

Abstract
Elevated plasma lipoprotein(a) [Lp(a)] levels constitute an independent risk factor for the development of atherosclerosis. However, the mechanism underlying Lp(a) atherogenicity is unclear. Recently, we demonstrated that Lp(a) may potentially be proatherogenic in transgenic rabbits expressing human apolipoprotein(a) [apo(a)]. In this study, we further investigated atherosclerotic lesions of transgenic rabbits by morphometry and immunohistochemistry. On a cholesterol diet, human apo(a) transgenic rabbits had more extensive atherosclerotic lesions of the aorta, carotid artery, iliac artery, and coronary artery than did nontransgenic littermate rabbits as defined by increased intimal lesion area. Enhanced lesion development in transgenic rabbits was characterized by increased accumulation of smooth muscle cells, that was often associated with the Lp(a) deposition. To explore the possibility that Lp(a) may be involved in the smooth-muscle cell phenotypic modulation, we stained the lesions using a panel of monoclonal antibodies against smooth-muscle myosin heavy-chain isoforms (SM1, SM2, and SMemb) and basic transcriptional element binding protein-2 (BTEB2). We found that a large number of smooth muscle cells located in the apo(a)-containing areas of transgenic rabbits were positive for SMemb and BTEB2, suggesting that these smooth muscle cells were either immature or in the state of activation. In addition, transgenic rabbits showed delayed fibrinolytic activity accompanied by increased plasma plasminogen activator inhibitor-1. We conclude that Lp(a) may enhance the lesion development by mediating smooth muscle cell proliferation and dedifferentiation possibly because of impaired fibrinolytic activity.
Lipoprotein(a) [Lp(a)] closely resembles low-density lipoprotein (LDL) in both lipid composition and the presence of apolipoprotein (apo) B-100 (apoB-100). Lp(a) is distinguished from LDL by the presence of an additional protein designated as apolipoprotein(a) [apo(a)], which is complexed to apoB-100 by disulfide linkage. 1,2 Plasma Lp(a) concentrations vary >1000-fold in the human population, ranging from <0.1 to >100 mg/dL. Lp(a) levels are under strict genetic control and are generally not affected by diet, exercise, or drug intervention. Since Lp(a) was discovered by Berg in 1963, 3 numerous cross-sectional and prospective studies have revealed that high plasma levels of Lp(a) are associated with human cardiovascular disease such as coronary heart diseases, stroke, and restenosis, 4-6 although several studies failed to detect such an association. 7,8 The involvement of Lp(a) in the pathogenesis of atherosclerosis was initially suggested by the presence of Lp(a) in human atherosclerotic lesions. 9,10
On the basis of a partial amino acid sequence, followed by a complete analysis of the nucleotide sequence of cloned apo(a) complementary DNA (cDNA), extensive homology was revealed between apo(a) and plasminogen. 11,12 This has led investigators to suggest that the proatherogenic association of high Lp(a) concentrations may result from its atherogenic potential as a cholesterol-rich lipoprotein particle after receptor-mediated uptake and/or the sequence mimicry between apo(a) and plasminogen, leading to an inhibition of plasminogen’s anti-thrombotic effects 13 and anti-proliferative effects on smooth muscle cells (SMCs) through the inhibition of conversion of the active form of transforming growth factor-β (TGF-β). 14-16 One possible mechanism is that Lp(a) may exert anti-fibrinolytic effects by competitively inhibiting the binding of plasminogen to cells and fibrin. 13 Thus far, in vitro data support these hypotheses, but whether or not these mechanisms are operating in vivo remains to be determined. The development of transgenic mice expressing human apo(a) provided an alternative means to study apo(a) functions. 17-20 However, unlike in humans, in which nearly all plasma apo(a) is associated with apoB-100, the human apo(a) in the transgenic mice circulates in a free form in the plasma rather than in association with the murine LDL. 20 Despite this, expression of human apo(a) resulted in increased aortic fatty streak formation when these apo(a) transgenic mice were fed a high-fat diet, 17,21,22 although other studies failed to reproduce the atherogenic effect of the apo(a) in transgenic mice expressing either human apo(a) alone or both human apo(a) and apoB. 23,24 These results may suggest that a certain strain of mouse with a different genetic background may be resistant to the atherogenic effects of apo(a).
To define the metabolic and pathological consequences of Lp(a), our laboratory generated transgenic rabbits expressing human apo(a) and showed that human apo(a) is efficiently assembled into Lp(a) in the plasma. 25 Recently, we reported that Lp(a) substantially increases the development of atherosclerosis (surface area involvement defined by Sudan IV staining) in transgenic rabbits fed a cholesterol-rich diet. 26 To explore the mechanism(s) underlying Lp(a) atherogenicity, the present study was designed with three goals in mind: 1) to determine the influence of Lp(a) on SMC proliferation and dedifferentiation, namely, phenotype changes, during atherosclerosis in transgenic rabbits; 2) to disclose whether the TGF-β pathway may be involved in the lesion formation; and 3) to determine whether Lp(a) inhibits the fibrinolytic process in transgenic rabbits. Our results revealed that Lp(a) may enhance the lesion development by stimulating SMC proliferation and dedifferentiation possibly through impaired fibrinolytic activity.
Materials and Methods
Rabbits
A total of 11 (5 male and 6 female) transgenic and 18 (10 male and 8 female) nontransgenic littermates at the age of 4 to 5 months were fed a diet containing 0.3% cholesterol and 3% soybean oil by weight for 16 weeks as described previously. 26 All rabbits were housed in a single cage in a pathogen-free animal facility on a 12-hour dark/12-hour light cycle and had free access to food and water. All animal experiments were performed with the approval of the Animal Research Committee of the University of Tsukuba.
Analysis of Plasma Lipids and Lipoproteins
Plasma total cholesterol and triglycerides were measured using enzymatic assays (Wako Chemicals, Osaka, Japan). Plasma lipoproteins and plasma levels of human apo(a) were determined by an enzyme-linked immunosorbent assay using human recombinant apo(a) as a standard. 27,28
Quantitative Analysis for Histology and Immunohistochemistry
Rabbits were euthanized with an overdose injection of sodium pentobarbital solution (Abbott Laboratories, North Chicago, IL). A thoracotomy and laparotomy were performed to expose the heart and the entire arterial tree, which included both common carotid arteries, the whole aorta, and both common iliac arteries (Figure 1) ▶ . In this study, we attempted to make histologically quantitative and qualitative analyses on each part of the arterial wall by measuring the intimal lesion area and quantifying the cellular components (SMC versus macrophages). For these purposes, the representative lesion-prone segments of the aorta, carotid artery, and iliac artery were cut in cross-sections as shown in Figure 1 ▶ , embedded in paraffin, and stained with hematoxylin and eosin and Elastica-van Gieson.
Figure 1.

Schematic illustration of the whole arterial tree of rabbits and selected lesion sites for section analyses. Number in parentheses indicates the representative areas of each part of the artery selected for tissue section and measurement of intimal lesion areas. (1–3), Arch; (4–5), thoracic aorta; (6), abdominal aorta; (7–8), iliac arteries; (9–10), common carotid arteries.
