

Abstract
Mice lacking the intermediate filament protein desmin demonstrate abnormal mitochondria behavior, disruption of muscle architecture, and myocardial degeneration with extensive calcium deposits and fibrosis. These abnormalities are associated with cardiomyocyte hypertrophy, cardiac chamber dilation and eventually with heart failure. In an effort to elucidate the molecular mechanisms leading to the observed pathogenesis, we have analyzed gene expression changes in cardiac tissue using differential display polymerase chain reaction and cDNA atlas array methods. The most substantial changes were found in genes coding the small extracellular matrix proteins osteopontin and decorin that are dramatically induced in the desmin-null myocardium. We further analyzed their expression pattern both at the RNA and protein levels and we compared their spatial expression with the onset of calcification. Extensive osteopontin localization is observed by immunohistochemistry in the desmin-null myocardium in areas with massive myocyte death, as well as in hypercellular regions with variable degrees of calcification and fibrosis. Osteopontin is consistently co-localized with calcified deposits, which progressively are transformed to psammoma bodies surrounded by decorin, especially in the right ventricle. These data together with the observed up-regulation of transforming growth factor-β1 and angiotensin-converting enzyme, could explain the extensive fibrosis and dystrophic calcification observed in the heart of desmin-null mice, potentially crucial events leading to heart failure.
Ablation of desmin, the muscle-specific intermediate filament protein, by gene targeting in mice leads to transient cardiomyocyte hypertrophy and extensive cardiomyocyte death, followed by cardiac chamber dilation and heart failure. 1-3 Similarly, missense mutations of desmin have been identified in humans suffering from idiopathic dilated cardiomyopathy 4 as well as other more generalized myopathies with both skeletal and cardiac dysfunction. 5-8,9 The cellular and tissue pathology associated with cardiac dysfunction in desmin-null mice has been adequately addressed. 1,3,10,11 Briefly, mice lacking desmin demonstrate disruption of muscle architecture with mitochondrial abnormalities, including loss of normal positioning, extensive proliferation and clumping, as well as compromised respiratory function. These abnormalities are followed by myocardial degeneration with extensive fibrosis and dystrophic calcification. The molecular mechanisms underlining the development of these abnormalities are mainly unknown.
The inappropriate biomineralization occurring in soft tissues is defined as ectopic calcification. In the absence of a systemic mineral imbalance ectopic calcification is typically called dystrophic calcification and is commonly observed in injury, disease, and aging. 12,13 Although it can occur in all soft tissues, cardiovascular tissues seem particularly prone to dystrophic calcification. In arteries, calcification is correlated with atherosclerotic plaques with the known clinical consequences. Age-related dystrophic calcification in the human cardiovascular system can contribute significantly to cardiac dysfunction and is perhaps more prevalent than ischemic heart disease. 12 Despite the high prevalence and clinical significance, very little mechanistic data exist mainly because of lack of animal models.
Dystrophic calcification possesses several features of bone mineralization, including the presence of noncollagenous matrix proteins such as osteopontin, matrix Gla protein, osteocalcin, SPARC (osteonectin), and bone morphogenetic proteins, which all are thought to regulate also pathological calcification. 12,13 Indeed mice lacking matrix Gla protein, by gene targeting inactivation, have extensive calcification of arteries and valves, 14 thus supporting the idea that this protein is indeed a natural inhibitor of mineralization. Similar results have been obtained for the osteoprotegerin gene, a member of the transforming growth factor (TGF) receptor superfamily, known to regulate osteoclast differentiation. 15 Although dystrophic calcification in all of the above cases is restricted to the vascular system, in desmin-null mice the cardiac muscle is the target tissue and can only be compared to dystrophic cardiac calcinosis. This is an age-related cardiomyopathy that occurs in certain inbred mouse strains that can also lead to congestive heart failure. 16,17 There are also a few other cases in which mutations in sarcomeric proteins, among other cardiac abnormalities, demonstrated calcification but at minor levels. 18,19 The molecular mechanisms underlying ectopic calcium deposition at sites of inflammation and/or necrosis is a fundamental but poorly understood element of not only dystrophic cardiac calcinosis and desmin-null cardiomyopathy but for any tissue response to injury. Desmin-null mice could serve as a good model to unravel the molecular mechanisms of cardiovascular degeneration, calcification, and the development of heart failure in these animals.
Because of the complexity of the observed pathology of desmin-null hearts, it is anticipated that alterations in multiple processes should be responsible for the development of the observed cardiomyopathy. To address this issue we analyzed general gene expression changes in cardiac tissue of the desmin-null mice, using differential display polymerase chain reaction (PCR) and cDNA atlas array methods. The most substantial changes were found for genes coding for extracellular matrix proteins and especially for the small matricellular proteins osteopontin and decorin. 13 We connect their action to the extended inflammatory reaction first observed between the second and third week of the animal’s life because of pronounced cardiomyocyte death. These data, together with the observed up-regulation of TGF-β1 and angiotensin I-converting enzyme (ACE) could explain the extensive fibrosis and dystrophic calcification observed in the heart of desmin-null mice.
Materials and Methods
Animals
The procedures for the care and treatment of animals were according to institutional guidelines. Mice lacking desmin were generated by gene targeting via homologous recombination as previously described. 1 The mice used for this study were of the C57BL/6–129SV genetic background.
