

Abstract
Oxidative endothelial stress, leukocyte transmigration, and pulmonary thrombosis are important pathological factors in acute lung injury/acute respiratory distress syndrome (ALI/ARDS). Vascular immunotargeting of the H2O2-generating enzyme glucose oxidase (GOX) to the pulmonary endothelium causes an acute oxidative lung injury in mice. 1 In the present study we compared the pulmonary thrombosis and leukocyte transmigration caused by GOX targeting to the endothelial antigens platelet-endothelial cell adhesion molecule (PECAM) and thrombomodulin (TM). Both anti-PECAM and anti-TM delivered similar amounts of 125I-GOX to the lungs and caused a dose-dependent, tissue-selective lung injury manifested within 2 to 4 hours by high lethality, vascular congestion, polymorphonuclear neutrophil (PMN) sequestration in the pulmonary vasculature, severe pulmonary edema, and tissue oxidation, yet at an equal dose, anti-TM/GOX inflicted more severe lung injury than anti-PECAM/GOX. Moreover, anti-TM/GOX-induced injury was accompanied by PMN transmigration in the alveolar space, whereas anti-PECAM/GOX-induced injury was accompanied by PMN degranulation within vascular lumen without PMN transmigration, likely because of PECAM blockage. Anti-TM/GOX caused markedly more severe pulmonary thrombosis than anti-PECAM/GOX, likely because of TM inhibition. These results indicate that blocking of specific endothelial antigens by GOX immunotargeting modulates important pathological features of the lung injury initiated by local generation of H2O2 and that this approach provides specific and robust models of diverse variants of human ALI/ARDS in mice. In particular, anti-TM/GOX causes lung injury combining oxidative, prothrombotic, and inflammatory components characteristic of the complex pathological picture seen in human ALI/ARDS.
The pathogenesis of human acute lung injury (ALI) and the more severe variant, acute respiratory distress syndrome (ARDS) represents a complex interplay of pathological factors that may develop in response to diverse pulmonary and systemic insults. 2,3 It is thought that an initial lung insult (either vascular or epithelial) is then followed by secondary processes that amplify and modify the primary injury. Alveolar transmigration of white blood cells (WBCs) (particularly, neutrophils), as well as activation of coagulation and platelets leading to pulmonary thrombosis and fibrin deposition, are among the most important secondary pathological features of ALI/ARDS. 4-6 Pulmonary thrombosis and neutrophil transmigration can also be seen (although to a rather mild extent) in some animal models of ALI/ARDS (eg, endotoxemia, immune complex injury, cecal puncture, ischemia/reperfusion, hemorrhage/resuscitation, and skin burns). 7-10
The mechanisms of pulmonary thrombosis and WBC transmigration involve the generation of procoagulant and chemotactic factors 11,12 that induce a shift from an anti-inflammatory, anti-thrombotic endothelial surface to a proinflammatory, prothrombotic milieu. These changes occur because of alterations in endothelial entities such as the thrombomodulin-protein C system 13,14 and surface adhesion molecules. The specific molecular and cellular mechanisms responsible for pulmonary thrombosis and WBC transmigration in particular clinical settings remain to be better understood.
Because many studies have implicated oxidative endothelial injury in the initiation or/and propagation of ALI/ARDS, 15-17 we hypothesized that we could use the paradigm of vascular immunotargeting to develop a model in which a controlled and specific oxidative stress could be used to initiate ALI. We and others have established that immunoconjugates directed against endothelial cell antigens such as angiotensin-converting enzyme (ACE), platelet-endothelial cell adhesion molecule (PECAM-1), ICAM-1, and thrombomodulin (TM), preferentially accumulated in the lungs in intact animals because the pulmonary vasculature appears to be a primary target after intravenous injection. 18-21 We therefore conjugated glucose oxidase (GOX, an enzyme generating H2O2 from glucose) with monoclonal antibodies directed against the endothelial antigens and documented that GOX conjugates bound to endothelial cells, entered the cells, and caused oxidative stress in cell culture. 22-24 Moreover, we found that vascular immunotargeting of GOX to the pulmonary endothelium could be used to generate models of specific oxidative vascular lung injury in mice. Thus, we documented that anti-PECAM/GOX, but not control IgG/GOX conjugates, induced acute injury in the lungs, but not in other organs, after intravenous injection in mice. 1 In our initial study we found that anti-PECAM/GOX induced significant lung injury in mice characterized by evidence of oxidative stress and increase in pulmonary permeability. 1 However, this model did not induce WBC transmigration into the alveolar space or result in substantial pulmonary thrombosis, as is evident in most forms of severe lung injury in humans.
Because PECAM-1 is involved in WBC transmigration, 25 we reasoned that its blockage by anti-PECAM/GOX might compromise the process. This consideration led to a hypothesis that the effects of a GOX conjugate(s) might depend on the properties of the particular endothelial antigen used as the anchor for immunotargeting. Thus, we postulated that both oxidative stress induced by H2O2 generation and inhibition of a specific endothelial protein caused by a GOX conjugate in the pulmonary vasculature may dictate the pathological features of the lung injury induced by GOX immunotargeting. The goal of present work was to test this hypothesis, and in doing so, establish a robust and specific murine model of human ALI/ARDS featuring pulmonary thrombosis and alveolar PMN transmigration.
To accomplish this goal, we compared the targeting and effects of GOX conjugated with monoclonal antibodies directed against two distinct endothelial antigens, PECAM-1 and TM. Similar to PECAM-1, TM is a transmembrane endothelial glycoprotein expressed at high levels on the surface of the pulmonary endothelium. 26 TM suppresses intravascular thrombosis by converting thrombin from a procoagulant into an anti-coagulant enzyme. 13 We therefore postulated that TM inhibition by the conjugate would not inhibit leukocyte transmigration and would predispose pulmonary vasculature toward thrombosis. The results presented in this article support this hypothesis and show that GOX immunotargeting to PECAM and TM provided robust, yet clearly different variants of pulmonary oxidative vascular injury in mice. Importantly, anti-TM/GOX, but not anti-PECAM/GOX, induced an ALI that was accompanied by neutrophil transmigration into the alveolar compartment and intravascular thrombosis. Therefore, immunotargeting of anti-TM/GOX conjugates offers a model of murine lung injury with remarkable similarities to the acute phase of human ALI/ARDS.
Materials and Methods
Reagents
The following materials were used in the study: fatty acid-free bovine serum albumin from Boehringer-Mannheim-Roche (Indianapolis, IN), dimethyl formamide, rat IgG, biotinylated glucose oxidase (b-GOX), components of buffer solutions from Sigma (St. Louis, MO), O-phthalaldehyde and Z-Phe-His-Leu from Serva (Heidelberg, Germany), streptavidin and 6-biotinylaminocaproic acid N-hydroxysuccinimide ester (BxNHS) from Calbiochem (San Diego, CA). Protein concentration was determined by BioRad microassay kit (Hercules, CA). Monoclonal antibody (mAb) 390 is produced in rat and reacting with murine PECAM-1, whereas mAb 34 is produced in rat against murine TM. Both anti-TM and anti-PECAM monoclonal antibodies were characterized previously. 18,20 A mAb raised in rats against human creatine kinase was used as control IgG (ATCC hybridoma, CKMM 14.15; ATCC, Manassas, VA).
Conjugation of GOX to Anti-TM and Anti-PECAM
Immunoglobulins were modified by a biotinylating reagent, BxNHS, to produce biotinylated derivatives (b-IgG, b-anti-PECAM, or b-anti-TM) as described previously. 19 Biotinylated GOX was labeled with 125I using Iodogen-coated tubes. To construct the tri-molecular heteropolymer complex b-anti-TM/SA/b-GOX or IgG/SA/b-GOX, as well as conjugates with other biotinylated enzymes (eg, b-catalase) we used a two-step procedure established in our laboratory. 1,20 Briefly, SA and b-GOX were mixed at a molar ratio SA:b-GOX = 5 and incubated for 1 hour on ice. This ratio is optimal for conjugation of SA/b-GOX complex with biotinylated immunoglobulins. The complex was then incubated with b-anti-TM, b-anti-PECAM, or b-IgG, to form b-anti-TM/SA/b-GOX b-anti-PECAM/SA/b-GOX conjugates, or their nonimmune counterpart, b-IgG/SA/b-GOX. These conjugates are indicated as anti-TM/GOX, anti-PECAM/GOX, and IgG/GOX in the text. Enzymatic activity of GOX conjugated with either the immune or the nonimmune carrier did not differ from that of the initial preparation of biotinylated GOX (∼100 U/mg).