Analysis of relative distribution of SMCs and macrophages in the lesions was performed with a computer-assisted MacSCOPE image analysis system 26 after immunohistochemical staining for HHF35 and RAM11 monoclonal antibody (mAb), respectively, using a Histofine Sab-Po(M) kit (Nichirei Co., Tokyo, Japan) according to the manufacturer’s instructions. The whole glass slide was loaded in a Pathscan Enabler (Meyer Instrument, Inc., Houston, TX) and scanned at a resolution of 1012 pixels in 24-bit full color with use of a Polascan 35 Ultra scanner (Polaroid Co., Tokyo, Japan). A color threshold mask for immunostaining was defined to detect the red color by sampling and the same threshold was applied to all specimens. The percentage of the total area with positive color for each section was recorded and expressed as percent for each cellular distribution. The left coronary artery was used for the evaluation of coronary atherosclerosis as described. 29
Antibodies (Abs) used for this study were listed in Table 1 ▶ . In this study, we were specifically interested in SMC phenotypes in the lesions with use of a panel of Abs against smooth muscle myosin heavy chains (SM1, SM2, SMemb) and basic transcriptional element binding protein-2 (BTEB2), which mediates the transcriptional regulation of the SMemb gene during phenotypic modulation of SMCs. 30-32 In addition, we also attempted to evaluate the relative changes in the contents of plasminogen and plasminogen activator inhibitor 1 (PAI-1), TGF-β (both latent and active forms), and TGF-β receptors I and II in the lesions. Evaluations of immunohistochemical staining were made by two independent trained observers blindly.
Table 1.
List of Antibodies Used for the Current Study
| Antibodies | Dilution | Species | Manufacturers |
|---|---|---|---|
| Apo(a) | ×200 | Mouse | Reference 25 |
| ApoB | ×200 | Goat | RockLand Inc., Gilbertsville, PA |
| RAM11 | ×12000 | Mouse | Dako Japan Inc., Tokyo, Japan |
| HHF-35 | ×50 | Mouse | Enzo Biochemicals, NY |
| Plasminogen/plasmin | ×100 | Mouse | American Diagnostica, Inc., Greenwich, CT |
| Plasminogen | ×100 | Mouse | TechnoClone, Vienna, Austria |
| Plasminogen | ×100 | Mouse | Advanced Immunochemical, Inc., Long Beach, CA |
| TGF-β1 | ×100 | Mouse | Biosource International, Camarillo, CA |
| LAP(TGF-β1) | ×100 | Goat | R&D Systems Inc., Minneapolis, MN |
| TGF-βRI(V22) | ×200 | Goat | Santa Cruz Biotech, Inc., Santa Cruz, CA |
| TGF-βRII(L21) | ×200 | Goat | Santa Cruz Biotech, Inc., Santa Cruz, CA |
| PAI-1 | ×200 | Mouse | American Diagnostica, Inc., Greenwich, CT |
| SM1 | ×3000 | Mouse | Yamasa, Inc., Tokyo, Japan |
| SM2 | ×400 | Mouse | Yamasa, Inc., Tokyo, Japan |
| SMemb | ×3000 | Mouse | Yamasa Inc., Tokyo, Japan |
| BTEB2 | ×4000 | Mouse | Yamasa Inc., Tokyo, Japan |
HHF-35, smooth muscle α-actin; RAM11, rabbit monocyte/macrophages; SM1, smooth muscle-specific myosin heavy-chain isoform; SM2, myosin heavy-chain-specific to well-differentiated smooth muscle cells; SMemb, non-muscle myosin heavy chain; BTEB2, basic transcriptional element-binding protein-2; TGF-β, transforming growth factor-β; LAP, latency-associated peptide of TGF-β1; TGF-βR, TGF-β receptor; PAI-1, plasminogen activator inhibitor.
Euglobulin Clot Lysis Assay and PAI-1 Analysis
Plasma levels of total amount of PAI-1 in cholesterol-fed rabbits were measured by latex photometric immunoassay (SRL Inc., Tokyo, Japan). To reveal whether transgenic rabbit Lp(a) would inhibit the fibrinolytic process, rabbit plasma in the presence of sodium citrate was immediately frozen and delivered in dry ice to Dr. Maurhofer Olivier (Symphar Co., Geneva, Switzerland) for analysis. Plasma Lp(a) was isolated from Lp(a)-rich fractions of cholesterol-fed transgenic rabbits by ultracentrifugation at a density of 1.12 g/ml 33 and analyzed by the euglobulin clot lysis assay as described by others. 34 Recombinant human apo(a) prepared from transfected mammalian cells was used as a positive control.
Statistic Analysis
Plasma lipids were compared in the groups of rabbits by using the Student’s t-test, and the lesion analyses were compared using the Mann-Whitney U test for nonparametric analysis. In all cases, statistical significance was set at P < 0.05.
Results
Plasma Lipids and Lipoprotein Profiles
The average human apo(a) levels in transgenic rabbits were 8.1 ± 3.9 mg/dL in males and 8.8 ± 3.3 mg/dL in females; total cholesterol levels in plasma and body weight were identical between transgenic and control rabbits (Table 2) ▶ . Agarose gel electrophoresis analysis revealed that expression of human apo(a) did not lead to marked changes in lipoprotein profiles in transgenic rabbits compared to nontransgenic rabbits (Figure 2) ▶ . The lipoprotein profiles and Lp(a) distribution of cholesterol-fed rabbits were further studied by sequential density ultracentrifugation. 35 As shown in Figure 3A ▶ , in general, transgenic and nontransgenic rabbits exhibited a similar pattern of lipoprotein profiles; there was a marked increase in apoB-containing lipoproteins including lipoproteins with d < 1.006 (mainly β-very low-density lipoprotein) and also those with d = 1.006 to 1.02 (intermediate-density lipoproteins); and those with d = 1.02 to 1.04 (LDL) in both transgenic and nontransgenic rabbits. Basically, apoB contents in each density fraction of transgenic rabbits were almost identical to those of nontransgenic rabbits (Figure 3B) ▶ . Human apo(a) was concomitantly distributed in a range of apoB-containing lipoproteins: d < 1.006, d = 1.006 to 1.02, and d = 1.02 to 1.04 g/ml. This indicates that human apo(a) formed Lp(a) complexes with rabbit apoB (Figure 3C) ▶ even though their association was not completely covalent because of the difference in the cysteine residue site between rabbit and human apoB. 26
Table 2.
