

Abstract
Prolonged cold ischemia has been suggested as a factor that will exacerbate later graft arterial disease (GAD), a major limiting factor for long-term transplant survival. We therefore examined the effects of cold ischemia on GAD as well as adhesion molecule and cytokine expression in murine cardiac grafts. Mild GAD developed in isografts undergoing 4-hour cold ischemia. Relative to control isografts, cold ischemia induced transiently enhanced endothelial expression of intercellular adhesion molecule-1 (ICAM-1) at 4 hours post-transplant. There was also transiently-augmented gene expression of interleukin (IL)−1β, IL-6, and transforming growth factor-β in these cold-ischemic isografts. By 3 days post-transplantation, however, there were no longer any differences between control and cold ischemic isografts. Cold ischemia did not significantly affect the final grade of either parenchymal rejection or GAD in long-term (4 to 12 weeks) major histocompatibility complex (MHC) I- or MHC II-mismatched allografts molecules transplanted without immunosuppression. At early time points after cold ischemia (4 to 24 hours), allografts mismatched for MHC I and/or MHC II showed enhanced expression of ICAM-1 and cytokines comparable to that seen in isografts. By day 7 post-transplant, both control and cold ischemia allografts showed comparable expression of cytokines and adhesion molecules. Although prolonged cold ischemia can initiate mild GAD in isografts by transiently enhancing antigen non-specific inflammatory responses, it does not significantly augment subsequent alloresponses.
Progress in immunosuppressive therapy and management of acute allograft rejection has improved short-term survival of heart transplant patients. However, strategies for prevention and treatment of graft coronary artery disease (GAD) have proven elusive and GAD remains a major limiting factor for long-term graft survival. 1,2 Various immunological, infectious, and nonimmunologic factors may contribute to the development of GAD. 3-6 Characteristically, GAD affects the engrafted vessels and spares the host arteries. Although understanding of the pathogenesis of GAD is incomplete, two simple models can explain this selective involvement of the transplanted arteries: an immunological response directed against donor antigens, or a response to ischemic injury encountered during storage and transport postexplantation. 7 Thus, Gaudin et al showed that histologically proven ischemic injury during the perioperative period predicts the development of GAD in humans. 8 Another recent study 9 demonstrated the development of GAD in rat heart isografts following prolonged cold ischemia. The mechanisms for the development of GAD by cold ischemia/reperfusion are not fully understood. Several studies have demonstrated that warm ischemia and reperfusion resulted in increased cell adhesion molecule expression and stimulated reactive oxygen species and inflammatory cytokine production in a variety of organs, culminating in leukocyte accumulation and tissue destruction. 10-12 Much less is known about the effects of cold ischemia and reperfusion on early cytokine expression. It also remains controversial whether prolonged cold ischemia/reperfusion injury can aggravate GAD. 9,13 Particularly, it is uncertain whether early enhanced inflammation induced by prolonged cold ischemia can accentuate subsequent alloimmune responses, or whether ischemic injury and alloimmune responses may independently affect the development of GAD.
The present study used a heterotopic mouse heart transplant model to examine whether cold ischemia followed by reperfusion can induce GAD in isografts not subject to immunological injury, or augment GAD in major histocompatibility complex (MHC) I- or MHC II-mismatched allografts. We chose a four-hour ischemic period to correspond to the upper limit of cold ischemia typically permitted for clinical human heart transplantation. To gain mechanistic insight into the pathogenesis of transplantation complications, we further studied the effects of prolonged cold ischemia on the time course and magnitude of expression of inflammatory cytokines and cell adhesion molecules in isografts and in MHC I-, MHC II-, or in total-allomismatched allografts. The results indicate that cold ischemia transiently increases the expression of selected cytokines, as well as intercellular adhesion molecule-1 (ICAM-1), and may thereby contribute to the development of GAD. However, alloresponses in cardiac grafts occur largely after the effects of ischemic injury have already subsided, and the extent of acute parenchymal rejection or subsequent GAD are not significantly affected by prior cold ischemic injury.