Evaluation of Conjugate Size
Experiments with anti-PECAM/GOX that provided the data for our previous publication were performed with the conjugates of unknown size. 1 However, pilot experiments showed that intravenous injection of large (a mean diameter of 5 to 10 microns) 125I-GOX conjugates, including IgG/125I-GOX, provided an immediate uptake of 35 to 45% of injected 125I-GOX per gram of lung tissue, most likely because of embolization in the pulmonary capillaries, leading to a profound lung injury. To avoid this nonspecific effect, experiments described in the article have been performed with GOX conjugates with a mean diameter ranging from 120 to 250 nm. By varying the molar ratios between biotinylated GOX, SA, and biotinylated antibodies, we produced a series of the conjugates ranging from 100 to 10,000 nm diameter, determined by dynamic light scattering, as described elsewhere. 21 Briefly, conjugates were diluted in filtered, sterile water and analyzed by dynamic light scattering using either an ALV-500/E Multiple Tau-Digital correlator and Goniometer (ALV, Langen, Germany) or BI-90 Plus, particle size analyzer with BI-9000AT digital autocorrelator (Brookhaven Instruments Corp., Brookhaven, NY). Measurements were taken at 90o. Effective diameter was calculated as the mean of multiple determinations ± SD. The average particle size was calculated by means of the Stokes-Einstein equation from the diffusion coefficient obtained from a second order cumulative fit to the data. Our recent data show that 100- to 300-nm diameter is optimal for the specific, antigen-mediated targeting and internalization of the conjugates by endothelium, but precludes a nonspecific, mechanical uptake in the capillaries. 21
Evaluation of the Pulmonary Targeting of the Conjugates in Intact Mice
Normal BALB/c mice (Charles River, NJ) were injected with 1 to 5 μg of anti-TM/125I-GOX, anti-PECAM/125I-GOX, or IgG/125I-GOX in 100 μl of saline via tail vein. One hour later, animals were sacrificed; the internal organs were dissected, washed with saline, blotted dry, and weighed. Tissue radioactivity in blood and organs was determined in a γ-counter (Wallac-LKB). The results of 125I measurements in the organs were used to calculate the percent of injected dose per gram of tissue (%ID/g). The results of biodistribution studies of the radiolabeled anti-PECAM/GOX, anti-TM/GOX, and IgG/GOX revealed that all three conjugates display similar levels of the uptake in liver (30 to 40% ID/g), spleen (35 to 45% ID/g), heart (1 to 2%ID/g), and kidney (1.5 to 2.5% ID/g). This biodistribution pattern was similar to the previously reported absolute values of uptake in these organs of 125I-GOX and other radiolabeled enzymes (eg, 125I-β-GAL) conjugated with monoclonal antibodies directed against endothelial antigens PECAM-1, ACE, ICAM, TM, as well as control IgG. 1,19-21,23 Most likely, distribution of the immunoconjugates in the organs other than lungs reflects antigen-independent clearance such as the uptake by Fc-receptor-bearing phagocytes in the reticuloendothelial system and renal filtration. The only organ where the uptake of anti-PECAM/GOX and anti-TM/GOX was different from that of IgG/GOX was the lung. Accordingly, we focused on this organ and presented this data in the Results section.
Injection of GOX Conjugates and Characterization of the Lung Injury
To address pathological effect(s) caused by GOX/Ab in mice, we injected 15 to 100 μg of the immune or nonimmune conjugates in anesthetized BALB/c mice intravenously via tail vein in injection volumes not exceeding 150 μl. Animals were sacrificed by cervical dislocation at time points ranging from 1 to 24 hours after injection. 1 A global lung injury score (see details below) was assigned to each mouse by inspecting the lungs en bloc. The lungs were then harvested and allocated for histopathology; electron microscopy, measurements of myeloperoxidase (MPO) activity in lung tissue, wet-to-dry weight ratio, and for bronchoalveolar lavage (BAL) to determine BAL fluid protein concentrations and differential cell counts.
Lung injury was evaluated using an independent assessment of gross Acute Lung Injury Score (ALIS). ALIS was based on the macroscopic appearance of the redness (hemorrhage) and wetness (edema) of the lungs, ranging from 1 (normal) to 10 (severe hemorrhage and congestion). Representative examples are shown in Figure 4A ▶ . Lung injury was grouped by ALIS as follows: scores from 2 to 5, inclusively, were considered to represent relatively minor injury; scores of 9 or 10, reflecting very swollen and grossly bloody lungs, were categorized as severe injury; and intermediate scores between 6 and 8, defined by boggy lungs with modest amounts of pulmonary hemorrhage, were labeled as moderate injury.
Figure 4.
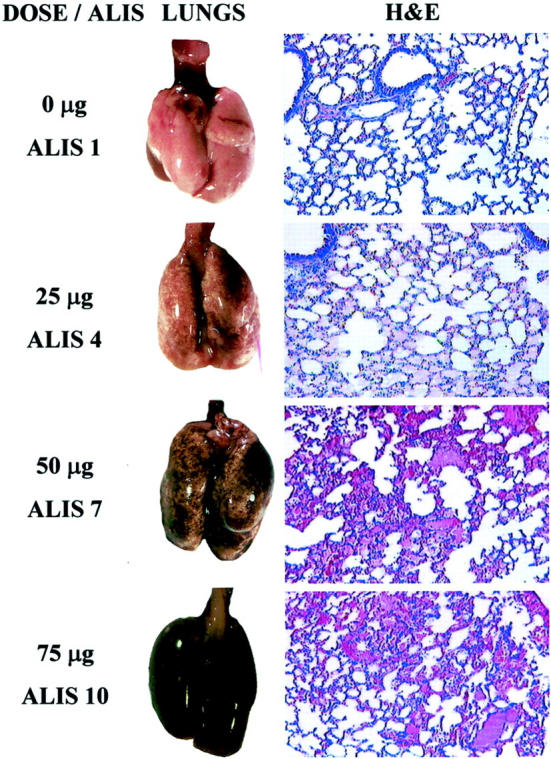
Dose-dependent lung injury after injection of anti-TM/GOX. Left column: The dose of anti-TM/GOX injected (0, 25, 50, and 75 μg) and a typical ALIS observed at this dose. Middle: The panels show typical gross morphological view of the lungs at postmortem. Right: The panels represent representative histopathological sections obtained from animals injected with the corresponding dose of anti-TM/GOX, stained with H&E (original magnification, ×100). Lungs from saline-injected control mice appeared evenly pink and without cellular infiltrates or vascular congestion on microscopic examination (ALIS 1, no detectable injury). Injection of 25 μg of anti-TM/GOX produced faintly red lungs (ALIS 4) with microscopic vascular congestion and mild edema. Injection of 50 μg of anti-TM/GOX resulted in frankly red and boggy lungs (ALIS 7) and microscopic signs of severe vascular congestion, proteinaceous fluid in the alveoli, moderate to severe alveolar hemorrhage, and inflammatory infiltration. Injection of 75 μg of anti-TM/GOX induced severe injury manifested by almost black hemorrhaged lung surface, tensely swollen lungs (ALIS 10), characterized microscopically by florid pulmonary edema, diffuse alveolar hemorrhage, and an intense acute inflammation.
For histopathological studies, the lungs were instilled before removal from the animal with 0.75 ml of buffered formalin through a 20-gauge angiocatheter placed in the trachea, immersed in buffered formalin overnight, and then processed for conventional paraffin histology. Sections were stained with hematoxylin and eosin (H&E), and examined by light microscopy. Internal organs were inspected, photographed, and processed for cryosectioning, routine histological processing, or electron microscopy.
Immunohistochemical Evaluation of Mouse Tissues
Immunohistochemistry analysis was done on 6-μm-thick frozen sections from OCT-embedded tissues or from 4-μm paraffin sections. The following antibodies were used: 1) rat anti-mouse PECAM-1, clone mAb 390; 2) rat anti-mouse TM, clone mAb 34; 18 3) a rabbit polyclonal antibody directed against iPF2α-III, an isoprostane (formerly known as 8-epi or 8-isoPGF2α; 27 4) rat anti-mouse CD41 directed against murine platelets (recognizes integrin αIIb chain); and 5) a rabbit polyclonal antibody to nitrotyrosine, NY, an indicator of protein nitration. 16,17 Visualization was achieved by the use of Vectastain kit or by Alkaline Phosphatase kit (Vector Laboratories, Burlingame, CA) using manufacturers protocols. When needed, Neutral Red was used as a counterstain (Sigma, St. Louis MO).
Evaluation of Lung MPO Content
MPO was assayed in lung tissue to assess the pulmonary accumulation of neutrophils. Mice were perfused through their left ventricle with 10 ml of phosphate-buffered saline (PBS), allowing the perfusate to exit from severed carotid arteries. Irrigation was continued until blood was no longer visible in the draining fluid. The lungs were then quickly removed and placed in K2HPO4 buffer at pH 7 on ice. The tissues were homogenized for 30 seconds in 1 ml of this buffer and centrifuged at 4000 rpm for 30 minutes at 4°C. The pellet was suspended in 1 ml of the same buffer and sonicated for 90 seconds on ice. The slurry was centrifuged at 2000 rpm for 10 minutes. A 0.1-ml aliquot of the supernatant, which contained any MPO present in lung parenchyma, was added to 0.3 ml of Hanks’ buffered saline solution (Life Technologies, Inc., Grant Island, NY) containing 25% bovine serum albumin (Sigma), 0.05 ml of 1.25 mg/ml O-dianisidine (ICN Pharmaceuticals, Costa Mesa, CA), and 0.05 ml of 0.05% H2O2. The reaction was stopped with 0.05 ml of 1% sodium azide (Sigma) after 15 minutes. The absorbance of the reaction product was assessed spectrophotometrically at 460 nm. Because the entire lung was homogenized and the same volume used for each assay, MPO units were expressed as the change in absorbance per lung; gram of tissue was not used as the reference denominator because GOX-induced pulmonary edema and hemorrhage confounded estimation of the premorbid lung weight.
Evaluation of BAL Fluid and Cell Differentials
BAL was performed by exposing and cannulating the trachea with a 20-gauge angiocatheter (Becton Dickinson, Sandy, UT), and then lavaging three times with 0.5 ml of PBS containing a protease inhibitor cocktail (Sigma) at 10 μl per ml. Recovery of infused fluid was >90%. The lavage fluid was spun at 2000 rpm for 3 to 4 minutes; the supernatant was collected, aliquoted, and frozen at −70°C, after which the cell pellet was suspended in PBS containing 5% serum. The cells were spun on a Shandon Cytospin-3 cell preparation system at 1500 rpm for 10 minutes and stained with a standard Diff-Quick Hemacolor kit (EM Diagnostic Systems, Gibbstown, NJ). Protein concentrations were later measured in the thawed supernatant of the BAL fluid using a standard BCA assay (Pierce Chemicals, Rockford, IL).