Comparison of the Extent of Atherosclerosis, the Average Plasma Cholesterol, and Body Weights in Transgenic and Control Rabbits
| Variables | Male | Female | ||
|---|---|---|---|---|
| Transgenic (n = 5) | Control (n = 10) | Transgenic (n = 6) | Control (n = 8) | |
| Total cholesterol (mg/dL) | 696 ± 92 | 722 ± 110 | 1315 ± 546 | 1234 ± 375 |
| Human apo(a) (mg/dL) | 8.1 ± 3.9 | ND† | 8.8 ± 3.3 | ND |
| Body weight (kg)** | 3.3 ± 0.3 | 3.3 ± 0.4 | 3.4 ± 0.3 | 3.5 ± 0.2 |
| Intimal lesion area (μm2) | ||||
| Arch | 160 ± 66* | 43 ± 24 | 121 ± 79* | 49 ± 23 |
| Thoracic | 10 ± 10* | 1 ± 4 | 18 ± 15* | 1 ± 2 |
| Abdominal | 52 ± 23* | 13 ± 5 | 24 ± 17 | 19 ± 15 |
| Iliac A. | 21 ± 9* | 2 ± 5 | 4 ± 5 | ND |
| Carotid A. | 18 ± 12* | 1 ± 3 | 4 ± 5 | ND |
Values are expressed as mean ± SD.
*P < 0.05 versus control rabbits; **, at the end of 16-week cholesterol-rich diet experiment.
†ND referes to not detectable.
The lesion area on cross-section was measured with image analysis system described in Materials and Methods.
Figure 2.

Plasma lipoprotein in rabbits on chow and cholesterol diets. Plasma (2 μl) from chow-fed rabbits (left) and cholesterol-fed rabbits for 16 weeks (right) was electrophoresed on a 1% agarose gel and stained for neutral lipids with Fat red 7B. Arrowheads indicate original position.
Figure 3.

Apolipoprotein distribution in transgenic and nontransgenic rabbit plasma on cholesterol diet for 16 weeks. The plasma lipoproteins from a fasting transgenic male rabbit (top) and a nontransgenic male rabbit (bottom) were isolated by sequential density ultracentrifugation. Lipoproteins (8 μl) were visualized with Fat red 7B (A), which stains neutral lipids. To analyze the distribution of apoB (B) and apo(a) (C), 2 μl of each lipoprotein fraction was resolved by 1% agarose gel electrophoresis. Immunoblotting was performed as described in Materials and Methods.
Quantification of Atherosclerotic Lesions
Table 2 ▶ summarizes the results of cross-sectional lesion area in transgenic and nontransgenic rabbits. In all parts of the aorta (except abdominal aorta in females) and iliac and carotid arteries, transgenic rabbits had significantly greater lesions than those of nontransgenic rabbits although female rabbits, both transgenic and nontransgenic, developed relatively fewer lesions than their male counterparts in each group. The reason for less effect of Lp(a) on the lesion formation in females than that in males may be in part, because of protective functions of estrogen. 36
It is worth noting that lesions were extensive in iliac and carotid arteries, resulting in a 10- and 18-fold increase in male transgenics compared to controls. In female control rabbits, no lesions were found in the selected sections (Table 2) ▶ .
Coronary artery atherosclerosis was evaluated by measuring the intimal thickness and lesion area of the left main trunk. As shown in Table 3 ▶ , male and female transgenic rabbits showed increased lesion size as compared to control rabbits, although the enhanced extent of lesions in female transgenic rabbits did not reach statistical significance. As in the case of the aortas and carotid and iliac arteries, female rabbits developed less coronary atherosclerosis than the male animals.
Table 3.
Comparison of Coronary Atherosclerosis in Transgenic and Control Rabbits
| Male | Female | |||
|---|---|---|---|---|
| Transgenic (n = 5) | Control (n = 10) | Transgenic (n = 6) | Control (n = 8) | |
| Intimal thickness (μm) | 345 ± 191* | 140 ± 111 | 160 ± 74 | 110 ± 83 |
| Lesion size (%) | 35.3 ± 17.3* | 10.3 ± 9.1 | 22.9 ± 14.5 | 15.9 ± 13.2 |
Intimal thickness was measured by calculating the thickest area within each coronary artery.
Lesion size was calculated by dividing total lesion area to lumen area. Values are expressed as mean ± SD.
*P < 0.05 versus control rabbits.
Histological and Immunohistochemical Staining
Histological observation revealed that atherosclerotic lesions in control rabbits were mainly composed of macrophage-derived foam cells and a relatively small number of SMCs (Figure 4A) ▶ . In apo(a) transgenic rabbits, however, the atherosclerotic lesions varied from foam cell-rich lesions to spindle-shaped, cell-rich lesions (Figure 4 ▶ ; B, C, and D), presumably SMCs that were either positive (from strong to weak staining intensity) or negative for smooth muscle α-actin mAb (see below).
Figure 4.
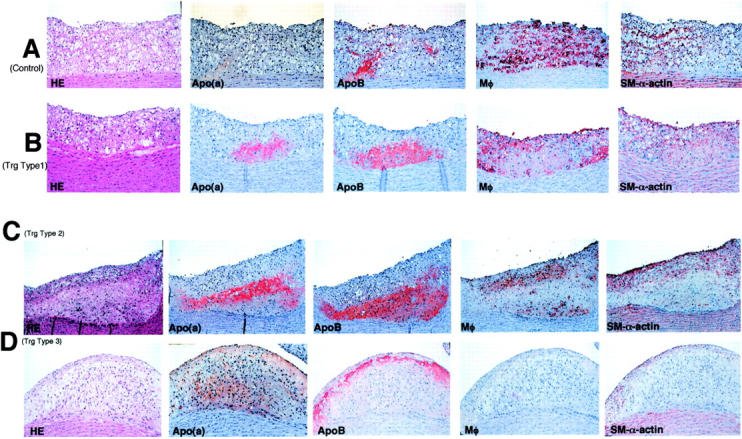
Histological and immunohistochemical analyses of aortic atherosclerosis in both transgenic and nontransgenic rabbits. Serial sections of paraffin-embedded specimens of the aorta arch were stained with either H&E or mAbs against apo(a), apoB, macrophage (Mφ), and smooth muscle α-actin. The top panel was from a control rabbit (A) and the lesions of transgenic rabbits were divided into three types according to the lesion features; type 1 lesion (B), type 2 lesion (C), and type 3 lesion (D) as described in the Results. Original magnification, ×40.
To evaluate the distribution of SMCs and macrophages in the lesions, we selected the aortic arch with the most extensive lesions in aorta and coronary artery of male rabbits and quantified the immunostaining area after immunohistochemical staining for HHF35 and RAM11 Abs. We found that the atherosclerotic lesions in transgenic rabbits contained relatively more SMCs and fewer macrophages than those in nontransgenic rabbits in both the aorta and coronary artery (Figure 5) ▶ . The ratios of SMCs to macrophages defined by each staining area in the lesions were 0.25 (control) versus 1 (transgenic) in the aortic arch and 0.8 (control) versus 1.5 (transgenic) in the coronary artery.
Figure 5.