Materials and Methods
Antibodies and Other Reagents
Antibodies for mouse ICAM-1, vascular cell adhesion molecule-1 (VCAM-1), E-selectin, and isotype- and class-matched immunoglobulin were purchased from PharMingen (San Diego, CA). Anti-mouse P-selectin antibody (10A10) was a generous gift of Dr. Michael A. Gimbrone, Jr. Normal goat serum, rabbit serum, and biotinylated secondary antibodies were from Vector Laboratories Inc. (Burlingame, CA) [α-32P]UTP was from Perkin Elmer Life Sciences, Inc. (Boston, MA).
Animals
Inbred male mice (9 to 12 weeks) of several strains were used in these experiments. C57BL/6 (B/6, H-2b) and BALB/c (B/c, H-2d) mice were obtained from Taconic Farms, Inc. (Germantown, NY). B6-C-H-2bm12KhEg (bm12, H-2bm12) mice MHC class II-mismatched from B/6 mice, and B6-C-H-2bm1ByJ (bm1, H-2bm1) mice MHC class I-mismatched from B/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Animals were maintained in the Harvard Medical School animal facilities and allowed ad libitum access to food and acidified water. Sentinel mice housed in the same room as experimental animals were consistently pathogen-free. All experiments conformed to animal care protocols approved by the institutional review group.
Heart Transplantation
Heterotopic heart transplantation was performed as previously described. 14,15 In brief, donor and recipient mice were anesthetized by inhalation of Metofan (Pittman-Moore, Mundelein, IL). Donor hearts were perfused with chilled, heparinized 0.9% saline or heparinized Stanford solution 16 via the inferior vena cava and harvested after ligation of the vena cava and pulmonary veins. The aorta and pulmonary artery of donor hearts were anastomosed to the abdominal aorta and inferior vena cava of recipient mice, respectively, using microsurgical technique. Ischemic time during the surgical procedure was routinely 30 minutes, and initial graft survival was greater than 90%.
Prolonged Cold Ischemia
For cold ischemia experiments, harvested donor hearts were perfused and stored in sterile saline (0.9% NaCl solution) for 4 hours at 4°C before transplantation. Preliminary experiments showed minimal histological evidence of myocardial necrosis at day 7 post-transplant in B/6 heart isografts following 4-hour cold ischemia in saline (data not shown). Different storage solutions may potentially offset the effects of prolonged cold ischemia on GAD. We therefore also tested the effects of cold ischemia on GAD development in the presence of Stanford solution, a storage medium reported efficacious for reducing GAD in human heart transplantation. 16 In this series of experiments, we evaluated GAD in MHC II- disparate grafts at 8 weeks and in MHC I-disparate grafts at 12 weeks. Stanford solution experiments, including non-ischemic controls, were performed as a distinct cohort from the saline experiments.
Histological Examination
Grafts were explanted at defined intervals of 0 hours, 4 hours, 24 hours, 3 days, 7 days, or 4, 8, or 12 weeks post-transplant, and sectioned into three transverse parts. The basal third was fixed in 10% phosphate-buffered formalin, embedded in paraffin, and 5 μ sections were stained with hematoxylin and eosin, or elastic tissue stains. The other transverse sections were either frozen in OCT compound (Ames Co., Division of Miles Laboratories, Elkhart, IN) for immunohistochemistry (see below) and/or analyzed by RNase protection assay (RPA; see below). The severity of parenchymal rejection and GAD was scored blindly by two independent observers (Y.F. and R.N.M.); scores uniformly fell within a range of one grade for the two observers, and were averaged. Parenchymal rejection was graded using a scale modified from the International Society for Heart and Lung Transplantation 17 (0, no rejection; 1, mild interstitial or perivascular infiltrate without necrosis; 2, focal interstitial or perivascular infiltrate with necrosis; 3, multifocal interstitial or perivascular infiltrate with necrosis; and 4, widespread infiltrate with hemorrhage and/or vasculitis). The severity of GAD was evaluated based on the lumenal narrowing by intimal hyperplasia, and was scored individually for each vessel (0, no or minimal [<10%] vascular occlusion; 1, 10 to 25% occlusion; 2, 25 to 50% occlusion; 3, 50 to 75% occlusion; 4, 75 to 100% occlusion). The final GAD score for each allograft heart was calculated by averaging the scores of all vessels; typically, the individual scores for >10 vessels on three to four transverse sections were averaged for each specimen.