Electron Microscopical Processing of the Lung Tissue
Electron microscopy was performed on epoxy resin-embedded tissues as described previously. 1 Briefly, the trachea was exposed and cannulated with a 20-gauge angiocatheter. The lungs were instilled with cold 0.75-ml Karnovsky’s fixative (2% paraformaldehyde and 2.5% glutaraldehyde; Electron Microscopy Sciences, PA) in 0.1 mol/L cacodylate buffer. They were immediately removed from the animal intact; the trachea was tied with suture and the entire tissue block immersed in the same fixative for 30 minutes on ice. The lungs were then cut with a blade in 1-mm 3 pieces, placed in fresh fixative, and left under vacuum (20 mmHg) overnight. After rinsing in cacodylate buffer, followed by rinsing in 0.05 mol/L maleate buffer, pH 5.2, the tissue was postfixed with 1% osmium tetroxide in 0.7 mol/L potassium ferrocyanide for 1 hour on ice. En bloc staining was done with uranyl acetate for 20 minutes followed by dehydration in a graded series of alcohols and embedded in Poly/bed 812 (Polysciences Inc.). Ultrathin sections (gold) were postcontrasted with 20% aqueous uranyl acetate (Amersham) for 3 minutes followed by aqueous 0.5%lead citrate for 3 minutes and viewed on a Hitachi H-600 transmission electron microscope (Nissey Sangyo) at 75 kV.
Statistical Analysis
Statistical differences among groups was determined using one-way analysis of variance. When statistically significant differences were found (P < 0.05) individual comparisons were made using the Bonferroni/Dunn test (Statview 4.0).
Results
Binding of Rat Monoclonal Antibodies Directed Against Murine Isoforms of TM and PECAM in the Mouse Lungs
To characterize the binding of anti-TM and anti-PECAM mAbs in lungs in situ, antibodies and control rat IgG were applied to frozen lung sections followed by labeled secondary anti-rat antibodies. Both anti-TM (Figure 1A) ▶ and anti-PECAM (Figure 1B) ▶ provided a similar pattern of strongly positive staining in the lung capillaries and larger vessels. Control rat IgG, used as a nonimmune counterpart in the study, provided no detectable staining in murine lungs (Figure 1C) ▶ .
Figure 1.

Binding of TM and PECAM antibodies in the murine lungs. Top: In situ immunostaining of lung frozen sections (A–C). Bottom: Detection of intravenously injected anti-TM and anti-PECAM mAb (D and E). Binding of anti-TM and anti-PECAM was revealed by secondary peroxidase-conjugated antibodies. Note that anti-PECAM and anti-TM binds to antigen both in situ and in vivo. Antigens are detected on pulmonary capillaries (see insets in bottom panels), and larger pulmonary vessels (V). Original magnifications: ×200 (A–F); ×500 (d, e).
To characterize binding of antibodies to the blood-accessible endothelial binding sites in vivo, a mAb or control rat IgG were injected intravenously. One hour later, lungs were harvested, embedded in OCT, sectioned, and exposed only to a labeled secondary anti-rat antibody (Figure 1, D to F) ▶ . Positive lumenal staining was seen in the pulmonary vessels in mice injected with either anti-TM (Figure 1D) ▶ or anti-PECAM (Figure 1E) ▶ . Both antibodies provided a similar immunostaining pattern, almost identical to that seen in situ. No staining with control IgG was observed (Figure 1F) ▶ . Therefore, anti-TM mAb 34 and anti-PECAM mAb 390 used in the present study represent good candidate carriers, which could provide a comparable immunotargeting of GOX to the pulmonary endothelium.
Pulmonary Targeting of Anti-TM/125I-GOX and Anti-PECAM/125I-GOX in Mice
To characterize the in vivo targeting, tissue levels of 125I-labeled conjugates were analyzed 1 hour after intravenous injection of a trace amount (1 to 5 μg/mouse) of anti-TM/125I-GOX, anti-PECAM/125I-GOX, or IgG/125I-GOX in intact mice. The conjugates had blood levels close to 3% ID/g and had similar levels in plasma, almost doubling that seen in blood, implying that they did not bind to blood cells at a significant level. Figure 2 ▶ shows that both anti-PECAM/GOX and anti-TM/GOX, but not IgG/GOX, accumulated in the lungs. The pulmonary uptake of anti-TM/125I-GOX and anti-PECAM/125I-GOX achieved 30% ID/g and was 10 times higher than that of IgG/125I-GOX. The lung-to-blood ratio of tissue radioactivity was close to 10 after injection of either anti-TM/125I-GOX or anti-PECAM/125I-GOX, thus indicating high selectivity of pulmonary targeting, similar to that observed with anti-angiotensin-converting enzyme and other affinity carriers. 28,29 Thus, anti-TM and anti-PECAM provide preferential pulmonary uptake of a conjugated cargo and provide comparable delivery of GOX to the pulmonary vasculature.
Figure 2.

Pulmonary targeting of GOX to TM and PECAM-1. The level of 125I in blood and lung 1 hour after intravenous injection of IgG/125I-GOX (filled bars), anti-TM/125I-GOX (open bars), and anti-PECAM/125I-GOX (hatched bars) in intact mice. The data are shown as mean ± SEM (n = 4). The pulmonary uptake of anti-TM/125I-GOX and anti-PECAM/125I-GOX was significantly higher than that of IgG/125I-GOX (P < 0.01).
ALI Caused by Anti-TM/GOX and Anti-PECAM/GOX in Intact Mice
To study in vivo effects of GOX immunotargeting to endothelium, we injected mice with the conjugates at doses ranging from 15 to 100 μg of GOX per mouse. To account for potential systemic and side effects of binding the conjugates to PECAM and TM, we injected animals with nonconjugated anti-PECAM or anti-TM, as well as the matching doses of control conjugates such as anti-PECAM/catalase and anti-TM/catalase. No mortality, tissue injury, or thrombosis was detected after injection of nonconjugated antibodies or control conjugates (not shown). In good agreement with our previous study, 1 injection of up to 100 μg of IgG/GOX (100- to 250-nm diameter) did not cause lung injury or animal mortality (not shown).
However, in sharp contrast with all other preparations, anti-TM/GOX and anti-PECAM/GOX injection caused dose-dependent lethality after intravenous injection in mice (Figure 3) ▶ . Thus, injection of 75 to 100 μg/mouse of either conjugate caused 95 to 100% mortality within 4 to 6 hours. Therefore, GOX targeting to either PECAM or TM induced a severe injury manifested within a few hours by high lethality in mice.
Figure 3.

Survival of mice injected with anti-TM/GOX (A) or anti-PECAM/GOX (B). Mice were injected with indicated doses of conjugate and the survival was determined during the 24 hours after injection. The figure presents cumulative results of several series of experiments. Number of animals for each conjugate dose is indicated.
Postmortem examination, and histological and electron microscopy studies revealed that neither anti-TM/GOX nor anti-PECAM/GOX caused overt injury in the organs except the lungs (not shown). In general, the effect of anti-TM/GOX in the organs was similar to that of anti-PECAM/GOX described in detail in the previous article. 1 Mild to modest renal injury manifested by edema in Bowman’s space in glomeruli was detected after injection of a high dose (75 to 100 μg/mouse) of any GOX conjugate. No detectable cardiac, hepatic, and splenic injury was noted on the postmortem histological examination after injection of any conjugate.
The lung was the major site of damage after anti-TM/GOX and anti-PECAM/GOX, whereas IgG/GOX did not cause lung injury (not shown). The overt dose-dependent pulmonary injury was seen 1 to 3 hours after injection of 30 to 70 μg of anti-TM/GOX and anti-PECAM/GOX (Figure 3) ▶ . Figure 4 ▶ shows results of a typical postmortem examination of murine lungs obtained after anti-TM/GOX conjugate injection. Injection of 25 μg induced patchy vascular congestion, slight edema, and modest leukocyte infiltration. Injection of 50 to 75 μg caused severe vascular congestion, diffuse hemorrhage, florid pulmonary edema and fluid exudation, accumulation of blood cells in the lungs, and a massive inflammatory infiltration (Figure 4 ▶ , right column). On gross examination, the lung injury was manifested by diffuse hemorrhage that, at high doses, resulted in a lung surface that appeared dark-brown (Figure 4 ▶ , lower panels in the middle column). To compare the amplitude of lung injury, we used a 10-point ALIS. The scoring was based on the postmortem gross examination of the lungs by at least two independent examiners. Scores ranging from 2 to 5, from 6 to 8, and from 9 to 10 were assigned to indicate a mild, moderate, and severe lung injury, respectively.
Figure 5 ▶ compares the lethality and the amplitude of lung injury (ALIS) caused by anti-TM/GOX and anti-PECAM/GOX. At high doses (75 μg and higher), the conjugates were equally potent. However, in the dose range from 20 to 70 μg anti-TM/GOX was more potent than anti-PECAM/GOX, both in terms of lethality (Figure 5A) ▶ and lung injury (Figure 5B) ▶ .
Figure 5.

Comparison of the injuring potency of anti-TM/GOX and anti-PECAM/GOX. The lethality (A) and severity of lung injury (ALIS, B) were determined after intravenous injection of indicated dose of anti-TM/GOX (circles) or anti-PECAM/GOX (squares) in intact mice. The ALIS data are shown as mean ± SEM for each group, with number of animals per group varying from 6 to 58. Note that in the intermediate doses range (20 to 70 μg of GOX/mouse), anti-TM/GOX causes higher lethality and inflicts more severe lung injury than anti-PECAM/GOX.
Characterization of Oxidative Vascular Injury in the Lungs Caused by GOX Targeting to TM or PECAM
In the next set of experiments, we compared lung injury induced in mice by an equally potent dose of the conjugates (ie, 25 to 35 μg of anti-TM/GOX versus 65 to 75 μg of anti-PECAM/GOX). Figure 6 ▶ shows that injection of either conjugate caused a marked elevation of pulmonary vascular permeability within 2 to 3 hours after injection. Thus, lung wet-to-dry ratio, an accepted parameter of lung edema, was significantly higher after injection of either conjugate than saline control (Figure 6A) ▶ . Both conjugates induced a 30- to 35-fold increase in protein level in the BAL, indicating a severe acute pulmonary edema (Figure 6B) ▶ .