Quantitative analysis for SMCs and macrophages within the atherosclerotic lesions of aortas (left) and coronary arteries (right) of transgenic and nontransgenic rabbits. The paraffin-embedded sections of aortic arch and coronary artery were immunohistochemically stained by mAbs against smooth muscle α-actin and Mφ, respectively, and immunostaining intensity was measured by Macscope image analysis as described in Materials and Methods. For each analysis, samples of seven transgenic and five nontransgenic rabbits were used. Data are expressed as the percentage of immunopositive area within the intima. *, P < 0.05 versus control rabbits.
To further elucidate whether apo(a) contributed to increased SMC proliferation in transgenic rabbits and whether there was a relationship between apo(a) deposition and lesion quality, we stained the lesions using mAb specific for human apo(a). We found that apo(a) deposition was frequently associated with the lesions in transgenic rabbits. In general, there were three kinds of patterns for apo(a) deposition in the lesion based on histological observation. We arbitrarily classified them as 1) foam cell-rich lesion that was primarily composed of foam cells similar to that in nontransgenic rabbits; 2) foam cell/SMC lesions that contained foam cells mainly on the top and spindle-shaped cells at the bottom; and 3) spindle-shaped, cell-rich lesions that were almost entirely of spindle-shaped cells (Figure 4 ▶ ; B, C, and D), and that were presumably immature SMCs (see below).
In the pattern 1 and 2 lesions of transgenic rabbits, apo(a) was either focally present or uniformly located beneath the foam cells (Figure 4, B and C) ▶ . The immunoreactive apo(a) was associated with apoB, suggesting that the apo(a) was directly from plasma Lp(a). Our previous study also excluded the possibility that apo(a) was produced de novo by arterial wall. 26 In the pattern 3 lesions, however, apo(a) was diffusely distributed around extracellular matrix and only partially co-localized with apoB (Figure 4D) ▶ . Regardless of the apo(a) deposition patterns, all intimal cells in apo(a)-containing areas were, almost invariably, not stained by mAb against macrophages. They were either negative or only weakly positive for anti-smooth muscle α-actin staining. We therefore, initially postulated that these intimal cells in apo(a)-containing areas may be those of immature or dedifferentiated SMCs.
To further examine the phenotype changes of these intimal SMCs, we performed immunohistochemical staining using a panel of mAbs for myosin heavy chains on a spindle cell-rich lesion. A representative lesion of aortic arch from a transgenic rabbit is shown in Figure 6 ▶ . In this lesion, apo(a) deposition was evident along the bottom of the lesion (Figure 6A) ▶ . Intimal cells at the center could not be stained by smooth muscle α-actin mAb but SMCs located in the superficial area and in the media were positive for smooth muscle α-actin mAb (Figure 6B) ▶ . SM1 antibody could detect both medial and almost all of the intimal SMCs, but SM2 antibody stained only mature SMCs from the media, suggesting that these intimal SMCs were immature (Figure 6, C and D) ▶ . To verify this contention, we further stained the lesions using SMemb mAb, a marker for activated and immature SMCs, and found that these intimal SMCs were positive for SMemb mAb (Figure 6E) ▶ . In parallel, we stained the lesions with mAb against BTEB2, a transcription factor for SMemb. As shown in Figure 6F ▶ , all intimal SMCs (note the cellular nuclei) but not medial SMCs were stained by BTEB2 mAb.
Figure 6.
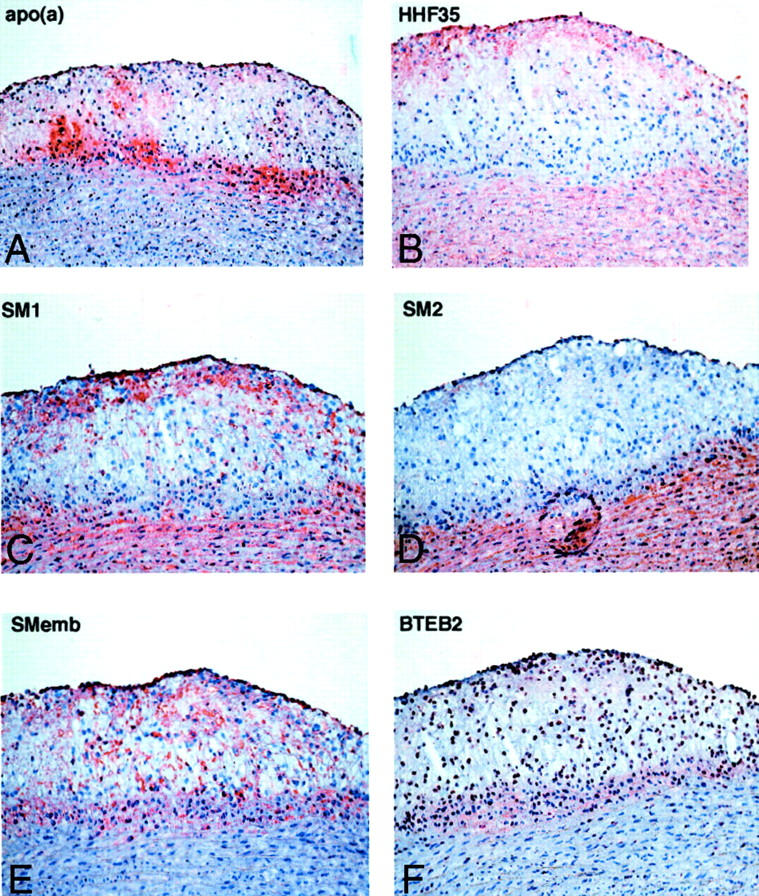
Smooth muscle cell phenotype changes in transgenic rabbits. Serial frozen sections were used for immunohistochemical staining. Human apo(a) was deposited in the lesions. There are many intimal cells at the center of the lesions. They are negative for smooth muscle α-actin(HHF35) and SM2, but they are positive for SM1 and SMemb. The nuclei are stained by BTEB2. Original magnifications: ×25 [apo(a)]; ×33 (HHF35, SM1, SM2, Smemb, and BTEB2).
Plasminogen and TGF-β Pathway
Previous studies using transgenic mice and cultured cells proposed that Lp(a) may compete with plasminogen to bind to fibrin or cell receptor, thereby impairing the plasmin generation and accordingly, delay the formation of the active form of TGF-β. 14-16 To investigate whether this mechanism was operating in human apo(a) transgenic rabbits, we performed immunohistochemical staining using three kinds of specific mAbs either against both plasminogen and plasmin or plasminogen only (Table 1) ▶ . As shown in Figure 7 ▶ , there was very weak staining for plasminogen in both transgenic and control rabbits, which did not enable us to make a direct comparison of these lesions between transgenic and control rabbits. Weak staining for plasminogen in the lesions was consistently confirmed by using all three kinds of antibodies listed in Table 1 ▶ . However, the tendency was that the immunoreactivity of the latent form of TGF-β1 was increased but the active form of TGF-β was reduced in type 3, SMC-rich lesions in transgenic rabbits compared to foam cell-rich lesions in both transgenic and control rabbits (Figure 7) ▶ . In such lesions, TGF-β receptor II predominated over TGF-β receptor I (Figure 8) ▶ .
Figure 7.