Isolation of RNA, Differential Display PCR, cDNA Arrays, and Northern Blots
Wild-type and desmin-null mice were anesthetized and blood-free hearts were pulverized into powder under liquid nitrogen. RNA was isolated usually from pools of 4 to 5 hearts using the Totally RNA isolation kit (Ambion, Austin, TX). PolyA RNA was isolated using oligo-dT cellulose (Ambion). The differential display PCR method was used essentially as described. 20 The mouse atlas 1.2 k (1185 genes) cDNA array (catalog no. 7853-1) analysis was performed by Clontech (Clontech, San Diego, CA), using pools of three hearts of 4-month-old desmin-null and wild-type animals. Northern blots were performed as previously described 2 using standard techniques. For the ACE probe we have isolated a 0.95-kb fragment of the mouse ACE cDNA (accession no., J04947; area, 2093 to 3044) by RT-PCR amplification and similarly for the TGF-β1 probe we have isolated a 1.54-kb fragment of the mouse cDNA (accession no., AJ009862; area, 414 to 1960). The mouse osteopontin cDNA was kindly provided by Dr. Larry Fisher, National Institute of Dental and Craniofacial Research, Bethesda MD. 21
Protein Extracts and Western Blots
For the osteopontin Western blot, pulverized tissue (same as in RNA isolation) was extracted either with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (2% SDS, 10% glycerol, 50 mmol/L Tris-HCl, pH 6.8) or with demineralizing buffer that contained all of the above plus 300 mmol/L ethylenediaminetetraacetic acid (EDTA). Bone extracts were prepared from the femur bone of 3-week-old animals. For the decorin Western blot, the pulverized tissue was extracted with guanidine buffer (6 mol/L guanidinine isothiocyanate, 50 mmol/L Tris-HCl, pH 7.3, 5 mmol/L EDTA). For the chondroitinase digestion the guanidine extracts were dialyzed against (50 mmol/L Tris-HCl, 30 mmol/L sodium acetate, pH 7.3, 5 mmol/L EDTA, 3 mmol/L phenylmethyl sulfonyl fluoride) and centrifuged at 13,000 × g for 20 minutes. The supernatant (∼5 μg of total protein) was incubated with 0.04 U of chondroitinase ABC from Sigma (catalog no. C3667; Sigma, St. Louis, MO) in a final volume of 40 μl of 0.5× phosphate-buffered saline (PBS) at 37°C for 3 hours.
The samples were analyzed by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed for decorin and osteopontin using the antibodies LF-113 and LF-123, respectively, in a 1:1000 dilution. The antibodies were kindly provided by Dr. Larry Fisher. 21
Immunofluorescence
Blood-free mouse hearts were immersed in OCT compound (Miles Inc., Torrance, CA) and frozen in liquid nitrogen. Frozen tissue sections (7-μm-thick) fixed with 4% paraformaldehyde in PBS were used for immunolabeling as previously described. 1 The anti-decorin (LF-113), anti-osteopontin (LF-123), and anti-collagen-αI (LF-67) antibodies were kindly provided by Dr. Larry Fisher and the anti-laminin antibody was from Sigma (catalog no. L9393). All of the above polyclonal antibodies were used at a 1:300 dilution. The appropriate secondary antibodies (Alexaflour-594 and Alexaflour-488) were from Molecular Probes (Eugene, OR) and used in a 1:800 dilution.
Histology
Routine histological analysis and hematoxylin-eosin staining was performed as previously described. 1 For immunohistochemical analysis 5-μm-thick paraffin sections, from tissues fixed overnight in 2% paraformaldehyde solution in PBS, were used. The anti-decorin (LF-113) and anti-osteopontin (LF-123) antibodies were used in a 1:800 dilution. Reagents for the immunoperoxidase labeling were from DAKO (Carpinteria, CA). Substitution of primary antibodies by normal rabbit IgG was used as a negative control. Von Kossa staining for calcium detection was performed as described. 22
Results
Expression of Genes Coding for Small Extracellular Matrix Proteins, Decorin, and Osteopontin Is Induced in the Heart of Desmin-Null Mice
To identify genes that are differentially expressed in the heart of desmin-null mice, a mouse cDNA array was screened with RNA isolated from hearts of 4-month-old wild-type and null animals. Nine percent of the 1185 cDNAs examined displayed at least a twofold difference between wild-type and null animals with 60% of the differentially expressed genes being up-regulated in the null heart. These differentially expressed cDNAs belong to several functional groups (data not shown), but the most substantial changes were found in genes coding for extracellular matrix proteins (Table 1) ▶ .
Table 1.
Induction of Fibrosis and Calcification Are Linked in the Heart of Desmin-Null Mice
| Des+ | Des− | Fold X | cDNAs |
|---|---|---|---|
| 1 | 42 | 42.0 | Osteopontin precursor (OP); bone sialoprotein 1 |
| 8 | 46 | 5.8 | Decorin; bone proteoglycan II (PG-S2) |
| 11 | 17 | 1.5 | Bone morphogenetic protein 1 (BMP1); biglycan |
| 1 | 4 | 4.0 | Osteoblast-specific factor 2 (OSF-2) |
| 5 | 17 | 3.4 | Fibronectin |
| 18 | 55 | 3.1 | Angiotensin-converting enzyme (ACE) |
General gene expression profile was determined in the heart of desmin-null and wild-type animals, using the mouse 1.2 k (1185 genes) Atlas Array from Clontech Lab. RNA was isolated from 4-month-old, wild-type (Des+) and desmin-null (Des−) animals. Data from extracellular matrix/cell structure proteins are presented. The numbers represent arbitrary units.
Osteopontin RNA, was markedly induced (42-fold) in the heart of desmin-null animals (Table 1) ▶ . The cDNA array screen results were confirmed by Northern blot analysis (Figure 1) ▶ , which indeed revealed a dramatic induction of osteopontin RNA in desmin-null hearts. The expression of osteopontin RNA in the heart of wild-type animal is undetectable, even for longer (5 days) exposures. Further analysis of the expression profile of osteopontin RNA at various ages shows maximum induction around 3 weeks of age and then a decline as animals age (compare 6- to 12-month-old animals in Figure 1 ▶ ). In contrast, no detectable expression of osteopontin RNA was observed in skeletal muscle (gastrocnemius) of both desmin-null and wild-type animals (data not shown).
Figure 1.

Osteopontin expression is dramatically induced in the heart of desmin-null animals. Expression profile of osteopontin mRNA in the heart of wild-type (+/+) and desmin-null (−/−) mice by Northern blot, reveals that osteopontin is induced early in the animal’s life (21 days) and declines as the animal ages. The expression of osteopontin mRNA was undetectable in wild-type animals. D, days; Mo, months. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a loading control (exposure, 12 hours).
The second highest increase (5.8-fold) in the heart of desmin-null animals revealed by the array screen (Table 1) ▶ was in the expression of decorin RNA that codes for a proteoglycan, another small extracellular matrix protein. 23 The up-regulation of decorin was initially observed by differential display PCR in which a fragment identical to the 1337- to 1763-bp region of the mouse decorin cDNA (accession no. X53928) was isolated. Differential expression of the decorin mRNA in desmin-null heart was also confirmed by Northern blot analysis (Figure 2) ▶ using the above fragment as a probe. The induction of the decorin mRNA in the heart of desmin-null animals is approximately threefold compared to the wild-type. This level of decorin induction could be observed as early as 9 days after birth and remains constant at least up to the age of 13 months.
Figure 2.

