

Abstract
CpG island methylation is an important mechanism for inactivating the genes involved in tumorigenesis. Gastric carcinoma (GC) is one of the tumors that exhibits a high frequency of aberrant CpG island methylation. There have been many reports suggesting a close link between Epstein-Barr virus (EBV) and the development of GC. However, little is known about the oncogenic mechanism of EBV in gastric carcinogenesis. Twenty-one cases of EBV-positive GC and 56 cases of EBV-negative GC were examined for aberrant DNA methylation of the CpG islands of 19 genes or loci and the differences in the methylation frequency between EBV-positive and -negative GCs were investigated to determine a role of aberrant methylation in EBV-related gastric carcinogenesis. The average number of methylated genes or loci was higher in EBV-positive GCs than in EBV-negative GCs (13.4 versus 7.8, respectively, P < 0.001). EBV-positive GCs showed methylation in at least 10 CpG islands (52.6% of the tested genes), whereas 62.5% of EBV-negative GCs showed methylation in <10 CpG islands. THBS1, APC, p16, 14-3-3 sigma, MINT1, and MINT25 were methylated at a frequency >90% in EBV-positive GCs. The methylation frequency difference in the respective CpG islands between EBV-positive and -negative GCs was statistically significant (P < 0.05). Among these genes or loci, the methylation frequency of p16 in the EBV-positive GCs was more than three times higher than in the EBV-negative GCs. The PTEN, RASSF1A, GSTP1, MGMT, and MINT2 were methylated in EBV-positive GCs at a frequency of more than three times that of the EBV-negative GCs. These results demonstrate a relationship between EBV and aberrant methylation in GC and suggest that aberrant methylation may be an important mechanism of EBV-related gastric carcinogenesis.
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus with well-established associations with endemic Burkitt’s lymphoma, 1 nasopharyngeal carcinoma, 2 and opportunistic B-cell lymphomas in immunodeficiency. 3 A subset of gastric carcinomas (GCs), known as lymphoepithelioma-like GC (LELC), are known to harbor the EBV genome in a high proportion of cases. 4-6 Furthermore, ∼6% of GCs without LELC features have been reported to contain EBV in cancer cells. 4-6 Based on findings such as the clonal nature of EBV in GC cells, 5 the presence of EBV in nearly all of the cancer cells in EBV-positive GC, and the presence of EBV in precancerous lesions of the stomach, 4,7 a causal role of EBV in gastric carcinogenesis has been suggested. However, the transforming oncoproteins of EBV such as latent membrane protein 1 (LMP1) and nuclear antigen 2 (EBNA2), are usually not expressed in EBV-positive GCs 5,8 and there is little evidence that p53 is targeted for inactivation by EBV. To date, little is known regarding the molecular mechanism of EBV-related gastric carcinogenesis.
EBV-positive GCs have been reported to show a higher frequency of p16 expressional loss than EBV-negative GCs. 9 Homozygous deletion or mutation of p16 is quite rare 10,11 but p16 promoter hypermethylation is common and well-correlated with p16 protein loss in primary GCs. 12,13 Considering these facts, it could be speculated that promoter hypermethylation may cause p16 inactivation in EBV-positive GCs and this event occurs more frequently in EBV-positive GCs than in EBV-negative GCs. Recently, CpG island methylator phenotype (CIMP) was proposed in colon, 14 stomach, 15 and pancreas cancers. 16 It is characterized by widespread hypermethylation of CpG islands over the genome. CIMP-positive tumors in these organs showed p16 promoter hypermethylation much more frequently than CIMP-negative tumors. In tumors of these organs, the presence of p16 promoter hypermethylation might indicate that the tumor is likely to be a CIMP-positive tumor. These findings led us to speculate that EBV-positive GCs might be CIMP-positive tumors and that the aberrant methylation process involves not only p16 but also other tumor suppressor genes and functions as an important mechanism for EBV-related gastric carcinogenesis. Recent studies demonstrating a close association between aberrant methylation and foreign viral DNA entry into host cells support this possibility. 17,18
To investigate the relationship between aberrant methylation and EBV-positive GCs, A candidate gene approach was used and 21 cases of EBV-positive GCs and 56 cases of EBV-negative GCs were examined for methylation of CpG islands, including 5 MINT loci and 14 genes undergoing epigenetic inactivation in a primary human tumor. The tested genes included those involved in cell-cycle regulation (p14, p16, 14-3-3 sigma, and COX2), signal transduction (APC, PTEN, and RASSF1A), DNA repair or protection (hMLH1, MGMT, and GSTP1), apoptosis (DAP-kinase), and angiogenesis (THBS1) or those related to metastasis and invasion (E-cadherin and TIMP-3). In addition, the EBV status was correlated with the clinicopathological features and microsatellite instability (MSI) status of GCs.