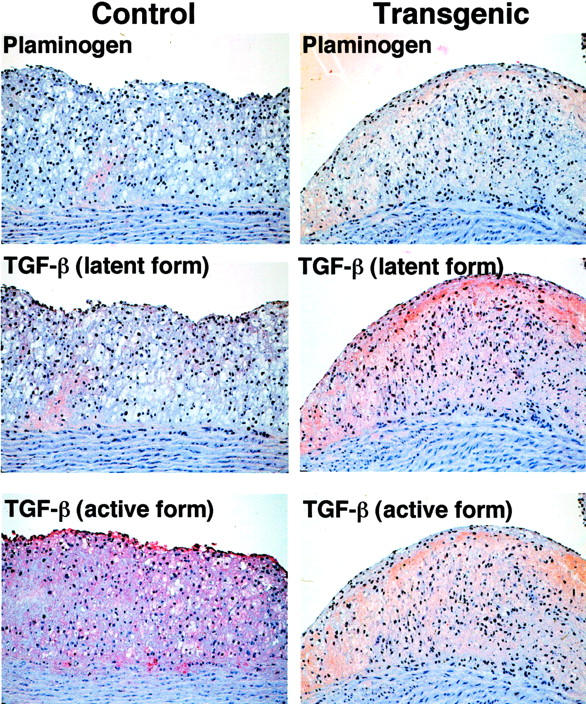
Immunohistochemical staining of plasminogen and TGF-β. Serial sections of Figure 4, A and D ▶ , were stained with mAbs against plasminogen, latent type of TGF-β, and active type of TGF-β. Plasminogen was barely stained in both transgenic and control rabbits. In SMC-rich lesions of transgenic rabbits, staining intensity for the latent type of TGF-β was relatively increased, whereas the active type of TGF-β was reduced compared to foam cell-rich lesions of control rabbits. Original magnification, ×40.
Figure 8.
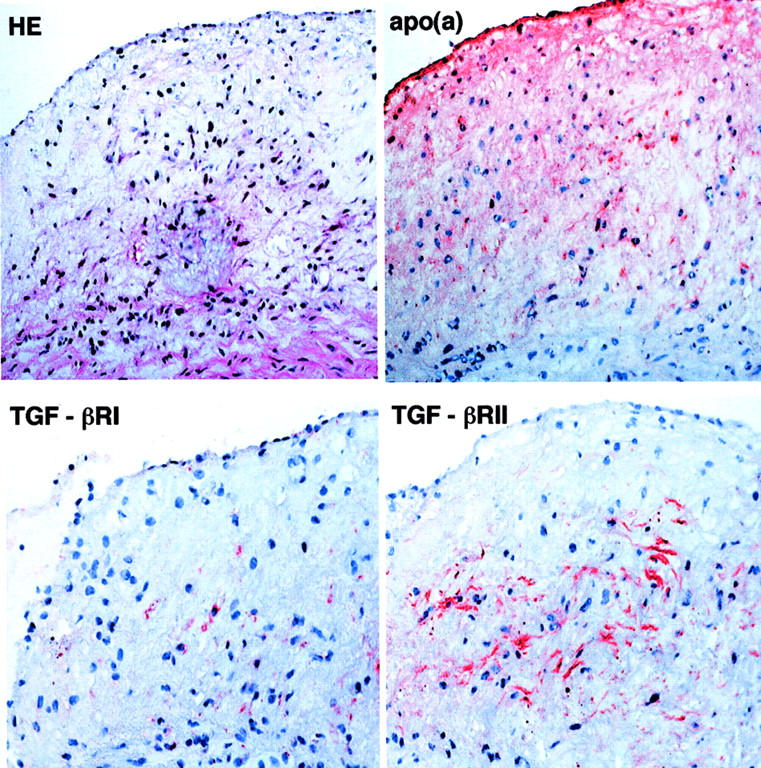
Distribution of type I and II TGF-β receptors in transgenic rabbits. Serial frozen sections were used for histological and immunohistochemical staining. The lesion is composed of immature SMCs associated with apo(a) deposition. Immunostaining intensity for type II receptor is stronger than that for type I receptor. Original magnification, ×160.
Fibrinolytic Activity and PAI-1
We further examined the effect of isolated Lp(a) from transgenic rabbit plasma on euglobulin clot lysis time. As shown in Figure 9 ▶ , Lp(a) from transgenic rabbits apparently delayed the clot lysis at the dose of 0.8 ∼ 6.4 mg/dL compared to control rabbit plasma. Furthermore, transgenic rabbits had higher levels of total plasma PAI-1 than did nontransgenic rabbits (Figure 10) ▶ . Immunohistochemical staining showed that in the lesions, PAI-1 was produced almost by all kinds of vascular cells including endothelial cells, SMCs, and macrophages (Figure 10) ▶ .
Figure 9.

Effect of transgenic rabbit plasma Lp(a) on fibrinolysis in vitro using rabbit euglobulin clot assay. Euglobulin clot assay was performed in microtiter wells and their lysis time was monitored by measurement of turbidity at 340 nm. The representative data of euglobulin clot assay is shown. Clots containing indicated contents of transgenic rabbit Lp(a) showed delayed time for the clot lysis compared to control rabbit plasma fraction.
Figure 10.