Preliminary experiments demonstrated that GAD develops more rapidly in MHC II-disparate grafts relative to MHC I-disparate grafts, frequently exhibiting complete lumenal occlusion by 8 weeks. Conversely, post-transplant intervals of 12 weeks are typically required to develop moderate-to-severe GAD in MHC I-disparate grafts. Because the composition of cellularity versus matrix deposition in GAD varies as the lesions mature, 15 we evaluated GAD at two time points. MHC II-disparate grafts were therefore assessed at 4 and 8 weeks post-transplant, while MHC I-disparate grafts were examined at 8 and 12 weeks. The expression of cell adhesion molecules following immunohistochemical staining was evaluated blindly by two observers (Y.F. and R.N.M): −, not present or focal weak staining; +, diffuse or strong staining; these evaluations were 100% concordant.
RNase Protection Assay
Total RNA was prepared by guanidinium thiocyanate/phenol/chloroform/isoamylalcohol isolation method using TRIZOL (Gibco BRL). Fifteen micrograms of total RNA for each sample was analyzed quantitatively for cytokine mRNA expression by RPA. Multiprobe RNase protection assay kit mCK-3b (containing DNA templates for tumor necrosis factor (TNF)-β, lymphotoxin (LT)-β, TNF-α, interleukin (IL)−6, interferon (IFN)-γ, IFN-β, transforming growth factor (TGF)-β1, TGF-β2, TGF-β3, macrophage migration-inhibitory factor (MIF), L32 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) and mCK-2b (containing DNA templates for IL-12p35, IL-12p40, IL-10, IL-1α, IL-1β, IL-1 receptor antagonist (Ra), IL-18, IL-6, IFN-γ, MIF, L32, and GAPDH) were obtained from PharMingen. RPA was performed following the manufacturer’s instructions. In brief, the DNA templates were used to synthesize the [α-32P]UTP-labeled RNA probes in the presence of GCAU mixture and T7 RNA polymerase. Each RNA sample was hybridized with the labeled RNA probes by overnight incubation at 56°C, followed by digestion with RNase A and T1 mixture for 45 minutes at 30°C. The samples were treated by proteinase K, extracted with Tris-saturated chloroform-isoamylalcohol-phenol solution, and then ethanol-precipitated in the presence of ammonium acetate. The protected RNA duplexes were dried, redissolved in loading buffer and, after denaturing at 90°C, resolved on a 5% acrylamide-urea sequencing gel. 32P-labeled probes were used as molecular size markers. The gel was absorbed to filter paper, dried under vacuum, and exposed on film (BioMax- MR; Kodak, Rochester, NY) and phosphorimager screen. The volume integrations of mRNA-protected 32P-labeled probe fragments were analyzed by bioimaging analyzer (Molecular Dynamics, Sunnyvale, CA). Finally, volume integrations of the protected bands for each cytokine were normalized against the bands for GAPDH in the corresponding lane. Total RNA samples from naive hearts of donor strain were used as normal heart controls.
Immunohistochemical Staining
Four-μm-thick frozen sections were fixed in acetone for 5 minutes at 4°C. To reduce nonspecific binding, the sections were first incubated with 5% normal goat or rabbit serum for 30 minutes at room temperature (RT). Hamster monoclonal antibody against mouse ICAM-1 and rat monoclonal antibodies against mouse VCAM-1, P-selectin, and E-selectin were used as primary antibodies. Sections were incubated with primary antibodies for 90 minutes at RT, washed in phosphate buffered saline (PBS), and incubated with biotinylated secondary antibodies (goat anti-hamster IgG[H + L] or rabbit anti-rat IgG[H + L]) for 45 minutes at RT. After washing in PBS, the sections were incubated with an alkaline phosphatase-conjugated avidin-biotin complex (Vectastain ABC-AP kit; Vector Labs) and washed. Alkaline phosphatase activity was visualized by incubating in substrate solution (Fast Red; Sigma Chemical Co., St. Louis, MO). Sections were counterstained with hematoxylin. Naive hearts of the donor strain were used as normal heart samples. Sections of mouse placenta were used for positive control staining of P-selectin and E-selectin.