Materials and Methods
Tumor Samples and DNA Extraction
Two hundred and thirty-three GC cases surgically excised at the Asan Medical Center, Seoul, Korea, between 1996 and 1998 were examined for the presence of EBV using EBV RNA in situ hybridization. Among these, 21 cases (9%) were EBV-positive. Of the EBV-negative cases, 56 consecutive cases were selected as controls. The control samples were characterized previously for p16 and hMLH1 promoter methylation, both p16 and hMLH1 protein expression, and MSI status. 12,19 The archival materials were histologically examined and portions of tumors where tumor cells comprised >50% of the cells were scraped from 20-μm-thick paraffin sections. Uninvolved mucosa was used as a normal control. Genomic DNA was extracted from the samples using the classical method of phenol/chloroform/isoamylalcohol and proteinase K.
EBV-Encoded RNA (EBER) in Situ Hybridization
The EBV RNA in situ hybridization was performed using a fluorescein-conjugated peptide nucleic acid probe complimentary to a portion of the small EBERs 1 and 2. Five-μm thick sections on slides coated with poly-l-lysine were routinely deparaffinized, dehydrated, and predigested with 3 μg/ml proteinase K, and then hybridized for 2 hours at 37°C. Anti-fluorescein antibody-alkaline phosphatase was used with a NBT/BCIP kit (DAKO, Copenhagen, Denmark) to detect the EBER signals. Counterstaining was done with 0.3% hematoxylin.
Bisulfite Modification and Methylation-Specific Polymerase Chain Reaction (MSP)
The bisulfite treatment was performed as described previously. 20 Briefly, 1 μg of genomic DNA was treated with sodium bisulfite to convert unmethylated cytosines to uracil and leave 5-methyl cytosines unchanged. The modification was performed for 16 hours at 55°C and then the modified DNA was purified and eluted into 50 μl of TE buffer.
MSP was used to examine the methylation status of 14 genes and 5 cancer-specific MINT clones (APC, COX2, DAP-kinase, E-cadherin, GSTP1, hMLH1, MGMT, PTEN, p14, p16, RASSF1A, 14-3-3 sigma, THBS1, TIMP-3, MINT1, MINT2, MINT12, MINT25, and MINT31). The primer sequences of each CpG island are described in Table 1 ▶ . MSP was performed in 25-μl reaction volumes containing 1× polymerase chain reaction (PCR) buffer (16.6 mmol/L (NH4)2SO4/67 mmol/L Tris/pH 8.8/6.7 mmol/L MgCl2/10 mmol/L β-mercaptoethanol), dNTPs (each at 1 mmol/L), primers (10 pmol each), and bisulfite-modified DNA (30 to 50 ng). The reactions were hot-started at 97°C for 5 minutes before adding 0.75 U of Taq polymerase (Takara Shuzo Co., Kyoto, Japan). The amplifications were performed in a Thermal cycler (Perkin-Elmer, Foster City, CA) for 33 cycles, followed by a final 10-minute extension. The annealing temperature for the primers is given in Table 1 ▶ . The PCR products were electrophoresed on a 2.5% agarose gel and visualized under UV illumination after staining with ethidium bromide.
Table 1.
Primer Sequences and PCR Conditions for MSP Analysis
| Primer name | Forward primer (5′-3′) | Reverse primer (5′-3′) | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| APC | M: TATTGCGGAGTGCGGGTC | M: TCGACGAACTCCCGACGA | 98 | 55 |
| COX2 | M: TTAGATACGGCGGCGGCGGC | M: TCTTTACCCGAACGCTTCCG | 161 | 61 |
| DAP-kinase | M: GGATAGTCGGATCGAGTTAACGTC | M: CCCTCCCAAACGCCGA | 98 | 60 |
| E-cadherin | M: TTAGGTTAGAGGGTTATCGCGT | M: TAACTAAAAATTCACCTACCGAC | 116 | 57 |
| GSTP1 | M: TTCGGGGTGTAGCGGCGTC | M: GCCCCAATACTAAATCACGACG | 91 | 59 |
| hMLH1 | M: TATATCGTTCGTAGTATTCGTGT | M: TCCGACCCGAATAAACCCAA | 153 | 60 |
| MGMT | M: TTTCGACGTTCGTAGGTTTTCGC | M: GCACTCTTCCGAAAACGAAACG | 81 | 59 |
| MINT1 | M: GGGTTGAGGTTTTTTGTTAGC | M: CTACTTCGCCTAACCTAACG | 102 | 64 |
| MINT2 | M: AATCGAATTTGTCGTCGTTTC | M: AAATAAATAAATAAAAAAAAACGCG | 88 | 60 |
| MINT12 | M: TTGGGAGTTTATTTAGGTCG | M: ACAACGATCTTCCGAATTTA | 152 | 55 |
| MINT25 | M: GTTCGTTAGAGTAATTTTGCG | M: TTATAACTAACGAAACACCGC | 128 | 55 |
| MINT31 | M: TTGAGACGATTTTAATTTTTTGC | M: AAAACCATCACCCCTAAACG | 100 | 62 |
| PTEN | M: TTCGTTCGTCGTCGTCGTATTT | M: GCCGCTTAACTCTAAACCGCAACCG | 206 | 62 |
| p14 | M: GTGTTAAAGGGCGGCGTAGC | M: AAAACCCTCACTCGCGACGA | 122 | 60 |
| p16 | M: TTATTAGAGGGTGGGGCGGATCGC | M: GACCCCGAACCGCGACCGTAA | 150 | 65 |
| U: TTATTAGAGGGTGGGGTGGATTGT | U: CAACCCCAAACCACAACCATAA | 151 | 60 | |
| RASSF1A | M: GTGTTAACGCGTTGCGTATC | M: AACCCCGCGAACTAAAAACGA | 93 | 60 |
| THBS1 | M: TGCGAGCGTTTTTTTAAATGC | M: TAAACTCGCAAACCAACTCG | 74 | 62 |
| TIMP-3 | M: CGTTTCGTTATTTTTTGTTTTCGGTTTTC | M: CCGAAAACCCCGCCTCG | 116 | 59 |
| 14-3-3 sigma | M: TGGTAGTTTTTATGAAAGGCGTC | M: CCTCTAACCGCCCACCACG | 104 | 56 |
M, methylated-specific primers; U, unmethylated primers.