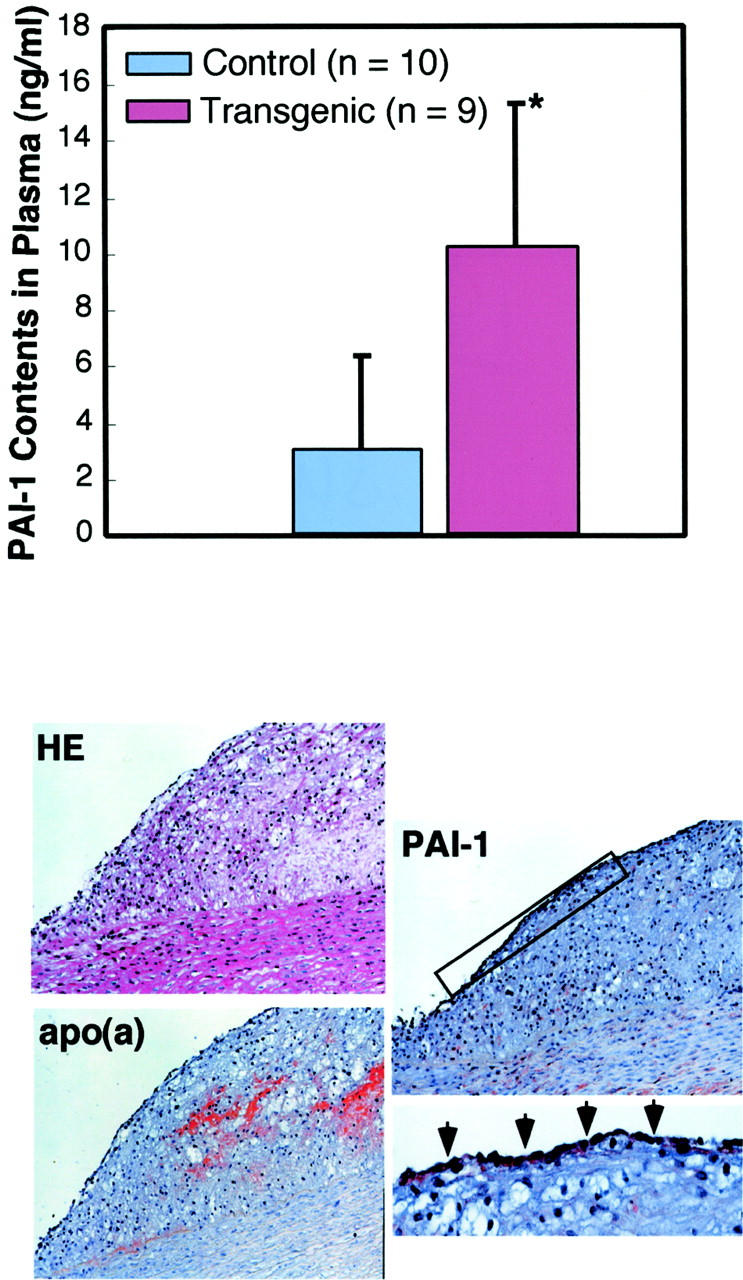
Total amount of plasma PAI-1 (top) and demonstration of PAI-1 in the lesions (bottom). Plasma levels of total amount of PAI-1 were significantly (*, P < 0.05) higher in transgenic rabbits than those in control rabbits. In the apo(a)-containing lesions of transgenic rabbits, PAI-1 is produced by intimal cells and medial SMCs (original magnification, ×33). Inset on the bottom right shows PAI-1 production by endothelial cells with arrowheads (original magnification, ×66).
Discussion
Development of Atherosclerosis in Transgenic Rabbits
The results of this study provide further evidence that Lp(a) increases the susceptibility to atherosclerosis in transgenic rabbits on a cholesterol-rich diet. Transgenic rabbits showed uniform enhancement of the lesion size in all parts of the aortic tree. Compared to nontransgenic rabbits, in which the lesions were mainly macrophage-derived foam cells, the lesions of transgenic rabbits were predominantly enriched in SMCs, suggesting that Lp(a) promotes the proliferation of SMCs during the lesion development of atherosclerosis. All these features were not documented in the previous studies using transgenic mice in which human apo(a) was unable to bind to murine apoB to form Lp(a) in plasma. 20 On a high-fat diet, human apo(a) transgenic mice developed more fatty streaks (rather than other types of lesions) in the aortic sinus. 17 Expression of human apo(a) in transgenic rabbits, however, aggravated atherosclerosis in all arterial beds examined, and furthermore, the lesions were characterized by a marked proliferation of SMCs. Especially noteworthy in this study was the significant increase of lesions in muscular arteries, carotid, iliac, and coronary arteries, in transgenic rabbits compared to control rabbits. This diffuse spatial distribution may mimic the situation in humans in that high levels of Lp(a) lead to the increased incidence of coronary, peripheral vascular, and cerebrovascular diseases in patients. 2
Apo(a) Deposition and SMC Proliferation in Atherosclerosis
We investigated the relationship between apo(a) deposition and lesion quality. In the lesions of transgenic rabbits, human apo(a) was mainly deposited in extracellular matrix but not apparently associated with foam cells. Consistent with our previous observation, Lp(a) may not, if at all, contribute to the formation of foam cells. Of particular note, apo(a)-deposited areas contained numerous immature SMC-like cells based on the finding that the lesions were negative or only weakly positive for smooth muscle α-actin staining. This notion has been further strengthened by the fact that these SMCs are negative for SM2 but positive for SM1 and SMemb and a transcript factor for SMemb, BTEB2 mAbs. SM1 is constitutively expressed at all developmental stages of SMCs, whereas SM2 is a marker of mature SMCs and appears only after birth. 30,31 On the other hand, SMemb is an embryonic isoform of myosin heavy chain, the expression of which is prominent in fetal aorta and normally down-regulated during development. 30 SMemb expression is up-regulated in immature or activated SMCs such as neointimal SMCs resulting from vascular injury. 32 Taken together, these results indicate that intimal SMCs in transgenic rabbits are either immature or in the state of dedifferentiation and resemble those of neointimal synthetic-type SMCs from balloon injury or restenosis after percutaneous transluminal coronary angioplasty. 32 We thus suggest that Lp(a) may not only stimulate the migration and proliferation of medial SMCs toward the intima, but also mediate the phenotypic alteration of SMCs toward an immature state.
Lp(a) and TGF-β Pathway
Several previous studies have demonstrated that human Lp(a) and apo(a) stimulate the proliferation of SMCs in vitro through the inhibition of plasminogen activation and consequent reduction of plasmin. 14 In this context, reduced generation of plasmin leads to inhibition of latent TGF-β activation, which is an inhibitor of SMC growth. Grainger et al 14 also showed that in transgenic mice expressing human apo(a) at the levels of 3.8 mg/dL, there was approximately threefold less active plasmin in the vessel wall than that in normal mice and the active form of TGF-β was significantly reduced. 15 In the current study, we also attempted to examine this hypothesis in transgenic rabbits using immunohistochemical staining. Unfortunately, we found that the total contents (as observed by immunostaining intensity) of plasminogen in the lesions were extremely low and seemingly uncomparable between transgenic and control rabbits. Immunohistochemical staining revealed that latent form of TGF-β tended to be comparatively increased and accompanied by concomitant reduction of active form of TGF-β in SMC-rich lesions in transgenic rabbits but not in other lesions of transgenic and nontransgenic rabbits, suggesting that the presence of apo(a) deposition in the arterial wall might, if at all, lead to inhibition of TGF-β activation. However, one should be cautious in interpreting this observation in terms of Lp(a)-plasminogen-TGF-β1-SMC growth scenario derived from immunohistochemical staining because 1) the cellular components in such apo(a)-containing areas (SMC-rich) of transgenic rabbits were substantially different from those of other types of lesions (macrophage-derived foam cell-rich) in transgenic and control rabbits; and 2) the net amounts of autocrine/paracrine production of TGF-β1 from different types of cells (SMC versus macrophages) in the lesions may not be directly compared. Moreover, increased type II/type I TGF-β receptor ratio as shown in Figure 8 ▶ is also not in favor of the Lp(a)-plasminogen-TGF-β1-SMC hypothesis. It is possible that Lp(a) may stimulate growth of SMCs via the independent mechanisms from the inhibition of TGF-β activation. 37 Therefore, it is still uncertain whether Lp(a) has a direct effect on SMC proliferation and dedifferentiation either dependent on or independent of the inhibition of TGF-β activation or both.