Decorin, ACE, and TGF-β1 mRNAs are up-regulated in the heart of desmin-null mice. mRNA was isolated from the hearts of 2.5-month-old wild-type (+/+) and desmin-null (−/−) animals and analyzed by Northern blot using the corresponding cDNA probes. Shown are representative results for each mRNA. GAPDH was used as a loading control (exposure, 3 days).
Another gene linked to fibrosis, ACE, a key regulator of the renin-angiotensin system 24 was induced (3.1-fold, Table 1 ▶ ) in desmin-null hearts. ACE induction was also confirmed by Northern blot analysis (Figure 2) ▶ . The two ACE messages observed, sizes 4.9 and 4.2 kb, are commonly found in Northern blots of somatic tissues. 25 Given the established connection between decorin, TGF-β1, and fibrosis 26,27 we wanted to see how the expression of TGF-β1 is modulated in the desmin-null heart. Although the cDNA array screen did not reveal any difference, Northern blot analysis did show increase in the expression of TGF-β1 RNA in desmin-null hearts (Figure 2) ▶ . The increase was ∼2.5-fold in 3-week-old and 4-month-old animals.
Analysis of Decorin and Osteopontin Expression by Western Blots
To further examine whether the observed increase in osteopontin and decorin RNA levels was accompanied by similar changes at the protein level, we performed Western blot analysis. Murine osteopontin has a predicted molecular weight of 35 kd. However, in SDS-PAGE it shows anomalous migration with an apparent molecular weight of 45 to 75 kd, 28 because of posttranslational modifications, depending on the tissue of origin. Western blot analysis of cardiac extracts from 4-month-old desmin-null animals using anti-osteopontin antibody, showed the presence of high amounts of the protein in a high-molecular weight complex form (Figure 3A) ▶ . This complex is mainly retained in the stacking gel and has an apparent molecular weight ranging from ∼130 to >200 kd. Even after treatment of the samples with demineralizing buffer for 12 hours, the complex remains the same indicating that osteopontin most possibly is in a stable polymeric form. 29 Extracts from wild-type hearts were negative for osteopontin protein. Two minor bands of ∼40- to 50-kd molecular weight were also detected in both wild-type and desmin-null cardiac extracts at similar intensity. These could result from either cross-reactivity of the antibody that has been occasionally seen in preparations of different tissues 28 or could be basic levels of nonmodified or degraded osteopontin (the antibody used recognizes the carboxy half of the molecule). Bone extracts were used as a positive control revealing a major band of ∼65-kd molecular weight and a diffused high-molecular weight band, remained on the top of the gel (Figure 3A) ▶ as in the case of desmin-null heart extracts.
Figure 3.

Expression of osteopontin and decorin proteins is elevated in the heart of desmin-null (−/−) animals compared to wild-type (+/+). Heart extracts from 4-month-old animals were electrophoresed in a 9% SDS-PAGE and immunoblotted for osteopontin (A) and decorin (B). Osteopontin could be detected only in extracts from desmin-null heart as a high-molecular weight complex trapped in the stacking gel. Bone extracts were used as a positive control. For the decorin immunodetection, heart extracts were digested with (ABC+) chondroitinase ABC or not (ABC−) before analysis by SDS-PAGE. Note that in the undigested extracts decorin runs as a diffused band centering ∼90 kd and is significantly up-regulated in the hearts of desmin-null animals. After chondroitinase ABC digestion the proteoglycan is converted to an ∼48-kd band clearly overexpressed in the desmin-null animal whereas in the wild-type only a faint band is observed.
Western blot analysis revealed that decorin was also significantly increased (approximately threefold) in the heart of desmin-null animal when compared to the wild type (Figure 3B) ▶ . Heart extracts from 4-week-old, 4-month-old, and 8-month-old animals gave similar results (only data from 4-month-old animals are shown). In SDS-PAGE analysis of cardiac extracts, decorin runs as a diffused band with an apparent molecular weight of 85 to 105 kd. After digestion with chondroitinase ABC, decorin migrates at ∼48 kd (Figure 3B) ▶ , indicating the existence of a glycosaminoglycan chain as expected. The predicted molecular weight of the core protein is ∼38 kd and usually is modified with one glycosaminoglycan chain and two or three N-linked oligosaccharides. 23 Analysis of protein extracts from skeletal muscle (tongue and gastrocnemius) did not show any obvious differences in decorin expression between wild-type and desmin-null animals (data not shown).
Localization of Osteopontin, Decorin, Collagen, and Laminin in Cardiac Tissue Sections
Immunohistochemical and immunofluorescence analysis was performed to determine the relationship in the spatial and temporal distribution of the different matrix proteins and the pattern of calcium deposits. Extensive osteopontin staining was first observed at the age of 3 weeks in the right ventricle of desmin-null animals (Figure 4, A and B) ▶ in areas with pronounced myocyte death, acute inflammatory infiltrate, and calcium precipitation with a gritty appearance (Figure 4, C and D) ▶ . With the progression of the pathology the extensive myocyte death leads to further tissue remodeling with replacement of cardiomyocytes with fibrosis and accumulation of calcium precipitates in psammoma body structures (Figure 5) ▶ . These structures of concentric calcium laminations were positive for osteopontin staining, and were observed very frequently. In the right ventricle degeneration and calcification can reach up to 80% of the myocardium thickness. In some cases, after work overload, the damage is so extensive that it leads to rapture of the cardiac wall. 9,11 Various degrees of osteopontin staining and calcium precipitation could be also detected in areas with features of chronic inflammation (Figure 6) ▶ , such as presence of lymphocytes, macrophages, and fibroblast-like cells. These areas could be found spontaneously in different regions of the cardiac tissue, such as the ventricles (Figure 6A) ▶ , the interventricular septum (Figure 6C) ▶ , and the papillary muscles. Immunostaining of cardiac tissue sections of wild-type animals for osteopontin were negative in all corresponding cases checked (not shown). In all cases studied, osteopontin seemed to co-localize with calcium deposits (Figures 4, 5, and 6) ▶ ▶ ▶ , suggesting that this protein plays a crucial role during calcification. Comparison of osteopontin and decorin localization revealed that decorin does not co-localize with osteopontin inside calcified areas, but it is abundant in the immediate surrounding fibrotic region (Figure 5C) ▶ .
Figure 4.