Sequencing Analysis
To test whether there was adequate bisulfite modification and to rule out false amplification, the MSP products were sequenced to determine the methylation status of each CpG site of the specific CpG islands. The MSP products were retrieved from the gels then purified and sequenced using both a ABI Prism Dye Terminator Cycle Sequencing Kit (Perkin-Elmer) and an ABI Prism 377 DNA Sequencer (Perkin-Elmer).
Immunohistochemistry
Tissue sections were deparaffinized in xylene, rehydrated in graded alcohol, and then washed in water. Antigen retrieval was performed using microwave irradiation in 10 mmol/L of citrate buffer (pH 6.0). The endogenous peroxidase activity and nonspecific protein binding was blocked by incubation with 3% H2O2 and 10% normal goat serum, respectively. Sections were incubated with antibodies to hMLH1 protein (clone G168-728, dilution 1:50; Pharmingen, San Diego, CA), p16 (SC1661, dilution 1:100; Santa Cruz, Santa Cruz, CA), LMP1 (clones CS1-4, dilution 1:25; DAKO, Copenhagen, Denmark), EBNA2 (clone PE2, dilution 1:50; DAKO), and BZLF1 (clone BZ.1, dilution 1:50; DAKO) at 4°C overnight. After reacting them with biotinylated secondary anti-mouse antibodies, the antigen-antibody reactions were visualized using streptavidin-horseradish peroxidase conjugate (DAKO LSAB kit, Los Angeles, CA) and diaminobenzidine as a chromogen. The slides were counterstained with hematoxylin.
MSI Analysis
To analyze MSI status of 21 EBV-positive GCs, 10 microsatellite loci were used: BAT26, BAT40, D2S123, D2S136, D3S1067, D5S299, D9S165, D9S171, TP53, and D17S250. The standard PCR was performed with a 25-μl reaction mixture containing 10 pmol of the primers, 1.5 mmol/L MgCl2, 0.2 mmol/L of each deoxynucleotide triphosphate (dNTP), 0.75 U of the Taq polymerase (Takara Shuzo Co.). The reaction mixture was subjected to 35 PCR cycles with primer annealing at between 45°C to 60°C for 1 minute. Denaturation of the PCR products, gel electrophoresis, and silver staining were performed as previously described. 21 To avoid PCR artifacts, all positive tests were duplicated. Tumors with an instability in 30% or more of the examined loci were scored as being MSI-positive.
Results
EBERs in Situ Hybridization and Clinicopathological Features
EBV was positive in 21 cases (9%) on the EBER in situ hybridization. All tumor cells in EBER-positive cases tested positive for EBERs regardless of the histological grade (Figure 1) ▶ . Normal gastric epithelial cells surrounding the tumor did not test positive for EBERs. Table 2 ▶ summarizes the comparison of the clinicopathological features between EBV-positive and -negative GCs. Among 21 EBV-positive GC cases, 9 were LELCs. The patients with EBV-positive GCs were aged 50 years in average, which is lower than the average age (56 years) of patients with EBV-negative GCs (P = 0.043). EBV-positive GCs showed a predilection toward the body or cardia (17 of 21, 81%), in contrast to the antral dominance of EBV-negative GCs (34 of 56, 60.7%; P = 0.02). Male to female ratio did not differ in EBV-positive and -negative GCs.
Figure 1.

In situ hybridization for EBERs showing diffuse positivity in tumor cell nuclei from a moderately differentiated adenocarcinoma (left) and a poorly differentiated carcinoma with lymphoid stroma (LELC) (right).
Table 2.
Clinicopathological Features of EBV-Positive and -Negative GCs
| EBV-positive GCs (n = 21) | EBV-negative GCs (n = 56) | P value* | |
|---|---|---|---|
| Age (years) | 50.2 | 56.6 | 0.043 |
| M:F ratio | 17:4 | 41:15 | >0.05 |
| Location | |||
| Antrum | 4 (19%) | 34 (60.7%) | 0.02 |
| Body/cardia | 17 (81%) | 22 (39.3%) | |
| Histology† | |||
| WD | 0 | 3 (5.4%) | 0.019 |
| MD | 5 (23.8%) | 17 (30.4%) | |
| PD | 16 (76.2%) | 21 (37.5%) | |
| SRC | 0 | 9 (16.1%) | |
| MUC | 0 | 6 (10.7%) | |
| LELC‡ | 9 (42.9%) | 0 | <0.001 |
*Age was compared using Student’s t test; histology was compared using chi-square test; the remainders, using two-tailed Fisher’s exact test.