Figure 6.
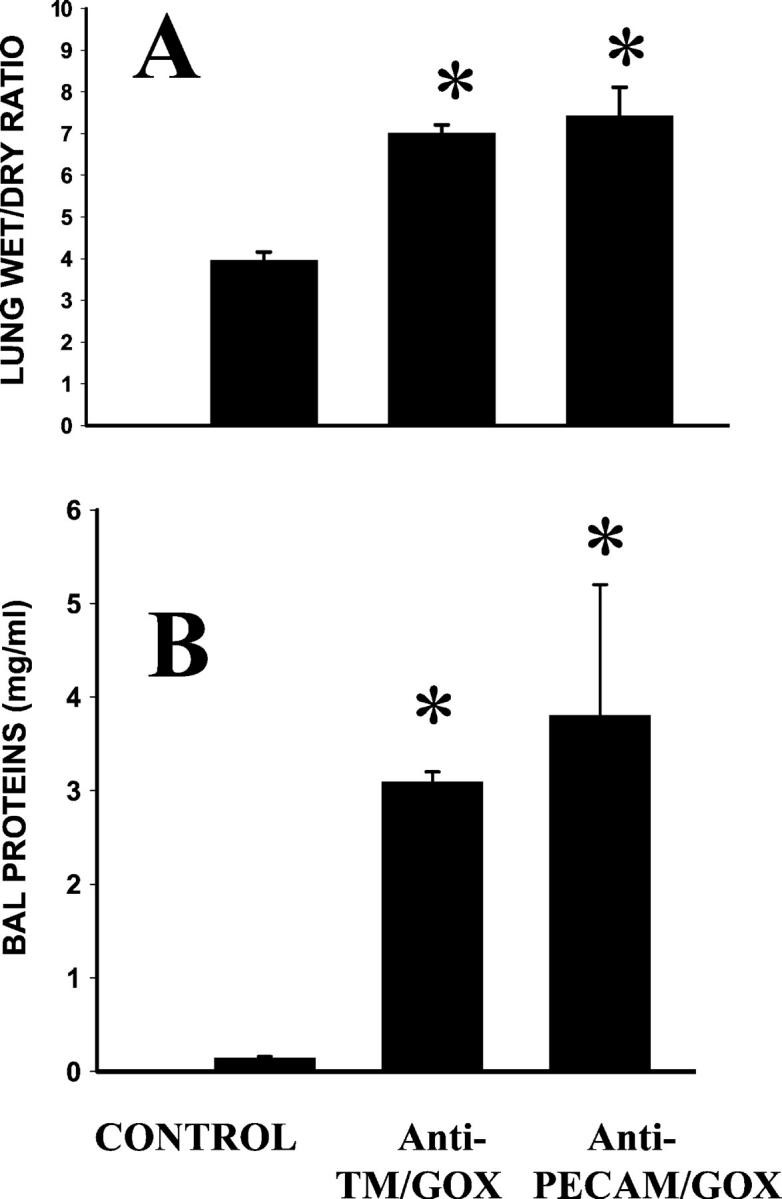
Anti-TM/GOX and anti-PECAM/GOX induce pulmonary edema in mice. The lung wet-to-dry ratio (A) and protein level in the BAL fluid (B) have been determined 4 hours after intravenous injection of saline (control) or equally potent doses of the conjugates [35 μg of anti-TM/GOX (n = 24) or 70 μg of anti-PECAM/GOX (n = 9)] in intact mice. The data are shown as mean ± SEM, P < 0.001 versus control (asterisk).
To identify products of tissue oxidation caused by H2O2 generated by the targeted GOX, we stained lungs harvested 4 hours after injection of either anti-TM/GOX or anti-PECAM/GOX with antibodies directed against iPF2α-III, (an F2 isoprostane), and nitrotyrosine. Isoprostanes are chemically stable, free radical-catalyzed products of arachidonic acid oxidation and thus the level of iPF2α-III reflects lipid peroxidation in the tissues. 27 Nitrotyrosine is a product of oxidative protein nitration that has been found in animal and human tissues in diverse pathological conditions associated with oxidative stress. 16,17 Figure 7 ▶ shows that injection of the conjugates caused positive immunostaining (visualized as a dark blue color) of the lung tissue by antibodies to iPF2α-III (left panels) and nitrotyrosine (middle panels). Therefore, both anti-TM/GOX and anti-PECAM/GOX caused oxidative stress in the lungs detectable by accumulation of the products of oxidative modification of lipids and proteins.
Figure 7.

Detection of products of tissue oxidation in the lungs. Mice were injected with saline (control, top) or injuring doses of anti-PECAM/GOX (αPECAM, middle) or anti-TM/GOX (αTM, bottom). The lung tissue sections were stained with antibody directed against 8-epi iPF2a-III F2 isoprostane, a marker of lipid peroxidation (left column, 8-epi), antibody to nitrotyrosine, a marker of protein oxidative nitration (middle column, Nitro-Y), or control rabbit IgG (control, right column). The positive immunostaining was revealed by secondary antibody conjugated with alkaline phosphatase (blue color), Neutral Red counterstaining was used to mark individual cells. Original magnification, ×200.
The results presented in this section indicate that both anti-TM/GOX and anti-PECAM/GOX cause severe vascular oxidative injury in the pulmonary vasculature, manifested by overt edema and accumulation of products of lipid and protein oxidation in the lungs.
Pulmonary Neutrophil Dynamics after Injection of Anti-TM/GOX or Anti-PECAM/GOX
H&E staining of lung tissue sections revealed that the conjugates caused a marked pulmonary sequestration of WBCs in mice (Figure 8A) ▶ . Increased numbers of leukocytes, especially polymorphonuclear neutrophils (PMN), were noted in the lung tissue 2 to 3 hours after injection of either anti-TM/GOX (Figure 8b) ▶ or anti-PECAM/GOX (Figure 8c) ▶ , but not saline (Figure 8a) ▶ . To obtain a more quantitative and specific measure of pulmonary sequestration of PMN, we determined levels of MPO in the lung homogenates and found that at equally potent doses, both conjugates induced pulmonary sequestration of PMN of a similar magnitude (Figure 8B) ▶ .
Figure 8.

Anti-TM/GOX and anti-PECAM/GOX cause pulmonary leukocyte sequestration. A: H&E sections from paraffin-embedded lung tissues harvested 4 hours after injection of saline control (a), or equally injuring doses of anti-TM/GOX (b) or anti-PECAM/GOX (c). B: Analysis of MPO in the lung tissue homogenates after injection of a marginally effective dose of either conjugate (left pair, low dose: 15 mg of anti-TM/GOX and 30 mg of anti-PECAM/GOX) or highly injuring dose of either conjugate (right pair, high dose: 30 mg of anti-TM/GOX and 75 mg of anti-PECAM/GOX). The data in B are shown as mean ± SEM, dotted line represents baseline value.
However, analysis of BAL fluids obtained from mice injected with equally potent doses of the conjugates revealed that anti-TM/GOX and anti-PECAM/GOX caused strikingly different effects in terms of leukocyte transmigration into the alveolar space. Thus, anti-TM/GOX caused a rapid influx of leukocytes in the alveolar space that peaked within 1 to 2 hours after injection (Figure 9A) ▶ . After an initial peak, leukocyte levels in the BAL declined, but remained markedly elevated during 24 hours after injection of anti-TM/GOX (the entire time interval tested in the study). By sharp contrast, no significant WBC influx was detected in the BAL at any time during the 24 hours after injection of anti-PECAM/GOX (Figure 9) ▶ . Differential analysis of BAL cytospins revealed that neutrophils comprised 20% of leukocytes migrated into the airways and alveolar compartments after injection of anti-TM/GOX, whereas no significant elevation of PMN over the basal level could be detected after anti-PECAM/GOX injection (Figure 9, B and C) ▶ .
Figure 9.

Leukocyte dynamics in the lung differ after injection of anti-TM/GOX and anti-PECAM/GOX. Total WBC counts in the BAL fluid (A) reveals that only anti-TM/GOX (squares), but not anti-PECAM/GOX (circles) cause WBC influx into the alveoli. B: Typical BAL cytospin preparations at an original magnification of ×500 after injection of saline control (a), anti-PECAM/GOX (b), or anti-TM/GOX (c). C shows the number of PMN neutrophils in BAL obtained 4 hours after injection of the indicated conjugates. The data are shown as means ± SEM.
Inspection of tissue sections at a high magnification revealed that the total number of PMN in the lung tissue was elevated approximately fourfold after injection of equally injuring doses of either anti-TM/GOX or anti-PECAM/GOX (40 to 50 and 75 to 80 μg of GOX, respectively) versus control saline-injected mice (Figure 10A) ▶ . This approach also permitted us to determine the number of PMNs in the pulmonary intravascular and alveolar compartments. Figure 10B ▶ shows that according to this method of analysis, more than 85% of PMNs were localized in the vascular space after anti-PECAM/GOX injection, versus more than 50% of PMN localized in the alveolar compartment after anti-TM/GOX injection (Figure 10B) ▶ . In control mice, ∼75% of PMN were found in the intravascular compartment. Figure 10C ▶ shows that the ratio of alveolar-to-vascular PMNs (normalized to the corresponding control value) was close to 0.6 after anti-PECAM/GOX injection versus 5 after anti-TM/GOX injection. This result indicates that anti-TM/GOX causes ∼10-fold higher alveolar PMN transmigration than anti-PECAM/GOX.
Figure 10.
