, 22:365-369 [DOI] [PubMed] [Google Scholar]
- 43.Acarin L, Gonzalez B, Castellano B: STAT3 and NFkappaB activation precedes glial reactivity in the excitotoxically injured young cortex but not in corresponding distal thalamic nuclei. J Neuropathol Exp Neurol 2000, 59:151-163 [DOI] [PubMed] [Google Scholar]
- 44.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL: Signaling and transcription in T help development. Annu Rev Immunol 2000, 18:451-494 [DOI] [PubMed] [Google Scholar]
- 45.Doherty PC, Allen JE, Lynch F, Ceredig R: Dissection of an inflammatory process induced by CD8+ T cells. Immunol Today 1990, 11:55-59 [DOI] [PubMed] [Google Scholar]
- 46.Griffin DE, Levine B, Tyor WR, Irani DN: The immune response in viral encephalitis. Semin Immunol 1992, 4:111-119 [PubMed] [Google Scholar]
- 47.Kennedy MK, Torrance DS, Picha KS, Mohler KM: Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol 1992, 149:2496-2505 [PubMed] [Google Scholar]
- 48.Benveniste EN: Cytokine actions in the central nervous system. Cytokine Growth Factor Rev 1998, 9:259-275 [DOI] [PubMed] [Google Scholar]
- 49.Popko B, Corbin JG, Baerwald KD, Dupree J, Garcia AM: The effects of interferon-gamma on the central nervous system. Mol Neurobiol 1997, 14:19-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong Y, Rohn WM, Benveniste EN: IFN-gamma regulation of the type IV class II transactivator promoter in astrocytes. J Immunol 1999, 162:4731-4739 [PubMed] [Google Scholar]
- 51.Lee SJ, Park JY, Hou J, Benveniste EN: Transcriptional regulation of the intercellular adhesion molecule-1 gene by proinflammatory cytokines in human astrocytes. Glia 1999, 25:21-32 [DOI] [PubMed] [Google Scholar]
- 52.Zhou ZHL, Chaturvedi P, Han YL, Aras S, Li YS, Kolattukudy PE, Ping D, Boss JM, Ransohoff RM: IFN-γ induction of the human monocyte chemoattractant protein (hMCP)-1 gene in astrocytoma cells: functional interaction between an IFN-γ-activated site and a GC-rich element. J Immunol 1998, 160:3908-3916 [PubMed] [Google Scholar]
- 53.Nguyen VT, Benveniste EN: Involvement of STAT1 and ets family members in interferon-gamma induction of CD40 transcription in microglia/macrophages. J Biol Chem 2000, 275:23674-23684 [DOI] [PubMed] [Google Scholar]
- 54.Lee SJ, Zhou T, Choi C, Wang Z, Benveniste EN: Differential regulation and function of Fas expression on glial cells. J Immunol 2000, 164:1277-1285 [DOI] [PubMed] [Google Scholar]
- 55.Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JEJ: Synergistic interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J 1990, 9:1105-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marra F, Choudhury GG, Abboud HE: Interferon-γ-mediated activation of STAT1α regulates growth factor-induced mitogenesis. J Clin Invest 1996, 98:1218-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohmori Y, Schreiber RD, Hamilton TA: Synergy between interferon-γ and tumor necrosis factor-a in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor κB. J Biol Chem 1997, 272:14899-14907 [DOI] [PubMed] [Google Scholar]
- 58.Han Y, Rogers N, Ransohoff RM: Tumor necrosis factor-α signals to the IFN-γ receptor complex to increase Stat1α activation. J Interferon Cytokine Res 1999, 19:731-740 [DOI] [PubMed] [Google Scholar]
- 59.Lee CK, Smith E, Gimeno R, Gertner R, Levy DE: STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-γ. J Immunol 2000, 164:1286-1292 [DOI] [PubMed] [Google Scholar]