†Gastric carcinoma was classified, on the basis of a modification of the World Health Organization’s classification system, as well differentiated (WD), moderately differentiated (MD), poorly differentiated (PD), signet ring cell carcinoma (SRC), or mucinous carcinoma (MUC).
‡Lymphoepithelioma-like carcinoma.
Methylation of Multiple Genes in EBV-Positive GCs
Twenty-one EBV-positive GC cases and 56 EBV-negative GC cases were examined for the methylation status of the CpG islands of 14 cancer-related genes and 5 cancer-specific MINT loci. Figure 2 ▶ shows a representative example of MSP analysis. Sequencing of the representative MSP products of each CpG island exhibited conversion of all cytosines at non-CpG sites to thymine. All MSP products of each gene showed extensive methylation of CpG sites located inside the amplified genomic fragments. The results of bisulfite-genomic sequencing were consistent with those of MSP.
Figure 2.
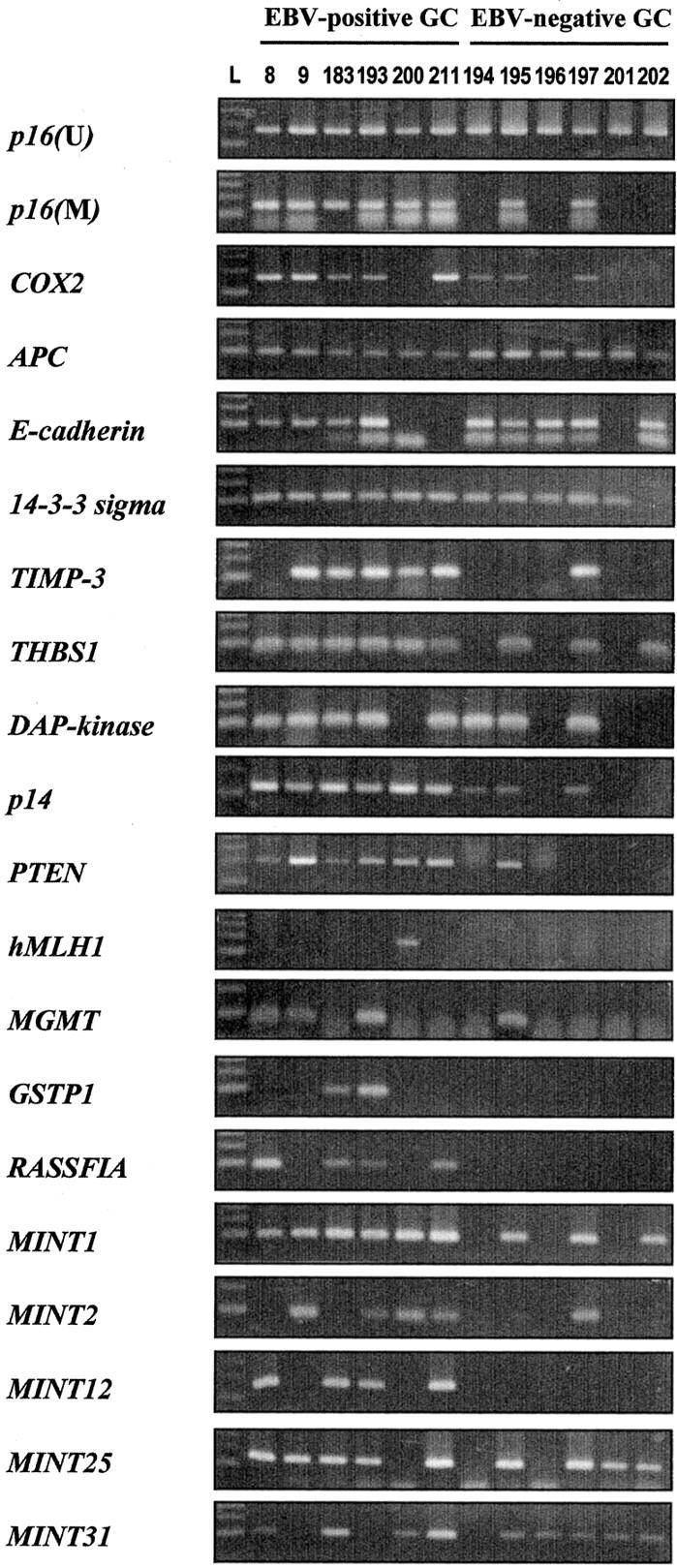
Representative samples of MSP analysis. p16 (U), unmethylated form of p16 gene; p16 (M), COX2, APC, E-cadherin, 14-3-3 sigma, TIMP-3, THBS1, DAP-kinase, p14, PTEN, hMLH1, MGMT, GSTP1, RASSF1A, MINT1, MINT2, MINT12, MINT25, and MINT31, the methylated forms of the respective CpG islands; L, 100-bp DNA ladder. Products that were amplified with primers specific to the unmethylated CpG islands of p16 after bisulfite modification were run as a control for DNA integrity.