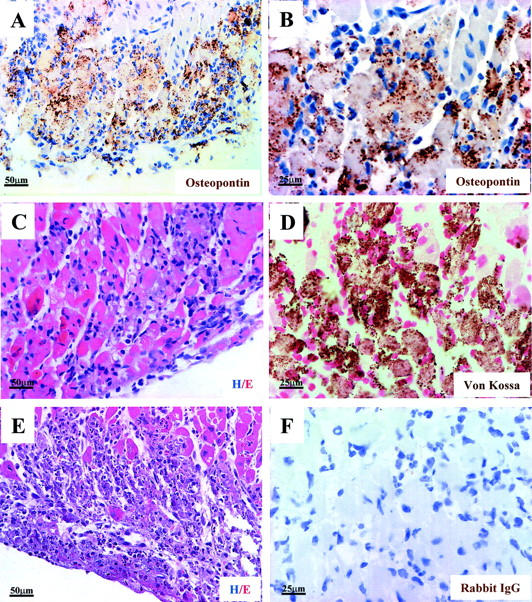
Extensive osteopontin localization is observed in the myocardium of desmin-null animals, in areas with acute inflammatory infiltrate, myocyte degeneration, beginning of calcium precipitation, and coagulative necrosis. A and B (higher magnification of A): Immunohistochemical localization of osteopontin in the right ventricle of a 3-week-old desmin-null animal. Wild-type animals were negative for osteopontin staining, (not shown). D: Von Kossa staining for calcium deposition indicates the beginning of calcium precipitation on the degenerating myocytes. C: H&E (Hem./Eos.) staining, indicating more clearly the infiltrating neutrophils-polymorphs, the degenerating myocytes, and the edema, typical characteristics of acute inflammatory reaction. E: Extended coagulative necrosis is more clearly observed a few days after the initial acute inflammatory infiltration. Degrading myocytes, necrotic cells, debris, and edema fluid are observed by H&E staining of a corresponding area in a desmin-null animal 5 days older than the one described in A–D. F: Negative control for the immunohistochemistry. Unrelated rabbit IgG was used as primary antibody. No staining is observed. A–D and F are from adjacent sections of a 3-week-old heart. For the immunoperoxidase staining (A, B, and F), diaminobenzidine was used as substrate and hematoxylin for nuclei counterstaining. Von Kossa staining for calcium deposition (D) is brown/black and counterstaining for nuclei by Kernechtrot is red.
Figure 5.
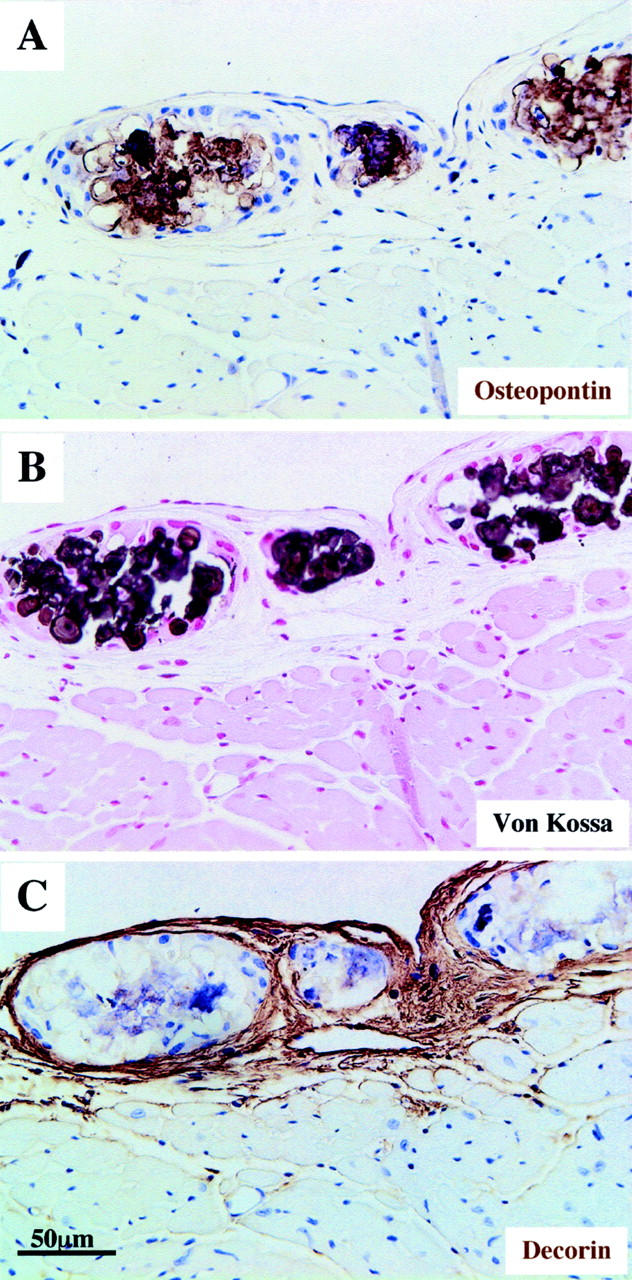
Immunohistochemical localization of osteopontin and decorin in advanced stage calcification. Osteopontin is detected in desmin-null animals in necrotic areas with extensive calcification and psammoma body morphology (A and B). These structures are large mineralized deposits with lamellated configurations surrounded by decorin-containing fibrotic tissue (C). A, B, and C are from serial sections in the outer surface of the right ventricle of a 6-month-old animal. Immunohistochemistry staining for osteopontin and decorin (A and C) and von Kossa staining (B) is same as in Figure 4 ▶ .
Figure 6.