- 60.Chen G, Hohmeier HE, Newgard CB: Expression of the transcriptional factor STAT-1a in insulinoma cells protects against cytotoxic effects of multiple cytokines. J Biol Chem 2001, 276:766-772 [DOI] [PubMed] [Google Scholar]
- 61.Fu XY, Schindler C, Improta T, Aebersold R, Darnell Jr JE: The proteins of ISGF-3. The interferon α-induced transcriptional activator, define a gene family involved in signal transduction. Proc Natl Acad Sci USA 1992, 89:7840–7843 [DOI] [PMC free article] [PubMed]
- 62.Leung S, Qureshi SA, Kerr IM, Darnell JE, Stark GR: Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol 1995, 15:1312-1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shuai K, Ziemlecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JE: Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature 1993, 366:580-583 [DOI] [PubMed] [Google Scholar]
- 64.Shuai K, Schindler C, Prezioso VR, Darnell Jr JE: Activation of transcription by IFN-γ: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 1992, 261:1808–1812 [DOI] [PubMed]
- 65.Matsumoto M, Tanaka N, Harada H, Kimura T, Yokochi T, Kitagawa M, Schindler C, Taniguchi T: Activation of the transcription factor ISGF3 by interferon-γ. Biol Chem 1999, 380:699-703 [DOI] [PubMed] [Google Scholar]
- 66.Kessler DS, Levy DE, Darnell Jr JE: Two-interferon-induced nuclear factors bind a single promoter element in interferon-stimulated genes. Proc Natl Acad Sci USA 1988, 85:8521–8525 [DOI] [PMC free article] [PubMed]
- 67.Reich N, Evans B, Levy DE, Fahey D, Knight Jr E, Darnell Jr JE: Interferon-induced transcription of a gene encoding a 15-kDa protein depends on an upstream enhancer element. Proc Natl Acad Sci USA 1987, 84:6394–6398 [DOI] [PMC free article] [PubMed]
- 68.Lehtonen A, Matikainen S, Julkunen I: Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol 1997, 159:794-803 [PubMed] [Google Scholar]
- 69.Weihua X, Kolla V, Kalvakolanu DV: Interferon-γ-induced transcription of the murine ISGF3γ (p48) gene is mediated by novel factors. Proc Natl Acad Sci USA 1997, 94:103-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bluyssen HA, Muzaffar R, Vlieststra RJ, Van Der Made ACJ, Leung S, Stark GR, Kerr IM, Trapman J, Levy DE: Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc Natl Acad Sci USA 1995, 92:5645-5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jee Y, Kim G, Tanuma N, Matsumoto Y: STAT expression and localization in the central nervous system during autoimmune encephalomyelitis. J Neuroimmunol 2001, 114:40-47 [DOI] [PubMed] [Google Scholar]
- 72.Gisselbrecht S: The CIS/SOCS proteins: a family of cytokine-inducible regulators of signaling. Eur Cytokine Netw 1999, 10:463-470 [PubMed] [Google Scholar]
- 73.Hilton DJ, Nicholson SE: The SOCS proteins: a new family of negative regulators of signal transduction. J Leukoc Biol 1998, 63:665-668 [DOI] [PubMed] [Google Scholar]
- 74.Polizzotto MN, Bartlett PF, Turnley AM: Expression of “suppressor of cytokine signalling” (SOCS) genes in the developing and adult mouse central nervous system. J Comp Neurol 2000, 423:348-358 [PubMed] [Google Scholar]
- 75.Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SBH, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M: The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA 1999, 96:2071-2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Starr R, Hilton DJ: Negative regulation of the JAK/STAT pathway. BioEssays 1999, 21:47-52 [DOI] [PubMed] [Google Scholar]
- 77.Yasukawa H, Sasaki A, Yoshimura A: Negative regulation of cytokine signaling pathways. Annu Rev Immunol 2000, 18:143-164 [DOI] [PubMed] [Google Scholar]