Aberrant methylation was significantly more frequent in EBV-positive GCs than in EBV-negative GCs (Figure 3) ▶ . All of the EBV-positive GCs showed methylation in at least 10 CpG islands tested (52.6% of the tested CpG islands), whereas 62.5% of EBV-negative GCs showed methylation in <10 CpG islands. The methylation index (number of methylated genes per tested loci) averaged 0.7 (13.3 of 19) and 0.4 (7.8 of 19) in EBV-positive and EBV-negative GCs, respectively, showing a marked difference (P < 0.001). The methylation status had no relationship with the histological features of EBV-positive GCs, namely LELCs or non-LELCs.
Figure 3.
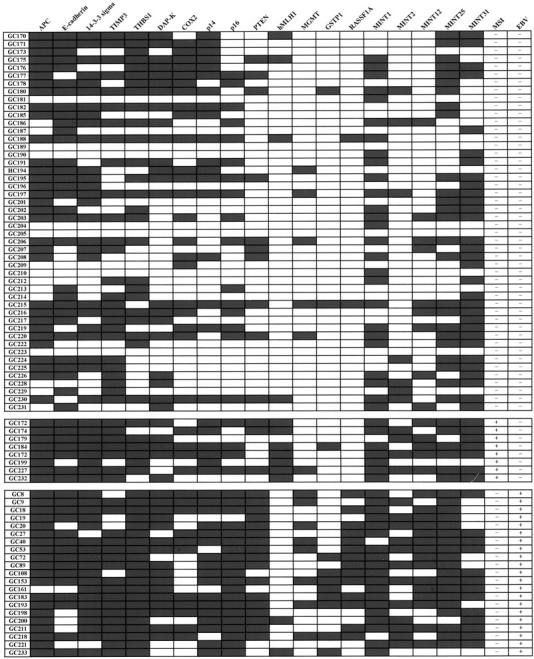
Methylation profile of 19 CpG islands and MSI status in EBV-negative GCs without MSI (top), EBV-negative GCs with MSI (middle), and EBV-positive GCs (bottom). Filled boxes, the presence of methylation; open boxes, the absence of methylation. MSI+, a tumor with MSI in >30% of the tested loci; MSI−, a tumor with MSI <30% of the tested loci. The average number of methylated CpG islands was 7.1, 12, and 13.4 in EBV-negative and MSI-negative GCs, EBV-negative and MSI-positive GCs, and EBV-positive GCs, respectively. The methylation frequency was significantly higher in the latter two groups than in the former group and the difference of the methylation frequency between the latter two groups is statistically not significant.
APC, E-cadherin, MINT31, 14-3-3 sigma, TIMP-3, MINT1, MINT25, THBS1, DAP-kinase, COX2, and p14 were methylated in >40% of GCs regardless of the EBV status (Table 3) ▶ . The methylation frequencies for the CpG islands tested except for COX2, E-cadherin, hMLH1, TIMP-3, and Mint31 were significantly higher in EBV-positive GCs than in EBV-negative GCs. In contrast, hMLH1 was methylated in the EBV-positive GCs less frequently than in EBV-negative GCs (9.5% versus 19.6%, P > 0.05). Both groups showed a similar COX2, E-cadherin, TIMP-3, and MINT31 methylation frequency. THBS1, APC, p16, 14-3-3 sigma, MINT1, and MINT25 were methylated at a frequency >90% in EBV-positive GCs. The difference of the methylation frequency in p16, PTEN, and RASSF1A between EBV-positive and -negative GCs was >50%, showing greatest in RASSF1A. GSTP1 and MGMT were methylated at frequencies <11% in EBV-negative GCs but EBV-positive GCs were methylated at more than three times the rate of the EBV-negative GCs.
Table 3.
Methylation Frequencies and Methylation Indices in EBV-Positive and -Negative GCs
| EBV-positive GC (n = 21) | EBV-negative GC (n = 56) | P value* | |
|---|---|---|---|
| THBS1 | 21 (100%) | 31 (55.4%) | <0.001 |
| APC | 20 (95.2%) | 40 (71.4%) | 0.030 |
| p16 | 20 (95.2%) | 20 (35.7%) | <0.001 |
| 14-3-3 sigma | 19 (90.5%) | 35 (62.5%) | 0.024 |
| p14 | 17 (81.0%) | 24 (42.9%) | 0.004 |
| TIMP-3 | 17 (81.0%) | 34 (60.7%) | 0.112 |
| DAP-Kinase | 16 (76.2%) | 27 (48.2%) | 0.039 |
| PTEN | 16 (76.2%) | 14 (25%) | <0.001 |
| E-cadherin | 15 (71.4%) | 38 (67.9%) | 1.000 |
| RASSF1A | 14 (66.7%) | 2 (3.6%) | <0.001 |
| COX2 | 11 (52.4%) | 26 (46.4%) | 0.799 |
| GSTP1 | 8 (38.1%) | 3 (5.4%) | 0.001 |
| MGMT | 8 (38.1%) | 6 (10.7%) | 0.016 |
| hMLH1 | 2 (9.5%) | 11 (19.6%) | 0.496 |
| MINT1 | 19 (90.5%) | 33 (58.9%) | 0.013 |
| MINT25 | 19 (90.5%) | 31 (55.4%) | 0.004 |
| MINT12 | 15 (71.4%) | 14 (25.0%) | <0.001 |
| MINT31 | 13 (61.9%) | 38 (67.9%) | 0.787 |
| MINT2 | 12 (57.1%) | 11 (19.6%) | 0.002 |
| Methylation index† (mean± SD) | 0.70 ± 0.13 | 0.41 ± 0.24 | <0.001 |
*The methylation frequency of each tested gene or locus was compared between EBV-positive and -negative GCs, using a two-tailed Fisher’s exact test. The methylation index was compared using a Student’s t test.