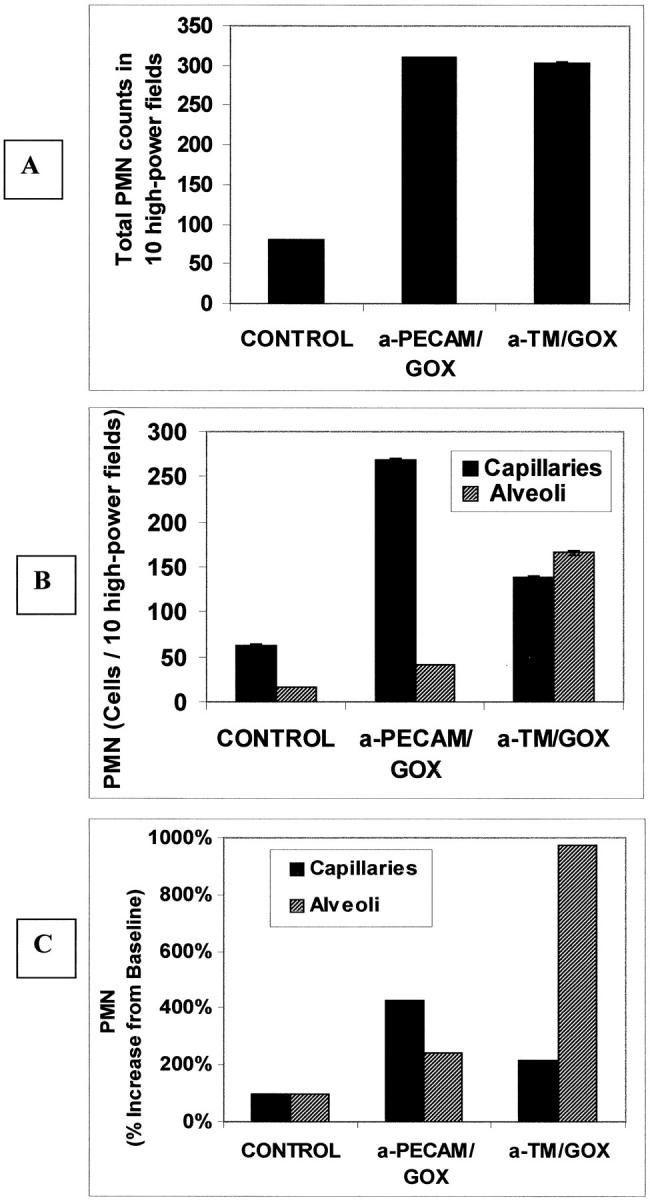
Quantitative evaluation of neutrophil compartmental localization in control and GOX/Ab-treated lungs. PMN were evaluated in 10 high-power light-microscopic fields (×100 oil objective) from H&E lung sections. A: Total number of neutrophils in whole-lung sections from untreated baseline (control) animals and maximally injured, GOX-immunoconjugate treated (ALIS = 10) mice. B: PMN in capillaries (black) or alveoli (checkered) in control or GOX-treated mice. C: Percent increase of neutrophil numbers in the two compartments after targeted GOX treatment over baseline, untreated mice (means ± SEM).
Ultrastructural examination of the lung tissue in a transmission electron microscope revealed that lungs harvested from mice injected with anti-TM/GOX or anti-PECAM/GOX possess sharply distinct morphological features. Numerous PMNs that were found in both vascular and alveolar in the lungs of anti-TM/GOX-injected mice retained their granular structure (Figure 11A) ▶ . The inset in Figure 11A ▶ shows the detailed view of a typical event in the lung tissue observed after anti-TM/GOX injection: a migrating neutrophil disrupts endothelial and epithelial cells and protrudes from the vascular into alveolar space. In contrast, after anti-PECAM/GOX injection, numerous PMNs that were found within the pulmonary vascular compartment showed lack of dense granules (Figure 11B ▶ , inset). Thus, alveolar PMN accumulation represents a hallmark of the transmigration-competent anti-TM/GOX model, whereas intravascular PMN activation is characteristic of the transmigration-frustrated anti-PECAM/GOX model.
Figure 11.
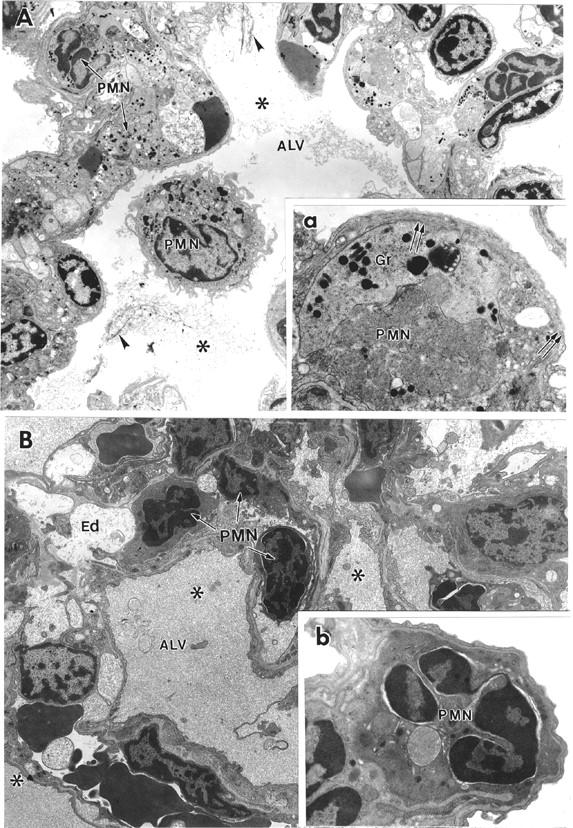
Different ultrastructural features of lung injury induced by anti-TM/GOX and anti-PECAM/GOX in mice. Transmission electron micrographs show typical morphological patterns of the lungs after injection of anti-TM/GOX (A) or anti-PECAM/GOX (B). A: Lower power (original magnification, ×4,400): visible blebbing of epithelial cells, swelling of endothelium and proteinaceous material (asterisk) with fibrin strands (arrowheads) in the alveoli. Capillaries are packed with PMN neutrophils and platelets. Inset (a, high power ×15,000) shows leukocytes retaining granules (g) in extravascular space and focal damage of endothelial cell (double arrows). B: Low power (original magnification, ×4,400): severe interstitial edema (Ed), proteinaceous alveolar material, numerous PMNs trapped in vessels contain no visible granules and condensed, pycnotic nuclei. Inset (b, high power ×15,000) shows neutrophil in capillary, with condensed nuclear chromatin, dense cytoplasm, and loss of granules.
Pulmonary Thrombosis in the TM/GOX Model
In addition to differences in the distribution of neutrophils between the two models, morphological analysis also revealed a much larger amount of platelet deposition and intravascular thrombosis in the lungs after TM/GOX injection, both at light and electron microscopy levels (Figure 12A) ▶ . In contrast, deposition of thrombi and platelets were rarely seen in the lungs after anti-PECAM/GOX injection (Figure 12B) ▶ . A semiquantitative analysis of intravascular thrombi revealed 46.6 ± 6.6 microthrombi versus 1.6 ± 0.33 microthrombi per 10 high-power fields after injection of equally injuring doses of anti-TM/GOX versus anti-PECAM/GOX, respectively.
Figure 12.
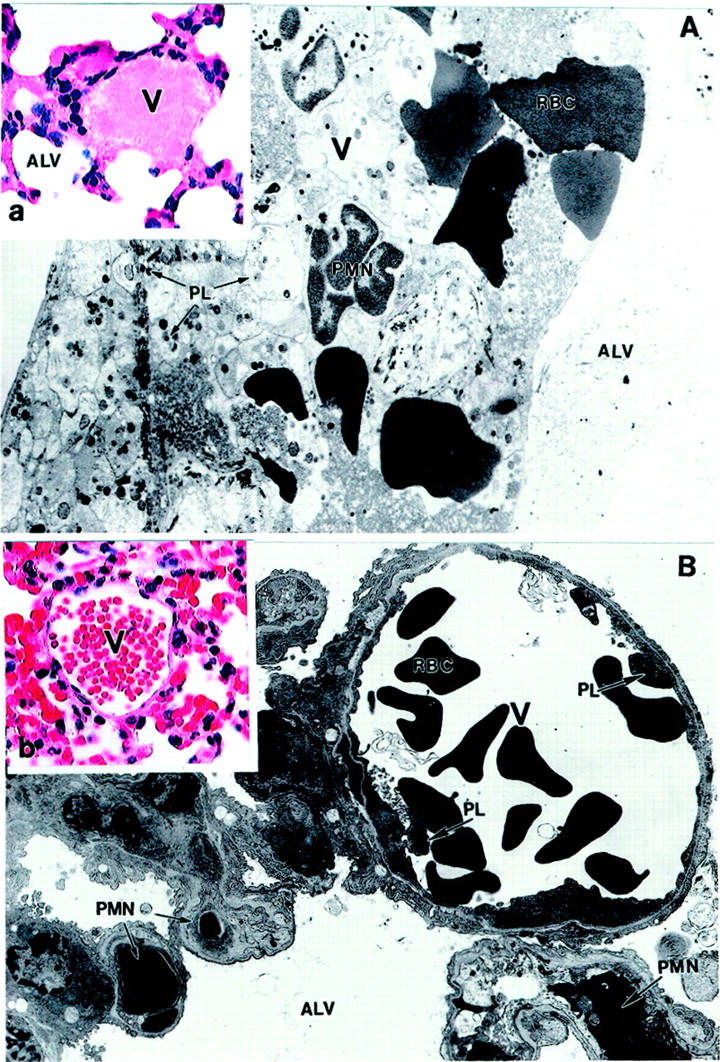
Anti-TM/GOX causes profound pulmonary thrombosis. Transmission electron micrographs and H&E staining (insets; original magnifications, ×500) of lung tissue after anti-TM/GOX (A, a) and anti-PECAM/GOX (B, b) injection in mice. A, a: A venule occluded by platelet (PL) thrombus with entrapped PMNs and red blood cells (RBCs). B, b: Absence of thrombi in either arteriole or capillaries. Multiple degranulated PMNs are visible in capillaries. Original magnifications: ×4000 (A); ×7700 (B); ×250 (a and b). Alv, alveolus; V, venule; PL, platelet.