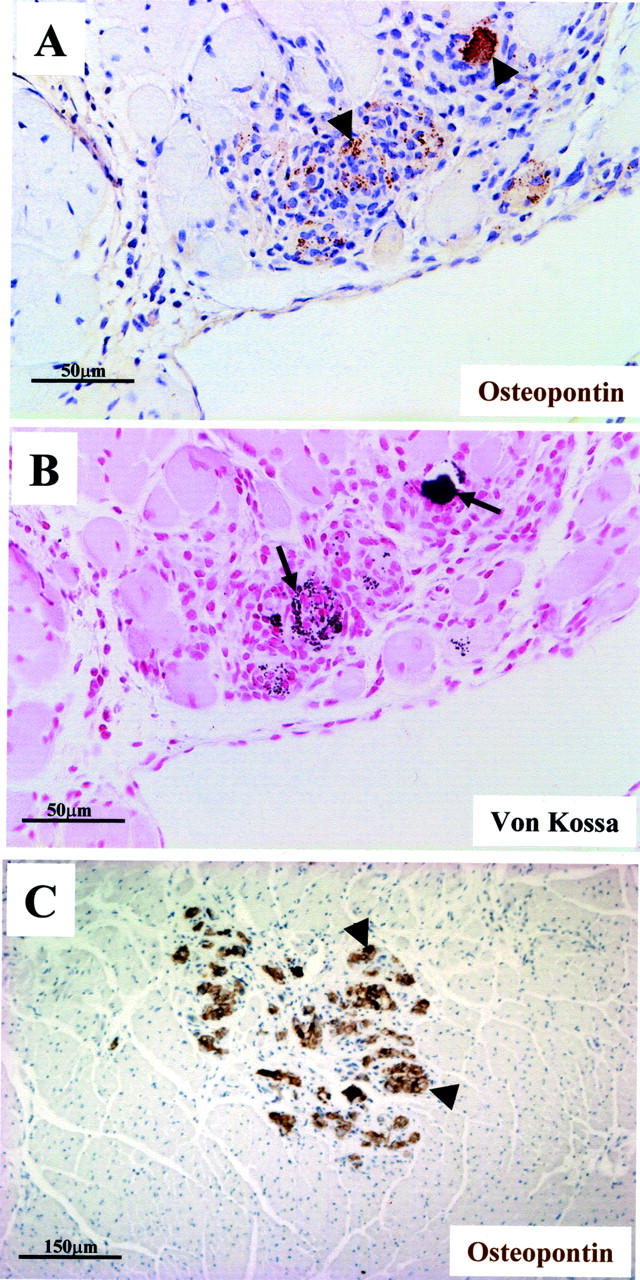
Osteopontin co-localizes with calcium in areas with chronic inflammation. Immunohistochemical localization of osteopontin (arrowheads) in a hypercellular area of infiltrate in the right ventricle of desmin-null animal (A) together with mild granular calcium deposits as indicated by a serial section stained by von Kossa (B, arrows). Osteopontin (arrowheads) is also detected in calcified regions of other parts of the myocardium such as the septum (C). A and B are from a 6-month-old animal and C from a 2-month-old animal. Staining procedure is same as in Figure 4 ▶ .
Immunofluorescence localization of decorin in the heart of desmin-null animals gave more intense staining in the epicardium (not shown) and very strong and extensive staining in fibrotic areas (Figure 7B) ▶ in contrast to the wild-type animals, where decorin is localized mainly in the endomysium (Figure 7A) ▶ . Extended areas of decorin staining could be found in different compartments of cardiac tissue as early as in 4 weeks after birth and throughout the entire life of the animal.
Figure 7.
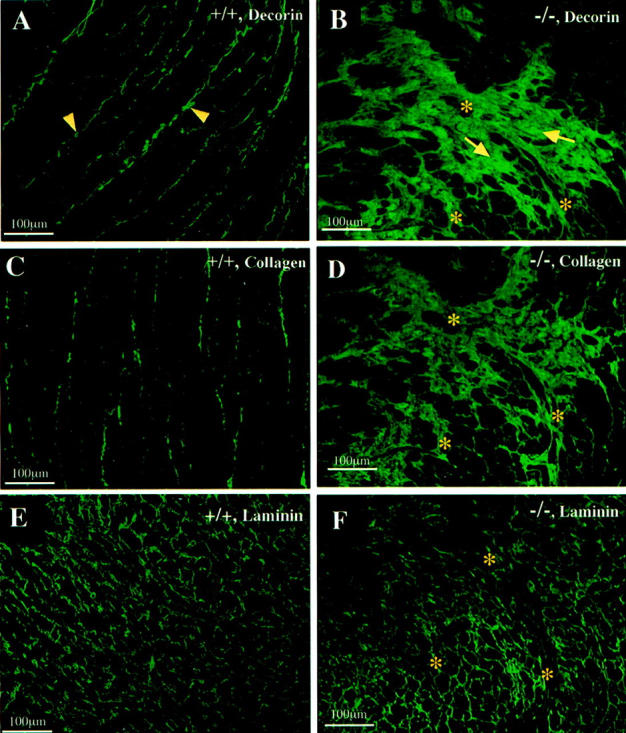
Abnormal accumulation of decorin and collagen but not laminin in the myocardium of desmin-null animal. Immunofluorescence localization of decorin, collagen, and laminin was performed in cardiac tissue from wild-type (A, C, and E) and desmin-null (B, D, and F) animals. Extensive decorin staining could be seen in fibrotic areas (arrows) of desmin-null-only myocardium (B) but not in wild-type animal (A), where only staining of the endomysium (arrowheads) could be detected. B, D, and F: Staining of serial sections of desmin-null myocardium, for decorin, collagen, and laminin. Note that laminin shows normal pattern and is not a component of the fibrotic areas (compare with the wild-type in E). Decorin (B) has a similar staining pattern as collagen (D) and co-localizes with it in fibrotic areas of desmin-null myocardium. Asterisks in B, D, and F indicate corresponding spots in serial sections.
Immunofluorescence localization of collagen-α1(I) gave intense staining in the heart of desmin-null animals in fibrotic (Figure 7D) ▶ and perivascular areas, with a pattern very similar to decorin. On the other hand immunofluorescence localization of laminin (α1,β1,γ1), an abundant basement membrane protein that envelopes individual cardiomyocytes, did not reveal any obvious difference between wild-type (Figure 7E) ▶ and desmin-null animals (Figure 7F) ▶ . The data revealed that laminin does not contribute to the extended fibrotic lesions in the heart of desmin-null animals (compare Figure 7 ▶ ; B, D, and F).
Discussion
In an effort to elucidate the molecular mechanisms by which the lack of desmin leads to the observed cardiac pathology, we have screened RNA from wild-type and desmin-null hearts for changes in gene expression. It was very encouraging to find that the most dramatic changes observed in gene expression were for osteopontin and decorin, molecules linked to fibrosis, calcification, and cardiomyocyte death, the hallmarks of the desmin-null heart pathology.