Lp(a) and Fibrinolysis
The inhibition of plasminogen activation by Lp(a) has also been suggested to contribute to the development of vascular lesions by inhibiting fibrinolysis. It is of interest that we found that transgenic rabbits had higher plasma levels of PAI-1 than did nontransgenic rabbits and Lp(a) isolated from transgenic plasma inhibited the fibrinolytic process, suggesting that Lp(a) does possess prothrombogenic properties. Furthermore, Lp(a) may enhance the production of PAI-1 from the arterial wall thereby exacerbating this process. 38 Although the biological significance and relevance of this anti-fibrolytic effect of Lp(a) to atherosclerosis remains to be defined, these results suggest that apo(a) transgenic rabbits may be potentially susceptible to thrombosis and may be specifically useful for studying pathophysiology of thrombosis.
Transgenic rabbits had similarly high levels of total cholesterol as did control rabbits on a cholesterol-rich diet (Table 2) ▶ . However, atherogenic lipoproteins, β-very low-density lipoprotein, and intermediate-density lipoprotein from transgenic rabbits were markedly enriched in human apo(a) (Figure 3) ▶ , and, therefore, it is likely that the increased amount of human apo(a) in these atherogenic particles was the principal factor underlying the significant enhancement in the atherosclerosis and impaired fibrinolytic activity in these animals. Enrichment of apo(a) might increase the binding of these lipoproteins to proteoglycans with the arterial wall intima, 22 leading to the retention of atherogenic lipoproteins in the intima, thus accelerating atherogenesis. We recognize that the major atherogenic lipoproteins in cholesterol-fed rabbits are those of hepatic remnant lipoproteins, β-very low-density lipoprotein, whereas in humans, LDLs are the main atherogenic lipoproteins. It is necessary to have an appropriate animal (human-like) model in which both Lp(a) and LDL levels are elevated. Currently, we have generated WHHL transgenic rabbits that express higher levels of Lp(a) than normal transgenic rabbits and have elevated LDL levels, 28 as in human familial hypercholesterolemia. In humans, the risk of elevated Lp(a) concentrations is significantly increased in patients who also have high levels of LDL cholesterol. 39,40 Therefore, it may be particularly interesting to use WHHL transgenic rabbits to address several issues related to pathophysiological functions of Lp(a) in both atherosclerosis and thrombosis and also to test pharmacological intervention to lower Lp(a) to inhibit the progression of atherosclerosis.
In summary, we have shown that human apo(a) transgenic rabbits develop more extensive atherosclerosis than normal rabbits in the aorta, iliac artery, carotid artery, and coronary artery in response to a cholesterol-rich diet. Increased atherosclerosis in transgenic rabbits is associated with SMC proliferation and dedifferentiation, possibly related to impaired fibrinolytic activity. Although the molecular mechanism underlying this effect remains to be elucidated, the current results favor the notion that Lp(a) enhances the lesion progression of atherosclerosis by modifying the SMC phenotype.
Acknowledgments
We thank M. Olivier for performing the euglobulin clot lysis assay.
Footnotes
Address reprint requests to Jianglin Fan M.D., Ph.D., Department of Pathology, Institute of Basic Medical Sciences, University of Tsukuba, Tsukuba 305-8575, Japan. E-mail: j-lfan@md.tsukuba.ac.jp.
Supported by Grants-in-Aid for Scientific Research (10470046, 11470048, 11557016) from the Ministry of Education, Science, and Culture of Japan and the Japan Society for the Promotion of Sciences (JSPS-RFTF96I00202).
References
- 1.Utermann G: The mysteries of lipoprotein(a). Science 1989, 246:904-910 [DOI] [PubMed] [Google Scholar]
- 2.Utermann G: Lipoprotein (a). Scrive BA Sly WS eds. The Metabolic and Molecular Bases of Inherited Disease. 1995, :pp 1887-1912 McGraw-Hill, New York [Google Scholar]
- 3.Berg K: A new serum type system in man—the Lp(a) system. Acta Pathol Microbiol Scand 1963, 59:369-382 [DOI] [PubMed] [Google Scholar]
- 4.Scanu AM: Atherothrombogenicity of lipoprotein(a): the debate. Am J Cardiol 1998, 82:26Q-33Q [DOI] [PubMed] [Google Scholar]
- 5.Maher VM, Brown BG: Lipoprotein (a) and coronary heart disease. Curr Opin Lipid 1995, 6:229-235 [DOI] [PubMed] [Google Scholar]
- 6.Djurovic S, Berg K: Epidemiology of Lp(a) lipoprotein: its role in atherosclerotic/thrombotic disease. Clin Genet 1997, 52:281-292 [DOI] [PubMed] [Google Scholar]
- 7.Ridker PM, Hennekens CH, Stampfer MJ: A prospective study of lipoprotein(a) and the risk of myocardial infarction. JAMA 1993, 270:2195-2199 [PubMed] [Google Scholar]
- 8.Gurewich V, Mittleman M: Lipoprotein(a) in coronary heart disease. Is it a risk factor after all? JAMA 1994, 271:1025-1026 [PubMed] [Google Scholar]
- 9.Rath M, Niendorf A, Reblin T, Dietel M, Krebber HJ, Beisiegel U: Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989, 9:579-592 [DOI] [PubMed] [Google Scholar]
- 10.Jurgens G, Chen Q, Esterbauer H, Mair S, Ledinski G, Dinges HP: Immunostaining of human autopsy aortas with antibodies to modified apolipoprotein B and apoprotein(a). Arterioscler Thromb 1993, 13:1689-1699 [DOI] [PubMed] [Google Scholar]
- 11.McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM: cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330:132-137 [DOI] [PubMed] [Google Scholar]
- 12.Eaton DL, Fless GM, Kohr W, Mclean JW, Xu QT, Miller CG, Lawn RM, Scanu AM: Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci USA 1987, 84:3224-3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angles-Cano E, de la Pnena Diaz A, Loyau S: Inhibition of fibrinolysis by lipoprotein(a). Ann N Y Acad Sci 2001, 936:261-275 [DOI] [PubMed] [Google Scholar]
- 14.Grainger DJ, Kirschenlohr HL, Metcalfe JC, Weissberg PL, Wade DP, Lawn RM: Proliferation of human smooth muscle cells promoted by lipoprotein(a). Science 1993, 260:1655-1658 [DOI] [PubMed] [Google Scholar]
- 15.Grainger DJ, Kemp PR, Liu AC, Lawn RM, Metcalfe JC: Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature 1994, 370:460-462 [DOI] [PubMed] [Google Scholar]