Statistical Analysis
The incidence of GAD in isografts was compared between groups using χ 2 test. Values for histological grading of parenchymal rejection and GAD in allografts, and values for relative gene expression of inflammatory cytokines were expressed as mean ± SEM and compared using analysis of variance (ANOVA) followed by Fisher’s protected least significant difference (PLSD) post-hoc test.
Results
Prolonged Cold Ischemia Alone Causes GAD in Isografts
The majority of arteriopathy lesions in isografts were mild, with an average GAD score for each graft of less than 1. Therefore, to evaluate the effects of prolonged cold ischemia on GAD in isografts, we elected to compare the incidence of GAD between no-storage controls and 4-hour cold ischemia groups. Mild intimal thickening was observed by 8 weeks in the coronary arteries of isografts which had undergone 4-hour cold storage in saline (Figure 1B) ▶ , whereas isografts of the no-storage group did not show any histological features of GAD at 8 weeks (Figure 1, A and C) ▶ . The incidence of low-grade GAD in cold-ischemic isografts tended to increase at later time points (12 weeks), although there was no statistically significant difference in the incidence of GAD between no-storage group and 4-hour cold storage group at that time point. Notably, well-developed intimal lesions with severe luminal narrowing were seen only in the 4-hour cold ischemia group at 12 weeks (Figure 1D) ▶ . Four hours of cold ischemia in Stanford preservation solution tended to increase the extent of GAD relative to storage in saline, but the incidence of GAD was not significantly different at 12 weeks between Stanford solution and saline groups (Table 1) ▶ .
Figure 1.
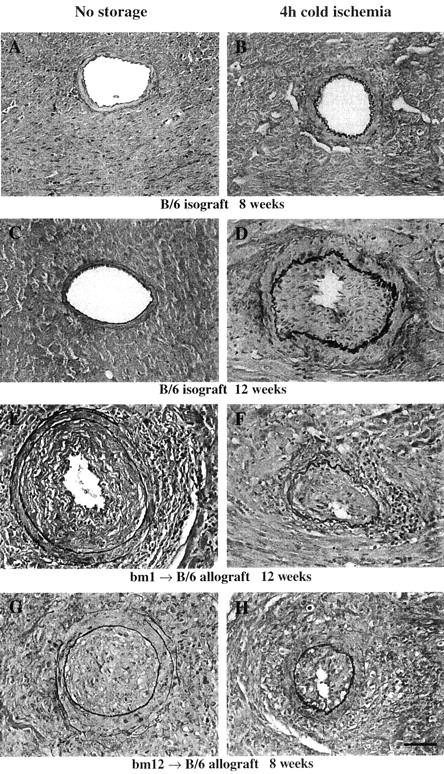
Elastin staining of the sections of mouse heart isografts and allografts. Scale bars, 50 μm. A: A representative section from a B/6 isograft in the no-storage group, 8 weeks after transplantation. Intimal lesions rarely developed in the isografts in the no-storage group. B: A representative section from a B/6 isograft following 4-hour cold ischemia in saline, 8 weeks after transplantation. Note the mild intimal thickening. C: A representative section from a B/6 isograft in the no-storage group, 12 weeks after transplantation. D: A representative section from a B/6 isograft following 4-hour cold ischemia in Stanford solution, 12 weeks after transplantation. The incidence of GAD tended to increase following 4-hour cold ischemia in either the saline and Stanford solution groups; GAD lesions from the 4-hour cold ischemia group showed moderate to severe intimal thickening at 12 weeks. E: A representative section from a bm1 to B/6 allograft in the no-storage group, 12 weeks after transplantation. F: A representative section from a bm1 to B/6 allograft in the 4-hour cold ischemia group, 12 weeks after transplantation. Similar severe GAD lesions developed in MHC class I disparate allografts without cold ischemia. G: A representative section from a bm12 to B/6 allograft in the no-storage group, 8 weeks after transplantation. H: A representative section from a bm12 to B/6 allograft in 4-hour cold ischemia group, 8 weeks after transplantation. Similar severe GAD lesions developed in MHC class II disparate allografts without cold ischemia.