To more specifically identify platelet deposition in the lungs, tissue sections were stained with a mAb directed against a murine platelet marker, CD41 (GP IIb) (Figure 13) ▶ . There was some basal specific CD41 staining in the pulmonary vessels of saline-injected mice (Figure 13B) ▶ , likely because of residual blood in the lung. The positive CD41 staining was more evident, however, in the lungs after anti-PECAM/GOX injection (Figure 13C) ▶ . This result may be attributed to entrapment of platelets in the congested pulmonary vessels. However, markedly more intense CD41 staining was documented in the lungs after anti-TM/GOX injection, presumably because of active intravascular pulmonary thrombosis (Figure 13D) ▶ .
Figure 13.

Detection of platelet thrombi in GOX-treated lungs. Mice were given injuring doses of anti-TM/GOX or anti-PECAM/GOX and lungs were harvested after 4 hours. Lung sections were processed for immunohistochemical platelet detection using an anti-CD 41 (anti IIb Ab). Platelets and platelet thrombi are shown as red-brown staining. A: Control, untreated tissues, given no primary Ab. B: Control untreated lung stained with anti-CD41. C and D: Lung sections treated with either anti-PECAM/GOX (C) or anti-TM/GOX (D), stained with anti-CD41. Original magnifications, ×250X.
Discussion
Investigation of the mechanisms and therapies of ALI/ARDS, a syndrome of diverse etiologies with significant morbidity and mortality, remains an area of active interest. 14,28,30 The pathology of the acute (exudative) phase of ARDS is characterized by a series of changes beginning with interstitial and intra-alveolar edema with a proteinaceous exudate containing cellular debris and by varying amounts of hemorrhage and fibrin deposition. Degenerative changes at this stage can be detected electron microscopically as well, seen as cytoplasmic swelling in capillary endothelial cells and alveolar epithelial cells with prominent interstitial edema. Hyaline membranes, a historic hallmark of the acute phase, are more prominent at the later stage of the acute phase, and are often accompanied by electron microscopically detectable sloughing of alveolar epithelial cells and denudation of the epithelial basement membrane.
The PMN sequestration and transmigration, as well as pulmonary thrombosis, represent important pathological features of ALI/ARDS. An interstitial inflammatory infiltrate consisting of neutrophils, macrophages, and red blood cells further augments thickening of the alveolar septae. 29,31 Alveolar transmigration of WBCs is a common event in ALI/ARDS 2,3,5,6,32-33 . Microthrombi are often present in alveolar capillaries and small pulmonary arteries. 5,6,17,34,35 It seems plausible to hypothesize that, at least in some versions of the syndrome, these hallmark manifestations of the syndrome result from primary pulmonary oxidative insult, 15-17 yet specific molecular and cellular mechanisms remain to be elucidated.
Throughout the years, a number of animal models have been developed for investigation of human ALI/ARDS. The models using primary airway insults, such as hyperoxia, 36,37 ozone exposure, 38 bacterial pneumonia, 39 repeated BAL with saline, 40 intratracheal instillation of hydrochloric acid, 41 or particulate nickel sulfate or other heavy metals 42 permit local initiation of the lung injury. However, events in the vascular compartment seem to be at least as important as those in the alveolar/airway compartment, in terms of the initiation of many (perhaps, the majority) variants of ALI/ARDS. The models based on the local insults delivered via the airways are thus of relatively limited use for investigation of intravascular initiation of the human syndrome.
The models using intravascular insults (eg, based on systemic activation of coagulation, complement, or leukocytes) arguably possess a higher similarity to variants of human ALI/ARDS induced by or associated with massive peripheral trauma, hemorrhage, or skin burns. The realization that distal organ injury may play an important role in the pathogenesis of ALI/ARDS, paved the way for the development of animal models of ALI such as hemorrhage/resuscitation; 14,43 ischemia/reperfusion of the intestines, 44 kidneys, 9 or skeletal muscle; 10,45 intravenous infusion of oleic acid; 46,47 intravenous infusion of endotoxin; 48-50 and skin burns. 7 These ALI/ARDS animal models are characterized by pulmonary neutrophil recruitment, thrombosis, and increased vascular permeability. However, the exact pathological events in the pulmonary vasculature culminating in ALI remain unclear in many of these models. In addition, the extent of the lung injury induced is often relatively mild and the overall condition of the animal is dominated by the systemic nature of the perturbation. Because many of these single-hit models offer a relatively minor lung injury that does not adequately reflect the severity of human ALI/ARDS, a number of so-called “two-hit models” were developed whereby the synergistic effects of two insults afford further exaggerated injury. Such models including cecal ligation puncture, leading to abdominal sepsis followed by acid aspiration or by hyperoxic exposure, 51,52 as well as hemorrhagic shock with resuscitation followed by intratracheal lipopolysaccharide administration, 53 may more closely resemble the human ARDS in which an initial insult primes the system followed by a second insult leading to organ damage. However, the added complexity to these models complicates the analysis of underlying mechanisms even beyond that in single-hit models.
Therefore, novel approaches using specific modes of pulmonary vascular injury that are thought to mimic important pathophysiological insults to the lung (eg, endothelial oxidative stress) could be useful for modeling of human ALI/ARDS in laboratory animals. An ideal ALI/ARDS model would use a controlled injury producing specific effects in vivo amenable for use in diverse animal species. This would allow studies in higher mammals (ie, pigs, primates) and in mice permitting studies in genetically altered animals.
Our data indicate that immunotargeting of GOX (an enzyme that generates a single reactive oxygen species, H2O2) to pulmonary endothelium using specific anti-endothelial cell antibodies provides such a model. In the present study, we systematically characterized the pathological features of the lung injury caused by GOX targeted to PECAM-1 or TM. Consistent with preferential pulmonary accumulation of anti-TM/GOX and anti-PECAM/GOX, both conjugates induced acute, dose-dependent, tissue-selective lung injury characterized by an increase in vascular permeability, endothelial cell damage, hemorrhage, and accumulation of products of oxidative modification of lipids and proteins (summarized in Table 1 ▶ ). The robustness of the lung injury, by itself, represents an important advantage of the GOX immunotargeting model. In addition, both anti-PECAM/GOX and anti-TM/GOX caused accumulation of WBCs in the lungs. Most likely, H2O2 generated by the endothelium-associated GOX induces an oxidative stress in the lungs, activates endothelial cells, and causes pulmonary sequestration of WBCs. Pulmonary sequestration of WBCs represents a common event in diverse forms of ALI. 2,3
Table 1.
Parameters of Lung Injury in GOX Models of Lung Injury
| Immune conjugate injected intravenously in mice | Time/max injury | BAL WBC (cells/ml) | % PMN in BAL | BAL proteins |
|---|---|---|---|---|
| Anti-PECAM/GOX (dose: 50 to 75 μg) | 4 hours (n = 12) | 107% ± 21% (n = 12) | 1% ± 0 | 1,619% ± 285%* (n = 17) |
| Anti-TM/GOX (dose: 25 to 30 μg) | 4 hours (n = 18) | 442% ± 128* (n = 17) | 4.5% ± 0.1%‡ | 2,176% ± 495* (n = 18) |
| MPO | Lung wet/dry ratio | ALIS | Thrombi/10 high-power fields |
|---|---|---|---|
| 471% ± 81* (n = 9) | 152% ± 9 (n = 9) | 10 (n = 17) | 1.7 ± 0.3 (n = 30) |
| 375% ± 31* (n = 4) | 143% ± 5 (n = 24) | 10 (n = 18) | 46.7 ± 6.6 (n = 30) |
*P < 0.0001.
†P < 0.05.
‡Values taken at 24 hours after injection of anti-TM/GOX = 25.1% ± 11.7% PMN in BAL versus no change with anti-PECAM/GOX.
The values in the table represent percent increase from baseline untreated controls (average ± SEM).
However, certain important pathological features of anti-PECAM/GOX and anti-TM/GOX were strikingly different. Although both conjugates caused neutrophil accumulation in the lung tissues, only anti-TM/GOX injury was associated with neutrophil transmigration in the alveolar space. In addition, anti-TM/GOX lung injury was also associated with pulmonary thrombosis. We attribute these differences to inhibition of the target endothelial proteins, PECAM-1 and TM, by the conjugates.
PECAM-1, a constitutive surface adhesion molecule, mediates endothelial transmigration and extravasation of WBCs. 25,54 PECAM antibodies may suppress this function. Several groups have shown that PECAM antibodies attenuate neutrophil-mediated vascular injury in diverse animal models, 55,56 including one report showing inhibition of neutrophil transmigration into alveolar space after immune complex deposition in the lung. 54 Our present data suggest that PECAM blocking by the conjugate is a plausible explanation for the inhibition of WBCs extravasation observed after injection of anti-PECAM/GOX. In support of this notion, the results of pilot experiments showed that co-injection of nonconjugated PECAM antibody attenuates pulmonary neutrophils extravasation induced by anti-TM/GOX in mice (M. Christofidou, unpublished results).