†The methylation index is defined as the total number of genes or loci methylated divided by the total number of genes or loci tested.
Immunohistochemical Analysis and MSI Analysis
The methylation status was correlated with the protein expression in p16 and hMLH1 using immunohistochemistry. A close correlation was noted between p16 methylation and the loss of p16 protein (P < 0.001), and between hMLH1 methylation and the loss of hMLH1 protein in tumor cells (P < 0.001) (Table 4) ▶ . The loss of hMLH1 function by hMLH1 methylation was further confirmed by MSI analysis. Seven of the 13 cases with hMLH1 methylation were MSI-positive, whereas 1 of the 64 cases without hMLH1 methylation was positive (P < 0.001). EBV-positive GCs were all MSI-negative and showed hMLH1 methylation in two cases.
Table 4.
Immunohistochemistry Results
| Methylated | Unmethylated | P value* | |
|---|---|---|---|
| p16 immunostaining | |||
| Loss | 28 (75.7%) | 9 (24.3%) | <0.001 |
| No loss | 12 (30.8%) | 27 (69.2%) | |
| hMLH1 immunostaining | |||
| Loss | 7 (87.5%) | 1 (12.5%) | <0.001 |
| No loss | 6 (8.7%) | 63 (91.3%) |
*Two-tailed Fisher’s exact test.
None of the EBV-positive GCs demonstrated expression of viral proteins, LMP1, EBNA2, and BZLF1. p53 overexpression in >30% of tumor cells was detected in 3 (14.3%) of 21 EBV-positive GCs and in 22 of 56 (39.3%) EBV-negative GCs (P = 0.055, two-sided Fisher’s exact test).
Discussion
In the present study, EBV-positive GCs showed simultaneous methylation of multiple genes involved in several molecular pathways in gastric carcinogenesis, includingcell cycle regulation (p16, p14, 14-3-3 sigma, and COX2), DNA repair and protection (hMLH1, MGMT, and GSTP1), cell adherence and metastasis (E-cadherin and TIMP-3), angiogenesis (THBS1), apoptosis (DAP-kinase), and signal transduction (APC, PTEN, and RASSF1A). The average number of methylated MINT loci was significantly greater in EBV-positive GCs than in EBV-negative GCs. The aberrant methylation of several CpG islands scattered over the genome in EBV-positive GCs indicates that EBV-positive GCs constitute CIMP. It is well known that MSI-positive GCs caused by hMLH1 methylation is one of the CIMPs. 14,15 The present study showed concordant methylation of multiple genes or loci in MSI-positive GCs, all of which were EBV-negative. MSI-positive GCs displayed as much methylation as EBV-positive GCs. In addition, 25% of EBV- and MSI-negative cases were also hypermethylated in >10 genes or loci. Thus, EBV-positive GCs are a subset of the CIMP-positive GCs.
The p16 protein is a major inhibitor of cell cycle and loss of p16 protein promotes uncontrolled cell growth. p16 is a common target of inactivation in human cancers through multiple genetic or epigenetic mechanisms, including gene mutations, homozygous deletion, and CpG island methylation. In primary GCs, CpG island methylation seems to be a predominant mechanism of p16 inactivation. 11-13 In our study, most EBV-positive GCs showed p16 methylation that was closely correlated with p16 protein loss. The relationship between EBV and p16 protein loss was reported in EBV-positive undifferentiated nasopharyngeal carcinomas 22 although these tumors might have p16 inactivation through either hypermethylation or homozygous deletion of p16. RASSF1A is one of the major transcripts of RASSF1 (ras association domain family 1) that is frequently deleted in lung and breast cancer. 23,24 Recent studies have demonstrated that RASSF1A is frequently methylated in a variety of human cancers, including lung, breast, and nasopharyngeal carcinomas. 25-27 In our study, RASSF1A was methylated in 66.7% of EBV-positive GCs whereas in 3.6% of EBV-negative GCs, showing the greatest difference of the methylation frequency for the tested genes or loci between EBV-positive and -negative GCs. RASSF1A has been demonstrated to be frequently methylated in virus-associated cancers, including nasopharyngeal carcinomas 27 and SV40 (simian virus 40)-positive malignant mesotheliomas. 18
Although the exact causes of aberrant methylation in cancer remain to be proven, factors associated with aberrant methylation include changes in the local DNA structure 28 and heavy metal exposure. 29 When foreign virus DNA becomes inserted into the host genome, host cells tend to methylate not only the integrated foreign viral DNA but also the adjacent host DNA. 17,30 The methylation change is thought to be related to local DNA structural changes caused by the insertion of large amounts of foreign DNA. Our study showed a close association between EBV and aberrant methylation, similar to the relationship between SV40 and aberrant methylation in malignant mesothelioma. 18 These findings suggest that a viral oncogenic process might involve aberrant methylation resulting in inactivation of tumor suppressor genes. However, the local DNA structural change and ensuing aberrant methylation might not completely explain the methylation change observed in EBV-positive GCs because hypermethylation in relation to EBV is not a localized process restricted to a few peculiar genes but is a generalized process affecting many CpG loci scattered over the whole genome.