Osteopontin has been implicated in multiple diverse functions in both physiological and pathological processes as recently reviewed. 30,31 Although normally it is expressed in bone and at epithelia surfaces, 32 osteopontin is elevated during injury and inflammation in most tissues studied today, including calcification in atherosclerotic plaques, 33 T-cell response to infection, 34 wound healing, 35,36 and tumor growth. 37 These findings have suggested a role for osteopontin in modulating the inflammatory process, for example by stimulating macrophage infiltration. Also much like its role in bone, osteopontin’s ability to interact with hydroxyapatite crystals 38-40 probably serves both to interfere with bioapatite crystal growth through physical interactions, as well as to regulate host cell resorptive mechanisms of the ectopic calcification via receptor-mediated interactions. 31,41
Recent reports have shown expression of osteopontin by macrophages in cardiac tissue in response to myocardial necrosis caused by transdiaphragmatic freezing, 35 in cardiomyopathic Syrian hamster, 42 and in spontaneously hypertensive rats. 43 On the other hand in a single report cardiomyocytes have also been found as the primary source of osteopontin in a hypertrophy model of rat heart by renovascular hypertension and aortic banding. 44 These differences could be just the consequence of different time periods studied. It is possible that at early stages of cardiomyopathy myocytes may be the source of osteopontin mRNA but later interstitial nonmyocyte cells are taking over. In the present case the cell source of osteopontin was not revealed.
As described above, osteopontin is believed to act as an inhibitor of calcification. 39 However, in all cases tested in the desmin-null heart although osteopontin co-localizes with calcium deposits, the calcification does indeed progress overriding the inhibitory action of osteopontin or osteopontin is in a form that cannot anymore act as inhibitor of calcification. This could be linked to our observation that in desmin-null myocardium, osteopontin forms high-molecular weight complexes (Figure 3A) ▶ . These complexes, might represent polymeric forms of osteopontin, possibly covalently bound through the action of transglutaminase, as has previously been demonstrated. 29 This form of osteopontin has the increased ability to bind collagen, 45 thus it can serve in adhering together calcified deposits, the surrounding cells, and adhesive matrix. In such form it might be irreversibly bound to calcium deposits, thus promoting the isolation of these deposits by fibrotic tissue (Figure 5C) ▶ and eventually the formation of psammoma body structures.
The precise mechanism by which the absence of desmin leads to cardiomyocyte death and calcification, and the reasons why this does not happen that extensively in skeletal muscle, at the present, can be only speculated. Our recent studies have strongly suggested that desmin is very important for normal mitochondrial behavior and function. 11 There is plenty of evidence suggesting that impaired mitochondrial behavior and function could lead to cell death. 46 Because mitochondrial abnormalities are the earliest defects that have been observed in the desmin-null heart, we believe these defects are the main cause of death of these mice. The fact that heart muscle cells have the maximum content of desmin, 2% of total protein, compared to 0.35% of skeletal muscle cells, 47 and the highest volume density of mitochondria of all mammalian cells (36% in mice), 48 could easily explain why these cells are mostly affected. When compared to fast glycolytic muscle, slow oxidative skeletal muscle, which has also more mitochondria, is more affected by the absence of desmin. 11 However, the ability of skeletal muscle to regenerate could explain the lack of necrotic tissue accumulation and pronounced calcification. Other explanations are not excluded.
One of the many ways by which mitochondrial abnormalities could lead to cell death in desmin-null heart could be the disturbance of intracellular calcium homeostasis. Ultrastructural studies in dystrophic cardiac calcinosis (DDC) mice, which show many common features with the desmin-null calcification, 49 have shown that initial events of calcification include granular calcium deposition in or around mitochondria. 49,50 Mitochondria are able to take up large amounts of Ca+2 and buffer cytosolic Ca+2 levels. If this is compromised, excessive intracellular Ca+2 can contribute to cytotoxic events leading to formation of reactive oxygen species and cell death. 51 Cytotoxic events such as increased phosphate concentration 52 by overactivation of protein phosphatases and increased NO production by stimulated macrophages 53 can significantly induce the osteopontin expression. Another potential mechanism of osteopontin induction could involve angiotensin II, which can induce osteopontin expression in cardiac fibroblasts 54 and can directly increase both TGF-β1 and osteopontin in the heart. 55 The present cDNA array studies revealed increased ACE expression in the desmin-null hearts, which might contribute to osteopontin induction through angiotensin II. In turn, osteopontin could modulate calcium levels by different ways including calcium mobilization, 56,57 activation of Ca++ATPase pump, 58 Ca++ binding, 59 and by modulating hydroxyapatite crystal growth. 38 Thus, abnormalities in mitochondria apart from cytochrome c- and caspase-related prodeath events, can initiate and maintain the Ca++/osteopontin cycle described above, which could lead to the extensive calcification in desmin-null hearts.
From our study it is evident that another noncollagenous matrix protein, decorin, is a prominent component of fibrotic areas in the myocardium of desmin-null animals. Up-regulation of decorin and its co-localization with collagen in the desmin-null heart could correlate with its ability to participate in the assembly of fibrillar collagen. 60 Thus decorin up-regulation could be a counteraction to tissue necrosis, interfering with tissue remodeling by fibrosis. 26,27 Elevation in the expression of decorin was recently reported in two cases in myocardial infarction 61,62 and one in cardiac hypertrophy, 63 all of which displayed fibrosis. The two more recent cases were also elaborated by global gene expression analysis. 62,63 Decorin has the ability to bind and neutralize TGF-β1 and potentially abrogate its effect on tissue fibrosis. 26,27 On the other hand TGF-β1 has the potential to initiate the production of decorin. 64,65 Thus decorin in the heart of desmin-null mice could participate in a feedback loop that regulates TGF-β1 action. A potential mechanism by which induction of both osteopontin and decorin could be achieved in desmin-null hearts is by angiotensin II, directly or through TGF-β1, 55 as a result of the increased ACE expression found in desmin-null hearts (see Table 1 ▶ ).
In addition to osteopontin and decorin, molecules such as bone/cartilage proteoglycan I precursor (also named biglycan) and osteoblast-specific factor-2, initially thought to only participate in normal osteogenesis are also up-regulated in the heart of desmin-null mice (see Table 1 ▶ ) and in other cardiomyopathy models. 62,66 Except for osteoblast-specific factor-2, all of these molecules are also overexpressed in dystrophic vascular calcification, 12 suggesting that a common mechanism resembling osteogenesis may exist in vascular and cardiac calcification.
In conclusion, despite the complexity of the observed pathology in the desmin-null heart the data so far favor the possibility that the early observed mitochondria abnormalities, maybe through disturbance of Ca+2 homeostasis, can initiate a cycling cascade of events leading to extensive cell death and calcification. Among other interplayers of this cascade, the matricellular proteins osteopontin and decorin together with, or because of, increased TGF-β1 and ACE expression can modulate fibrosis and calcification and thus the development of dilated cardiomyopathy and heart failure.