In contrast, TM is not directly involved in neutrophil transmigration into the alveoli, hence anti-TM targeting did not compromise this important component of lung injury. TM (CD 141) is the endothelial cell-surface glycoprotein that suppresses coagulation by converting thrombin from a procoagulant enzyme into an anticoagulant one by activating plasma protein C that in turn degrades clotting factors V and VIII. 13,26 Intravascular administration of soluble recombinant or purified natural TM has been tested in animal studies as a way to suppress intravascular coagulation and pulmonary thrombosis associated with endotoxemia and sepsis. 48,49 Although the therapeutic value of this maneuver remains to be validated in clinical studies, an important role of a pathological deficiency of TM in pulmonary thrombosis seems to be well established. Thus, enhanced pulmonary deposition of fibrin and platelets has been reported in genetically modified TM−/− chimeric mice, especially when animals are challenged with hyperoxic pulmonary stress. 57
Abnormal coagulation and intravascular thrombosis are known landmarks of many forms of ALI/ARDS. 5,23,35 Massive trauma, hemorrhage/resuscitation, endotoxemia, and ischemia cause systemic activation of platelets and coagulation. On the other hand, local pulmonary insults, such as oxidative stress on hyperoxia or smoke inhalation, may compromise anti-thrombotic features of the pulmonary endothelium. Therefore, both systemic and local factors mediate pulmonary thrombosis and deposition of blood clots during ALI/ARDS. At least in part, these features of the human syndrome may be the result of functional insufficiency of the TM-protein C system, either because of endothelial injury or genetic factors. 14 Cytokines, oxidants, and activated neutrophils are known to inhibit TM in the endothelial cells by suppressing its synthesis, enhancing its degradation, and shedding from the plasma membrane. 58,59 Elevated levels of the soluble form of TM circulating in blood plasma, most likely reflecting endothelial injury and TM shedding, has been reported in many vascular and pulmonary disorders, including ALI/ARDS in human patients and animal models. 60,61 H2O2 directly inhibits TM. 58 In addition, antibodies can enhance TM internalization and block its interaction with thrombin and protein C. 62 In good agreement with these observations, we found that anti-TM/GOX caused a noticeable increase in intravascular thrombosis in the lungs, most likely because of inhibition of anti-thrombotic function of TM. Therefore, anti-TM/GOX immunotargeting approach combining H2O2 generation and TM suppression in the pulmonary endothelium may provide a novel model featuring several key components of human ALI/ARDS: oxidative stress, extravasation of leukocytes, and thrombosis.
The presented results indicate that vascular immunotargeting of GOX to endothelial antigens provides a set of animal models of oxidant-induced pulmonary injury that can be regulated in intensity, severity, and duration. Moreover, these experiments demonstrate that the functional activity of the antibody used to make the immunoconjugates can dictate the type of lung injury produced by the same initial oxidative insult. This feature permits even more precise dissection of the mechanisms of the lung injury. Thus, GOX targeting to TM results in a model of ALI accompanied by neutrophil extravasation and intravascular thrombosis that closely resembles human ALI/ARDS. Therefore, GOX immunotargeting-based models of ALI/ARDS may find applications in studies aimed at dissecting the pathways of oxidant-induced ALI/ARDS (including studies in genetically modified animals), finding new means to diagnose/predict the condition and testing of new therapeutic strategies for treatment of ALI/ARDS, such as specific anti-oxidant or/and anti-thrombotic interventions.
Acknowledgments
We thank Evguenia Argyris for her excellent technical support with the animal experiments and tissue/sample processing, Mildred Daise for culturing of the hybridomas and antibody purification, and Melanie Minda for her valuable support with the electron microscopy.
Footnotes
Address reprint requests to Dr. Vladimir R. Muzykantov, Institute of Environmental Medicine, University of Pennsylvania Medical Center, 1 John Morgan Building, 36th Street and Hamilton Walk, Philadelphia, PA 19104-6068. E-mail: muzykant@mail.med.upenn.edu.
Supported by the National American Heart Association (grant AHA no. 0030192N to M. C. S.), by an American Heart Association Established Investigator Grant, and Project 4 in the National Institutes of Health Specialized Center of Research in Acute Lung Injury (to V. R. M.).
References
- 1.Christofidou-Solomidou M, Pietra G, Solomides C, Argiris E, Harshaw D, FitzGerald G, Albelda S, Muzykantov V: Immunotargeting of glucose oxidase to endothelium in vivo causes oxidative vascular injury in the lungs. Am J Physiol 2000, 278:L794-L805 [DOI] [PubMed] [Google Scholar]
- 2.Ward P, Hanninghake G: Lung inflammation and fibrosis. Am J Respir Crit Care Med 1998, 157:S123-S129 [DOI] [PubMed] [Google Scholar]
- 3.Rinaldo J, Christman J: ARDS: pathogenesis. Fishman AP eds. Pulmonary Diseases and Disorders. 1998, :pp 2537-2548 McGraw-Hill, New York [Google Scholar]
- 4.Heinemann HO, Fishman AP: Nonrespiratory functions of mammalian lung. Physiol Rev 1969, 49:1-47 [Google Scholar]
- 5.Vesconi S, Rossi GP, Pesenti A, Fumagalli R, Gattinoni L: Pulmonary microthrombosis in severe adult respiratory distress syndrome. Crit Care Med 1988, 16:111-113 [DOI] [PubMed] [Google Scholar]
- 6.Car BD, Suyemoto MM, Neilsen NR, Slauson DO: The role of leukocytes in the pathogenesis of fibrin deposition in bovine acute lung injury. Am J Pathol 1991, 138:1191-1198 [PMC free article] [PubMed] [Google Scholar]
- 7.Mulligan MS, Till GO, Smith CW, Anderson DC, Miyasaka M, Tamatani T, Todd III RF, Issekutz TB, Ward PA: Role of leukocyte adhesion molecules in lung and dermal vascular injury after thermal trauma of skin. Am J Pathol 1994, 144:1008–1015 [PMC free article] [PubMed]
- 8.Shenkar R, Coulson WF, Abraham E: Anti-transformation growth factor-beta monoclonal antibodies prevent lung injury in hemorrhaged mice. Am J Respir Cell Mol Biol 1994, 11:351-357 [DOI] [PubMed] [Google Scholar]
- 9.Moore FA, Moore EE: Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am 1995, 75:257-277 [DOI] [PubMed] [Google Scholar]
- 10.Punch J, Rees R, Cashmere B, Oldham K, Wilkins E, Smith DJ: Acute lung injury following reperfusion after ischemia in the hind limbs of rats. J Trauma 1991, 31:760-765 [DOI] [PubMed] [Google Scholar]
- 11.Masubuchi T, Koyama S, Sato E, Takamizawa A, Kubo K, Sekiguchi M, Nagai S, Izumi T: Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol 1998, 153:1903-1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burns JA, Issekutz TB, Yagita H, Issekutz AC: The beta2, alpha4, alpha5 integrins and selectins mediate chemotactic factor and endotoxin-enhanced neutrophil sequestration in the lung. Am J Pathol 2001, 158:1809-1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esmon C: Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at vessel surface. FASEB J 1995, 9:946-955 [DOI] [PubMed] [Google Scholar]
- 14.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher Jr CJ: Recombinant human protein C worldwide evaluation in severe sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001, 344:699–709 [DOI] [PubMed]
- 15.Leff J, Parsons P, Day C, Taniguchi N, Jochum M, Fritz H, Moore F, Moore E, McCord J, Repine J: Serum antioxidants as predictors of adult respiratory distress syndrome in patients with sepsis. Lancet 1993, 341:777-780 [DOI] [PubMed] [Google Scholar]
- 16.Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S, Goodman RB, Hudson LD, Matalon S, Martin TR: Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001, 163:503-510 [DOI] [PubMed] [Google Scholar]
- 17.Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust III RF, Finkel B, Lanken PN, Ischiropoulos H: Plasma proteins modified by tyrosine nitration in acute respiratory distress syndrome. Am J Physiol 2000, 278:L961–L967 [DOI] [PubMed]
- 18.Kennel SJ, Falcioni R, Wesley JW: Microdistribution of specific rat monoclonal antibodies to mouse tissues and human tumor xenografts. Cancer Res 1991, 51:1529-1536 [PubMed] [Google Scholar]
- 19.Muzykantov V, Atochina E, Ischyropoulos H, Danilov S, Fisher A: Immunotargeting of anti-oxidant enzymes to the pulmonary endothelium. Proc Natl Acad Sci USA 1996, 93:5213-5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muzykantov V, Christofidou-Solomidou M, Balyasnikova I, Harshaw D, Schultz, Fisher AB, Albelda SM: Streptavidin facilitates internalization and pulmonary targeting of an anti-endothelial cell antibody (PECAM-1): a novel strategy for intraendothelial drug delivery. Proc Natl Acad Sci USA 1999, 96:2379–2384 [DOI] [PMC free article] [PubMed]
- 21.Wiewrodt R, Thomas AP, Cipelletti L, Christofidou-Solomidou M, Weitz DA, Feinstein SI, Schaffer D, Albelda SM, Koval M, Muzykantov V: Size-dependent immunotargeting of cargo materials to endothelial cells via poorly internalizable surface adhesion molecules. Blood 2002, 99:912-922 [DOI] [PubMed] [Google Scholar]