Methylation of the EBV genome may enable EBV-infected tumor cells to evade immune surveillance by suppressing a family of immunodominant viral antigens. 31 The pharmacological inhibition of methylation may lead to the expression of immunodominant viral antigens. 32 and expose the tumors to immune surveillance. Furthermore, the use of hypomethylating agents may restore the tumor-suppressive functions of the tumor suppressor genes that are silenced by aberrant methylation. Thus, the use of hypomethylating agents has promise for treating EBV-associated GCs.
In conclusion, CpG island hypermethylation of several genes or loci were examined and the methylation frequency between EBV-positive and -negative GCs was compared. EBV-positive GCs demonstrated significantly more frequent methylation of most CpG islands tested than EBV-negative GCs. More than half of the tested CpG islands were methylated in all of the EBV-positive GCs. These results indicate that EBV-positive GC constitutes CpG island methylator phenotype-positive GC and suggest that aberrant methylation might be an important mechanism of EBV-related gastric carcinogenesis.
Footnotes
Address reprint requests to Gyeong Hoon Kang, M.D., Department of Pathology, Seoul National University College of Medicine, 28 Yongon-dong, Chongno-gu, Seoul, 110-744, Korea. E-mail: ghkang@snu.ac.kr.
Supported in part by the Korea Science and Engineering Foundation (grant no. 1999-2-208-004-5), in part by a grant of the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (grant no. 01-PJ1-PG3-20800-0067), and in part by year (2001) BK21 project for Medicine, Dentistry, and Pharmacy, Seoul, Korea.
References
- 1.Epstein MA, Achong BG, Barr YM: Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1:702-703 [DOI] [PubMed] [Google Scholar]
- 2.zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, Santesson L: EBV-DNA in biopsies of Burkitt tumors and anaplastic carcinoma of the nasopharynx. Nature 1970, 228:1056-1058 [DOI] [PubMed] [Google Scholar]
- 3.Hanto DW, Frizzera G, Purtilo DT, Sakamoto K, Sullivan JL, Saemundsen AK, Klein G, Simmons RL, Najarian JS: Clinical spectrum of lymphoproliferative disorders in renal transplant recipients and evidence for the role of Epstein-Barr virus. Cancer Res 1981, 41:4253-4261 [PubMed] [Google Scholar]
- 4.Shibata D, Tokunaga M, Uemura Y, Sate E, Tanaka S, Weiss LM: Association of Epstein-Barr virus with undifferentiated gastric carcinomas with intense lymphoid infiltration: lymphoepithelioma-like carcinoma. Am J Pathol 1991, 139:469-474 [PMC free article] [PubMed] [Google Scholar]
- 5.Imai S, Koizumi S, Sugiura M, Tokunaga M, Uemura Y, Yamamoto N, Tanaka S, Sato E, Osato T: Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc Natl Acad Sci USA 1994, 91:9131-9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura S, Ueki T, Yao T, Ueyama T, Tsuneyoshi M: Epstein-Barr virus in gastric carcinoma with lymphoid stroma. Special reference to its detection by the polymerase chain reaction and in situ hybridization in 99 tumors, including a morphologic analysis. Cancer 1994, 73:2239-2249 [DOI] [PubMed] [Google Scholar]
- 7.Yanai H, Takada K, Shimizu N, Mizugaki Y, Tada M, Okita K: Epstein-Barr virus infection in non-carcinomatous gastric epithelium. J Pathol 1997, 183:293-298 [DOI] [PubMed] [Google Scholar]
- 8.Chapel F, Fabiani B, Davi F, Raphael M, Tepper M, Champault G, Guettier C: Epstein-Barr virus and gastric carcinoma in Western patients: comparison of pathological parameters and p53 expression in EBV-positive and negative tumors. Histopathology 2000, 36:252-261 [DOI] [PubMed] [Google Scholar]
- 9.Schneider BG, Gulley ML, Eagan P, Bravo JC, Mera R, Geradts J: Loss of p16/CDKN2A tumor suppressor protein in gastric adenocarcinoma is associated with Epstein-Barr virus and anatomic location in the body of the stomach. Hum Pathol 2000, 31:45-50 [DOI] [PubMed] [Google Scholar]
- 10.Igaki H, Sadaki H, Tachimori Y, Kato H, Watanabe H, Kimura T, Harada Y, Sugimura T, Terada M: Mutation frequency of the p16/CDKN2 gene in primary cancers in the upper digestive tract. Cancer Res 1995, 55:3421-3423 [PubMed] [Google Scholar]
- 11.Lee YY, Kang SH, Seo JY, Jung CW, Lee KU, Choe KJ, Kim BK, Kim NK, Koeffler HP, Bang YJ: Alterations of p16INK4A and p15INK4B genes in gastric carcinomas. Cancer 1997, 80:1889-1896 [DOI] [PubMed] [Google Scholar]