Acknowledgments
We thank Georgios Rassidakis, M.D., for his help in the interpretation of the histopathology data; Mr. Noah Weisleder for his critical comments; and Dr. Larry Fisher for supplying us with the osteopondin cDNA probe and the antisera LF-123, LF-69, and LF-113.
Footnotes
Address reprint requests to Dr. Yassemi Capetanaki, One Baylor Plaza Houston, TX 77030. E-mail: yassemic@bcm.tmc.edu.
Supported by National Institutes of Health grant AR39617 (to Y. C.).
References
- 1.Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y: Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 1996, 134:1255-1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y: The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol 1999, 31:2063-2076 [DOI] [PubMed] [Google Scholar]
- 3.Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C: Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol 1996, 175:362-366 [DOI] [PubMed] [Google Scholar]
- 4.Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R: Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100:461-464 [DOI] [PubMed] [Google Scholar]
- 5.Munoz-Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez-Figueras MT, Navas-Palacios JJ, Ariza A, Fuchs E: A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci USA 1998, 95:11312-11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC: Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998, 19:402-403 [DOI] [PubMed] [Google Scholar]
- 7.Sjoberg G, Saavedra-Matiz CA, Rosen DR, Wijsman EM, Borg K, Horowitz SH, Sejersen T: A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet 1999, 8:2191-2198 [DOI] [PubMed] [Google Scholar]
- 8.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG: Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000, 342:770-780 [DOI] [PubMed] [Google Scholar]
- 9.Capetanaki Y: Desmin cytoskeleton in healthy and failing heart. Heart Failure Rev 2000, 5:203-220 [DOI] [PubMed] [Google Scholar]
- 10.Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D: Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol 1997, 29:2107-2124 [DOI] [PubMed] [Google Scholar]
- 11.Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y: Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 2000, 150:1283-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giachelli CM: Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Pathol 1999, 154:671-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donley GE, Fitzpatrick LA: Vascular calcification. Trends Cardiovasc Med 1998, 8:199-206 [DOI] [PubMed] [Google Scholar]
- 14.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G: Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386:78-81 [DOI] [PubMed] [Google Scholar]
- 15.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998, 12:1260-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eaton GJ, Custer RP, Johnson FN, Stabenow KT: Dystrophic cardiac calcinosis in mice: genetic, hormonal, and dietary influences. Am J Pathol 1978, 90:173-186 [PMC free article] [PubMed] [Google Scholar]
- 17.Rings RW, Wagner JE: Incidence of cardiac and other soft tissue mineralized lesions in DNA-2 mice. Lab Anim Sci 1972, 22:344-352 [PubMed] [Google Scholar]
- 18.Fatkin D, Christe ME, Aristizabal O, McConnell BK, Srinivasan S, Schoen FJ, Seidman CE, Turnbull DH, Seidman JG: Neonatal cardiomyopathy in mice homozygous for the Arg403Gln mutation in the alpha cardiac myosin heavy chain gene. J Clin Invest 1999, 103:147-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG, Fischman DH: Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest 1999, 104:1235-1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consalez GG, Cabibbo A, Corradi A, Alli C, Sardella M, Sitia R, Fesce R: A computer-driven approach to PCR-based differential screening, alternative to differential display. Bioinformatics 1999, 15:93-105 [DOI] [PubMed] [Google Scholar]
- 21.Fisher LW, Stubbs JT, Young MF: Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand 1995, 266(Suppl):S61-S65 [PubMed] [Google Scholar]
- 22.Rungby J, Kassem M, Eriksen EF, Danscher G: The von Kossa reaction for calcium deposits: silver lactate staining increases sensitivity and reduces background. Histochem J 1993, 25:446-451 [DOI] [PubMed] [Google Scholar]
- 23.Iozzo RV: Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem 1998, 67:609-652 [DOI] [PubMed] [Google Scholar]
- 24.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M: Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci USA 2000, 97:931-936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein KE, Martin BM, Edwards AS, Bernstein EA: Mouse angiotensin-converting enzyme is a protein composed of two homologous domains. J Biol Chem 1989, 264:11945-11951 [PubMed] [Google Scholar]
- 26.Isaka Y, Brees DK, Ikegaya K, Kaneda Y, Imai E, Noble NA, Border WA: Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat Med 1996, 2:418-423 [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi Y, Mann DM, Ruoslahti E: Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346:281-284 [DOI] [PubMed] [Google Scholar]
- 28.Rittling SR, Feng F: Detection of mouse osteopontin by Western blotting. Biochem Biophys Res Commun 1998, 250:287-292 [DOI] [PubMed] [Google Scholar]
- 29.Kaartinen MT, Pirhonen A, Linnala-Kankkunen A, Maenpaa PH: Transglutaminase-catalyzed cross-linking of osteopontin is inhibited by osteocalcin. J Biol Chem 1997, 272:22736-22741 [DOI] [PubMed] [Google Scholar]
- 30.Giachelli CM, Steitz S: Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol 2000, 19:615-622 [DOI] [PubMed] [Google Scholar]
- 31.Denhardt DT, Giachelli CM, Rittling SR: Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol 2001, 41:723-749 [DOI] [PubMed] [Google Scholar]
- 32.Brown LF, Berse B, Van de Water L, Papadopoulos-Sergiou A, Perruzzi CA, Manseau EJ, Dvorak HF, Senger DR: Expression and distribution of osteopontin in human tissues: widespread association with luminal epithelial surfaces. Mol Biol Cell 1992, 3:1169-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzpatrick LA, Severson A, Edwards WD, Ingram RT: Diffuse calcification in human coronary arteries. Association of osteopontin with atherosclerosis. J Clin Invest 1994, 94:1597-1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patarca R, Saavedra RA, Cantor H: Molecular and cellular basis of genetic resistance to bacterial infection: the role of the early T-lymphocyte activation-1/osteopontin gene. Crit Rev Immunol 1993, 13:225-246 [PubMed] [Google Scholar]