- 22.Muzykantov VR, Sakharov DV, Sinitsyn VV, Domogatsky SP, Goncharov NV, Danilov SM: Specific killing of human endothelial cells by antibody-conjugated glucose oxidase. Analyt Biochem 1988, 169:383-389 [DOI] [PubMed] [Google Scholar]
- 23.Muzykantov V, Martynov A, Puchnina E, Danilov S: In vivo administration of glucose oxidase conjugated with monoclonal antibody to ACE: targeting into the lung. Am Rev Respir Dis 1989, 136:1464-1473 [DOI] [PubMed] [Google Scholar]
- 24.Gow A, Branco F, Cristofidou-Solomidou M, Schultz L, Albelda S, Muzykantov V: Immunotargeting of glucose oxidase: intracellular production of H2O2 and endothelial oxidative stress. Am J Physiol 1999, 277:271-281 [DOI] [PubMed] [Google Scholar]
- 25.Newman PJ: The biology of PECAM. J Clin Invest 1997, 99:3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esmon CT, Owen WG: Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc Natl Acad Sci USA 1981, 78:2249-2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pratico D, Iuliano L, Mauriello A, Spagnoli L, Lawson JA, Mclouf J, Violi F, FitzGerald GA: Localization of distinct F-2-isoprostanes in human atherosclerotic lesions. J Clin Invest 1997, 100:2028-2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson L, Steinberg K: ARDS: clinical features, management and outcome. Fishman AP eds. Pulmonary Diseases and Disorders. 1998, :pp 2548-2565 McGraw-Hill, New York [Google Scholar]
- 29.Ware LB, Matthay MA: The acute respiratory distress syndrome. N Engl J Med 2000, 342:1334-1349 [DOI] [PubMed] [Google Scholar]
- 30.Meyrick B: Pathology of the adult respiratory distress syndrome. Crit Care Clin 1986, 2:405-428 [PubMed] [Google Scholar]
- 31.Katzenstein AL: Acute lung injury patterns: diffuse alveolar damage and bronchiolitis obliterans-organizing pneumonias. LiVolsi V eds. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. 1997, :pp14-43 WB Saunders, Philadelphia [Google Scholar]
- 32.Pittet JF, Mackersie RC, Martin TR, Matthay MA: Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med 1997, 155:1187-1205 [DOI] [PubMed] [Google Scholar]
- 33.Chollet-Martin S: Polymorphonuclear neutrophil activation during the acute respiratory distress syndrome. Int Care Med 2000, 26:1575-1577 [DOI] [PubMed] [Google Scholar]
- 34.Malik A: Pulmonary microembolism and lung vascular injury. Eur Respir J Suppl 1990, 11:S499-S506 [PubMed] [Google Scholar]
- 35.Tomaschefski Jr JF, Davies P, Boggis C, Greene R, Zapol WM, Reid LM: The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol 1983, 112:112–126 [PMC free article] [PubMed]
- 36.Fox RB, Hoidal JR, Brown DM, Repine JE: Pulmonary inflammation due to oxygen toxicity: involvement of chemotactic factors and polymorphonuclear leukocytes. Am Rev Respir Dis 1981, 123:521-523 [DOI] [PubMed] [Google Scholar]
- 37.Fracica PJ, Knapp MJ, Crapo JD: Patterns of progression and markers of lung injury in rodents and subhuman primates exposed to hyperoxia. Exp Lung Res 1988, 14:S869-S885 [DOI] [PubMed] [Google Scholar]
- 38.Prows DR, Shertzer MJ, Daly MJ, Sidman CL, Leikauf GD: Genetic analysis of ozone-induced acute lung injury in sensitive and management. Nat Genet 1997, 17:471-474 [DOI] [PubMed] [Google Scholar]
- 39.Brigham K, Meyrick B: Endotoxin and lung injury. Am Rev Respir Dis 1986, 133:913-927 [PubMed] [Google Scholar]
- 40.Lachmann B, Robertson B, Vogel J: In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesth Scand 1980, 24:231-236 [DOI] [PubMed] [Google Scholar]
- 41.Rabinovici R, Neville LF, Abdullah F, Phillip DR, Vernick J, Fong KL, Hillegas L, Feuerstein G: Aspiration-induced lung injury: role of complement. Crit Care Med 1995, 23:1405-1411 [DOI] [PubMed] [Google Scholar]
- 42.Bouthillier L, Vincent R, Goegan P, Adamson IY, Bjarnason S, Stewart M, Guenette J, Potvin M, Kumarathasan P: Acute effects of inhaled urban particles and ozone: lung morphology, macrophage activity, and plasma endothelin-1. Am J Pathol 1998, 153:1873-1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warner RL, Lewis CS, Beltran L, Younkin EM, Varani J, Johnson KJ: The role of metalloelastase in immune complex-induced acute lung injury. Am J Pathol, 2001, 158:2139–2144 [DOI] [PMC free article] [PubMed]
- 44.Simpson R, Alon R, Kobzik L, Valeri CR, Shepro D, Hechtman HB: Neutrophil and nonneutrophil-mediated injury in intestinal ischemia-reperfusion. Ann Surg 1993, 218:444-454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seekamp A, Mulligan MS, Till GO, Smith CW, Miyasaka M, Tamatani T, Todd RF, Ward PA: Role of beta 2 integrins and ICAM-1 in lung injury following ischemia-reperfusion of rat hind limbs. Am J Pathol 1993, 143:464-472 [PMC free article] [PubMed] [Google Scholar]
- 46.Derks CM, Jacobovitz-Derks D: Embolic pneumopathy induced by oleic acid. A systematic morphologic study. Am J Pathol 1977, 87:143–158 [PMC free article] [PubMed]
- 47.Davidson KG, Bersten AD, Barr HA, Dowling KD, Nicholas TE, Doyle IR: Lung function, permeability, and surfactant composition in oleic acid-induced acute lung injury in rats. Am J Physiol 2000, 279:L1091-L1102 [DOI] [PubMed] [Google Scholar]
- 48.Uchiba M, Okajima K, Murakami K, Nava K, Okabe H, Takatsuki K: Recombinant human soluble thrombomodulin reduces endotoxin-induced pulmonary vascular injury via protein C activation in rats. Thromb Haemost 1995, 74:1265-1270 [PubMed] [Google Scholar]
- 49.Hasegawa N, Kandra T, Husari A, Veiss S, Hart W, Hedgpeth J, Wydko R, Raffin T: The effects of recombinant human thrombomodulin on endotoxin-induced multiple-system organ failure in rats. Am J Respir Crit Care Med 1996, 153:1831-1837 [DOI] [PubMed] [Google Scholar]
- 50.Czermak BJ, Breckwoldt M, Ravage ZB, Huber-Lang M, Schmal H, Bless NM, Friedl HP, Ward PA: Mechanisms of enhanced lung injury during sepsis. Am J Pathol 1999, 154:1057-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nemzek JA, Call DR, Ebong SJ, Newcomb DE, Bolgos GL, Remick DG: Immunopathology of a two-hit murine model of acid aspiration lung injury. Am J Physiol 2000, 278:L512-L520 [DOI] [PubMed] [Google Scholar]
- 52.Tani T, Fujino M, Hanasawa K, Shimizu T, Endo Y, Kodama M: Bacterial translocation and tumor necrosis factor-alpha gene expression in experimental hemorrhagic shock. Crit Care Med 2000, 28:3705-3709 [DOI] [PubMed] [Google Scholar]
- 53.Fan J, Marshall JC, Jimenez M, Shek PN, Zagorski J, Rotstein OD: Hemorrhagic shock primes for increased expression of cytokine-induced neutrophil chemoattractant in the lung: role in pulmonary inflammation following lipopolysaccharide. J Immunol 1998, 161:440-447 [PubMed] [Google Scholar]
- 54.Vaporciyan AA, Delisser HM, Yan HC, Mendiguren I, Thom SR, Jones ML, Ward PA, Albelda SM: Involvement of PECAM-1 in neutrophil recruitment in vivo. Science 1993, 262:1580-1582 [DOI] [PubMed] [Google Scholar]
- 55.Christofidou-Solomidou M, Nakada MT, Williams J, Muller WA, DeLisser HM: Neutrophil platelet endothelial cell adhesion molecule-1 participates in neutrophil recruitment at inflammatory sites and is down-regulated after leukocyte extravasation. J Immunol 1997, 158:4872-4878 [PubMed] [Google Scholar]
- 56.Nakada MT, Amin K, Christofidou-Solomidou M, O’Brien CD, Sun J, Gurubhagavatula I, Heavner GA, Taylor AH, Paddock C, Sun QH, Zehnder JL, Newman PJ, Albelda SM, DeLisser HM: Antibodies against the first Ig-like domain of human platelet endothelial cell adhesion molecule-1 (PECAM-1) that inhibit PECAM-1-dependent homophilic adhesion block in vivo neutrophil recruitment. J Immunol 2000, 164:452-462 [DOI] [PubMed] [Google Scholar]
- 57.Healy A, Hancock W, Christie P, Rayburn H, Rosenberg R: Intravascular coagulation activation in a murine model of thrombomodulin deficiency: effects of lesion size, age and hyperoxia on fibrin deposition. Blood 1998, 92:4188-4197 [PubMed] [Google Scholar]
- 58.MacGregor I, Perrie A, Donnely S, Haslett C: Modulation of human endothelial thrombomodulin by neutrophils and their release products. Am J Respir Crit Care Med 1997, 155:47-52 [DOI] [PubMed] [Google Scholar]
- 59.Moore KL, Esmon CT, Esmon NL: Tumor necrosis factor leads to the internalization and degradation of thrombomodulin from the surface of bovine aortic endothelial cells in culture. Blood 1989, 73:159-165 [PubMed] [Google Scholar]
- 60.Jansson J, Boman K, Brannstrom M, Nilsson T: High concentration of thrombomodulin in plasma is associated with hemorrhage: a prospective study in patients receiving long-term anticoagulant treatment. Circulation 1997, 96:2938-2943 [DOI] [PubMed] [Google Scholar]
- 61.Bajaj M, Triscomi S: Plasma levels of the three endothelium-specific proteins von Willebrand factor, tissue pathway inhibitor and thrombomodulin do not predict the development of ARDS. Intensive Care Med 1999, 25:1259-1266 [DOI] [PubMed] [Google Scholar]
- 62.Brisson G, Archipoff G, Hartmann M, Hanau D, Beretz A, Freyssinet J, Cazenave J: Antibodies to thrombomodulin induce receptor-mediated endocytosis in human saphenous vein endothelial cells. Thromb Haemost 1992, 68:737-743 [PubMed] [Google Scholar]