- 16.Grainger DJ, Metcalfe JC: Transforming growth factor-beta: the key to understanding lipoprotein(a)? Curr Opin Lipid 1995, 6:81-85 [DOI] [PubMed] [Google Scholar]
- 17.Lawn RM, Wade DP, Hammer RE, Chiesa G, Verstuyft JG, Rubin EM: Atherogenesis in transgenic mice expressing human apolipoprotein(a). Nature 1992, 360:670-672 [DOI] [PubMed] [Google Scholar]
- 18.Frazer KA, Narla G, Zhang JL, Rubin EM: The apolipoprotein(a) gene is regulated by sex hormones and acute-phase inducers in YAC transgenic mice. Nat Genet 1995, 9:424-431 [DOI] [PubMed] [Google Scholar]
- 19.Acquati F, Hammer R, Ercoli B, Mooser V, Tao R, Ronicke V, Michalich A, Chiesa G, Taramelli R, Hobbs HH, Muller HJ: Transgenic mice expressing a human apolipoprotein[a] allele. J Lipid Res 1999, 40:994-1006 [PubMed] [Google Scholar]
- 20.Chiesa G, Hobbs HH, Koschinsky ML, Lawn RM, Maika SD, Hammer RE: Reconstitution of lipoprotein(a) by infusion of human low density lipoprotein into transgenic mice expressing human apolipoprotein(a). J Biol Chem 1992, 267:24369-24374 [PubMed] [Google Scholar]
- 21.Liu AC, Lawn RM, Verstuyft JG, Rubin EM: Human apolipoprotein A-I prevents atherosclerosis associated with apolipoprotein[a] in transgenic mice. J Lipid Res 1994, 35:2263-2267 [PubMed] [Google Scholar]
- 22.Boonmark NW, Lou XJ, Yang ZJ, Schwartz K, Zhang JL, Rubin EM, Lawn RM: Modification of apolipoprotein(a) lysine binding site reduces atherosclerosis in transgenic mice. J Clin Invest 1997, 100:558-564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancini FP, Newland DL, Mooser V, Murata J, Marcovina S, Young SG, Hammer RE, Sanan DA, Hobbs HH: Relative contributions of apolipoprotein (a) and apolipoprotein-B to the development of fatty lesions in the proximal aorta of mice. Arterioscler Thromb Vasc Biol 1995, 5:1911-1916 [DOI] [PubMed] [Google Scholar]
- 24.Sanan DA, Newland DL, Tao R, Marcovina S, Wang J, Mooser V, Hammer RE, Hobbs HH: Low density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein (a). Proc Natl Acad Sci USA 1998, 95:4544-4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan J, Araki M, Wu L, Challah M, Shimoyamada H, Lawn MR, Kakuta H, Shikama H, Watanabe T: Assembly of lipoprotein (a) in transgenic rabbits expressing human apolipoprotein (a). Biochem Biophys Res Commun 1999, 255:639-644 [DOI] [PubMed] [Google Scholar]
- 26.Fan J, Shimoyamada H, Sun H, Marcovina S, Honda K, Watanabe T: Transgenic rabbits expressing human apolipoprotein(a) develop more extensive atherosclerotic lesions in response to a cholesterol-rich diet. Arterioscler Thromb Vasc Biol 2001, 21:88-94 [DOI] [PubMed] [Google Scholar]
- 27.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP: Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem 1995, 41:246-255 [PubMed] [Google Scholar]
- 28.Fan J, Challah M, Shimoyamada H, Shiomi M, Marcovina S, Watanabe T: Defects of the LDL receptor in WHHL transgenic rabbits lead to a marked accumulation of plasma lipoprotein[a]. J Lipid Res 2000, 41:1004-1012 [PubMed] [Google Scholar]
- 29.Hirata M, Watanabe T: Regression of atherosclerosis in normotensive and hypertensive rabbits. A quantitative analysis of cholesterol-induced aortic and coronary lesions with an image-processing system. Acta Pathol Jpn 1988, 38:559-575 [PubMed] [Google Scholar]
- 30.Aikawa M, Sivam PN, Kuro-o M, Kimura K, Nakahara K, Takewaki S, Ueda M, Yamaguchi H, Yazaki Y, Periasamy M: Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ Res 1993, 73:1000-1012 [DOI] [PubMed] [Google Scholar]
- 31.Aikawa M, Rabkin E, Voglic SJ, Shing H, Nagai R, Schoen FJ, Libby P: Lipid lowering promotes accumulation of mature smooth muscle cells expressing smooth muscle myosin heavy chain isoforms in rabbit atheroma. Circ Res 1998, 83:1015-1026 [DOI] [PubMed] [Google Scholar]
- 32.Watanabe N, Kurabayashi M, Shimomura Y, Kawai-Kowase K, Hoshino Y, Manabe I, Watanabe M, Aikawa M, Kuro-o M, Suzuki T, Yazaki Y, Nagai R: BTEB2, a Kruppel-like transcription factor, regulates expression of the SMemb/nonmuscle myosin heavy chain B (SMemb/NMHC-B) gene. Circ Res 1999, 85:182-191 [DOI] [PubMed] [Google Scholar]
- 33.Fless GM, ZumMallen ME, Scanu AM: Isolation of apolipoprotein(a) from lipoprotein(a). J Lipid Res 1985, 26:1224-1229 [PubMed] [Google Scholar]
- 34.Sangrar W, Bajzar L, Nesheim ME, Koschinsky ML: Antifibrinolytic effect of recombinant apolipoprotein(a) in vitro is primarily because of attenuation of tPA-mediated Glu-plasminogen activation. Biochemistry 1995, 34:5151-5157 [DOI] [PubMed] [Google Scholar]
- 35.Fan J, Ji Z-S, Huang Y, de Silva H, Sanan D, Mahley R, Innerarity T, Taylor J: Increased expression of apolipoprotein E in transgenic rabbits results in reduced levels of very low density lipoproteins and an accumulation of low density lipoproteins in plasma. J Clin Invest 1998, 101:2151-2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan L, Pervin S, Singh R, Rosenfeld M, Chaudhuri G: Estradiol inhibits leukocyte adhesion and transendothelial migration in rabbits in vivo: possible mechanisms for gender differences in atherosclerosis. Circ Res 1999, 85:377-385 [DOI] [PubMed] [Google Scholar]
- 37.Morishita R, Yamada S, Higaki J, Tomita N, Kida I, Aoki M, Moriguchi A, Hayashi S, Sakurabayashi I, Kaneda Y, Ogihara T: Conditioned medium from HepG2 cells transfected with human apolipoprotein(a) gene stimulates growth of human vascular smooth muscle cells: effects of overexpression of human apolipoprotein(a) gene. Hypertension 1998, 32:215-222 [DOI] [PubMed] [Google Scholar]
- 38.Etingin OR, Hajjar DP, Hajjar KA, Harpel PC, Nachman RL: Lipoprotein (a) regulates plasminogen activator inhibitor-1 expression in endothelial cells. A potential mechanism in thrombogenesis. J Biol Chem 1991, 266:2459-2465 [PubMed] [Google Scholar]
- 39.Armstrong VW, Cremer P, Eberle E, Manke A, Schulze F, Wieland H, Kreuzer H, Seidel D: The association between serum Lp(a) concentrations and angiographically assessed coronary atherosclerosis. Dependence on serum LDL levels. Atherosclerosis 1986, 62:249-257 [DOI] [PubMed] [Google Scholar]
- 40.Seed M, Hoppichler F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, Utermann G: Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med 1990, 322:1494-1499 [DOI] [PubMed] [Google Scholar]