- 12.Shim YH, Kang GH, Ro JY: Correlation of p16 hypermethylation with the p16 protein loss in sporadic gastric carcinomas. Lab Invest 2000, 80:689-695 [DOI] [PubMed] [Google Scholar]
- 13.Jang TJ, Kim DI, Shin YM, Chang HK, Yang CH: p16(INK4a) promoter hypermethylation of non-tumorous tissue adjacent to gastric cancer is correlated with glandular atrophy and chronic inflammation. Int J Cancer 2001, 93:629-634 [DOI] [PubMed] [Google Scholar]
- 14.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP: CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999, 96:8681-8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toyota M, Ahuja N, Suzuki H, Itoh F, Ohe-Toyota M, Imai K, Baylin SB, Issa JP: Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999, 59:5438-5442 [PubMed] [Google Scholar]
- 16.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JPJ, Hruban RH, Goggins M: Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res 2000, 60:1835-1839 [PubMed] [Google Scholar]
- 17.Resmus R, Kammer C, Heller H, Schmitz B, Schell G, Doerfler W: Insertion of foreign DNA into an established mammalian genome can alter the methylation of cellular DNA sequences. J Virol 1999, 73:1010-1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toyooka S, Pass HI, Shivapurkar N, Fukuyama Y, Maruyama R, Toyooka KO, Gilcrease M, Farinas A, Minna JD, Gazdar AF: Aberrant methylation and simian virus 40 tag sequences in malignant mesothelioma. Cancer Res 2001, 61:5727-5730 [PubMed] [Google Scholar]
- 19.Kang GH, Shim YH, Ro JY: Correlation of methylation of the hMLH1 promoter with lack of expression of hMLH1 in sporadic gastric carcinomas with replication error. Lab Invest 1999, 79:903-909 [PubMed] [Google Scholar]
- 20.Hermans JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang GH, Yoon GS, Lee HK, Kwon YM, Ro JY: Clinicopathologic characteristics of replication error-positive gastric carcinoma. Mod Pathol 1999, 12:15-20 [PubMed] [Google Scholar]
- 22.Shibosawa E, Tsutsumi K, Koizuka I, Hoshikawa M, Takakuwa T: Absence of nuclear p16 from Epstein-Barr virus-associated undifferentiated nasopharyngeal carcinomas. Laryngoscope 2000, 110:93-97 [DOI] [PubMed] [Google Scholar]
- 23.Lerman MI, Minna JD: The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res 2000, 60:6116-6133 [PubMed] [Google Scholar]
- 24.Sekido Y, Ahmadian M, Wistuba II, Latif F, Bader S, Wei M-H, Duh F-M, Gazdar AF, Lerman MI, Minna JD: Cloning of a breast cancer homozygous deletion junction narrows the region of search for a 3p21.3 tumor suppressor gene. Oncogene 1998, 16:3151-3157 [DOI] [PubMed] [Google Scholar]
- 25.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP: Epigenetic inactivation of a RAS association domain family protein from the lung tumor suppressor locus 3p21.3. Nat Genet 2000, 25:315-319 [DOI] [PubMed] [Google Scholar]
- 26.Dammann R, Yang G, Pfeifer GP: Hypermethylation of the CpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res 2001, 61:3105-3109 [PubMed] [Google Scholar]
- 27.Lo K-W, Kwong J, Hui AB-Y, Chan SY-Y, To K-F, Chan AS-C, Chow LS-N, Teo PML, Johnson PJ, Huang DP: High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 2001, 61:3877-3881 [PubMed] [Google Scholar]
- 28.Sandberg G, Schalling M: Effect of in vitro promoter methylation and CGG repeat expansion on FMR-1 expression. Nucleic Acids Res 1997, 25:2883-2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein CB, Costa M: DNA methylation, heterochromatin and epigenetic carcinogens. Mutat Res 1997, 386:163-180 [DOI] [PubMed] [Google Scholar]
- 30.Heller H, Kammer C, Wilgenbus P, Doerfler W: Chromosomal insertion of foreign (adenovirus type 12, plasmid, or bacteriophage lambda) DNA is associated with enhanced methylation of cellular DNA segments. Proc Natl Acad Sci USA 1995, 92:5515-5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robertson KD, Ambinder RF: Mapping promoter regions that are hypersensitive to methylation-mediated inhibition of transcription: application of the methylation cassette assay to the Epstein-Barr virus major latency promoter. J Virol 1997, 71:6445-6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masucci MG, Contreras-Salazar B, Ragnar E, Falk K, Minarovits J, Ernberg I, Klein G: 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt’s lymphoma line Rael. J Virol 1989, 63:3135-3141 [DOI] [PMC free article] [PubMed] [Google Scholar]