- 35.Murry CE, Giachelli CM, Schwartz SM, Vracko R: Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol 1994, 145:1450-1462 [PMC free article] [PubMed] [Google Scholar]
- 36.McKee MD, Nanci A: Secretion of osteopontin by macrophages and its accumulation at tissue surfaces during wound healing in mineralized tissues: a potential requirement for macrophage adhesion and phagocytosis. Anat Rec 1996, 245:394-409 [DOI] [PubMed] [Google Scholar]
- 37.Tuck AB, O’Malley FP, Singhal H, Harris JF, Tonkin KS, Kerkvliet N, Saad Z, Doig GS, Chambers AF: Osteopontin expression in a group of lymph node negative breast cancer patients. Int J Cancer 1998, 79:502-508 [DOI] [PubMed] [Google Scholar]
- 38.Boskey AL, Maresca M, Ullrich W, Doty SB, Butler WT, Prince CW: Osteopontin-hydroxyapatite interactions in vitro: inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner 1993, 22:147-159 [DOI] [PubMed] [Google Scholar]
- 39.Wada T, McKee MD, Steitz S, Giachelli CM: Calcification of vascular smooth muscle cell cultures: inhibition by osteopontin. Circ Res 1999, 84:166-178 [DOI] [PubMed] [Google Scholar]
- 40.Hunter GK, Kyle CL, Goldberg HA: Modulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem J 1994, 300:723-728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liaw L, Skinner MP, Raines EW, Ross R, Cheresh DA, Schwartz SM, Giachelli CM: The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. J Clin Invest 1995, 95:713-724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams EB, Halpert I, Wickline S, Davison G, Parks WC, Rottman JN: Osteopontin expression is increased in the heritable cardiomyopathy of Syrian hamsters. Circulation 1995, 92:705-709 [DOI] [PubMed] [Google Scholar]
- 43.Singh K, Sirokman G, Communal C, Robinson KG, Conrad CH, Brooks WW, Bing OH, Colucci WS: Myocardial osteopontin expression coincides with the development of heart failure. Hypertension 1999, 33:663-670 [DOI] [PubMed] [Google Scholar]
- 44.Graf K, Do YS, Ashizawa N, Meehan WP, Giachelli CM, Marboe CC, Fleck E, Hsueh WA: Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation 1997, 96:3063-3071 [DOI] [PubMed] [Google Scholar]
- 45.Kaartinen MT, Pirhonen A, Linnala-Kankkunen A, Maenpaa PH: Cross-linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J Biol Chem 1999, 274:1729-1735 [DOI] [PubMed] [Google Scholar]
- 46.Kroemer G, Reed JC: Mitochondrial control of cell death. Nat Med 2000, 6:513-519 [DOI] [PubMed] [Google Scholar]
- 47.Price MG: Molecular analysis of intermediate filament cytoskeleton—a putative load-bearing structure. Am J Physiol 1984, 246:H566-H572 [DOI] [PubMed] [Google Scholar]
- 48.Barth E, Stammler G, Speiser B, Schaper J: Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 1992, 24:669-681 [DOI] [PubMed] [Google Scholar]
- 49.Van Vleet JF, Ferrans VJ: Ultrastructural changes in inherited cardiac calcinosis of DBA/2 mice. Am J Vet Res 1987, 48:255-261 [PubMed] [Google Scholar]
- 50.McClure J, Pieterse AS, Pounder DJ, Smith PS: Myocardial fibre calcification. J Clin Pathol 1981, 34:1167-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter C: Nitric oxide and its congeners in mitochondria: implications for apoptosis. Environ Health Perspect 1998, 106(Suppl 5):S1125-S1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beck GR, Zerler B, Moran E: Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci USA 2000, 97:8352-8357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo H, Cai CQ, Schroeder RA, Kuo PC: Osteopontin is a negative feedback regulator of nitric oxide synthesis in murine macrophages. J Immunol 2001, 166:1079-1086 [DOI] [PubMed] [Google Scholar]
- 54.Ashizawa N, Graf K, Do YS, Nunohiro T, Giachelli CM, Meehan WP, Tuan TL, Hsueh WA: Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J Clin Invest 1996, 98:2218-2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kupfahl C, Pink D, Friedrich K, Zurbrugg HR, Neuss M, Warnecke C, Fielitz J, Graf K, Fleck E, Regitz-Zagrosek V: Angiotensin II directly increases transforming growth factor beta1 and osteopontin and indirectly affects collagen mRNA expression in the human heart. Cardiovasc Res 2000, 46:463-475 [DOI] [PubMed] [Google Scholar]
- 56.Denhardt DT, Lopez CA, Rollo EE, Hwang SM, An XR, Walther SE: Osteopontin-induced modifications of cellular functions. Ann N Y Acad Sci 1995, 760:127-142 [DOI] [PubMed] [Google Scholar]
- 57.Zimolo Z, Wesolowski G, Tanaka H, Hyman JL, Hoyer JR, Rodan GA: Soluble alpha v beta 3-integrin ligands raise [Ca2+]i in rat osteoclasts and mouse-derived osteoclast-like cells. Am J Physiol 1994, 266:C376-C381 [DOI] [PubMed] [Google Scholar]
- 58.Miyauchi A, Alvarez J, Greenfield EM, Teti A, Grano M, Colucci S, Zambonin-Zallone A, Ross FP, Teitelbaum SL, Cheresh D, Hruska KA: Recognition of osteopontin and related peptides by an alpha v beta 3 integrin stimulates immediate cell signals in osteoclasts. J Biol Chem 1991, 266:20369-20374 [PubMed] [Google Scholar]
- 59.Singh K, Deonarine D, Shanmugam V, Senger DR, Mukherjee AB, Chang PL, Prince CW, Mukherjee BB: Calcium-binding properties of osteopontin derived from non-osteogenic sources. J Biochem (Tokyo) 1993, 114:702–707 [DOI] [PubMed]
- 60.Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV: Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 1997, 136:729-743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IM: Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol 1999, 31:667–678 [DOI] [PubMed]
- 62.Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, Zheng Q, Protter AA, Schreiner GF, White RT: Altered patterns of gene expression in response to myocardial infarction. Circ Res 2000, 86:939-945 [DOI] [PubMed] [Google Scholar]
- 63.Hwang DM, Dempsey AA, Lee CY, Liew CC: Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics 2000, 66:1-14 [DOI] [PubMed] [Google Scholar]
- 64.Bassols A, Massague J: Transforming growth factor beta regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J Biol Chem 1988, 263:3039-3045 [PubMed] [Google Scholar]
- 65.Okuda S, Languino LR, Ruoslahti E, Border WA: Elevated expression of transforming growth factor-beta and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J Clin Invest 1990, 86:453-462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnatty SE, Dyck JR, Michael LH, Olson EN, Abdellatif M: Identification of genes regulated during mechanical load-induced cardiac hypertrophy. J Mol Cell Cardiol 2000, 32:805-815 [DOI] [PubMed] [Google Scholar]