Table 1.
Incidence of GAD in Isografts
| Time after transplantation | 4 weeks | 8 weeks | 12 weeks |
|---|---|---|---|
| Saline storage | |||
| No storage | 0 (n = 4) | 0 (n = 4) | 17 (n = 6) |
| 4-hour cold ischemia | 0 (n = 5) | 50 (n = 4) | 60 (n = 5) |
| Stanford solution | |||
| No storage | ND | ND | 25 (n = 4) |
| 4-hour cold ischemia | ND | ND | 80 (n = 5) |
Values are expressed as percent incidence of GAD. All of the averaged GAD scores applied for isografts were less than grade 1. Note the greater values of incidence of GAD (indicated by bold letters) in 4-hour cold ischemia groups relative to the no storage groups.
ND, not done.
Prolonged Cold Ischemia Does Not Affect the Severity of GAD and Parenchymal Rejection in Allografts
Both MHC I- (bm1 to B/6) and MHC II-mismatched (bm12 to B/6) allografts develop mild to severe GAD by 8 to 12 weeks post-transplant (Figure 1) ▶ . Because totally allomismatched (B/c to B/6) allografts undergo severe acute rejection and cease functioning at approximately 8 days, 18 they do not survive long enough to develop GAD in the absence of immunosuppression. Thus, B/c to B/6 allografts were not evaluated for GAD. Four-hour cold storage in either 0.9% saline or Stanford preservation solution did not significantly exacerbate or accelerate the development of GAD for either MHC I or MHC II antigen-mismatched allografts, compared with corresponding no-storage control groups (Tables 2 and 3) ▶ ▶ . Although the use of Stanford solution for donor heart perfusion and cold preservation resulted in significantly greater parenchymal rejection and GAD scores relative to grafts perfused in 0.9% saline (Table 3) ▶ , the Stanford and saline solution groups were transplanted as distinct cohorts at separate times, and are not strictly comparable. Inflammatory cell infiltration accompanied by more widespread myocyte necrosis (parenchymal rejection) occurred by day 3 in all allografts and became more prominent by day 7. In both MHC I- and MHC II-mismatched allografts, parenchymal rejection diminished at later stages (by 4 to 8 weeks), whereas GAD progressed by 4 to 8 weeks. 15 Prolonged cold ischemia did not significantly affect the severity nor change the time course of parenchymal rejection in any of the allograft combinations (Tables 2 and 3) ▶ ▶ .
Table 2.
GAD and Parenchymal Rejection Score in Allografts Stored in Saline
| Time after transplantation | 4 weeks | 8 weeks | 12 weeks | |||
|---|---|---|---|---|---|---|
| GAD | PR | GAD | PR | GAD | PR | |
| bm1 → B/6 | ||||||
| No storage | ND | ND | 1.21 ± 0.43 (7) | 1.93 ± 0.30 (7) | 0.93 ± 0.40 (7) | 1.63 ± 0.39 (8) |
| 4-hour cold ischemia | ND | ND | 1.92 ± 0.57 (6) | 1.86 ± 0.26 (7) | 0.64 ± 0.21 (7)* | 1.07 ± 0.13 (7) |
| bm12 → B/6 | ||||||
| No storage | 1.33 ± 0.63 (6) | 1.75 ± 0.50 (6) | 1.36 ± 0.51 (7) | 2.71 ± 0.26 (7) | ND | ND |
| 4-hour cold ischemia | 1.43 ± 0.58 (7) | 2.29 ± 0.53 (7) | 1.43 ± 0.40 (7) | 2.43 ± 0.23 (7) | ND | ND |
Values are expressed as mean ± SEM. PR, parenchymal rejection; GAD, graft coronary artery disease. The scales (1–4) are described in the Methods.
*p < 0.05 vs. 4-hour cold ischemia (bm1 to B/6) allograft group at 8 weeks.
ND, not done.
Table 3.
GAD and Parenchymal Rejection Score in Allografts Stored in Stanford Solution
| Time after transplantation | 8 weeks | 12 weeks | ||
|---|---|---|---|---|
| GAD | PR | GAD | PR | |
| bm1 → B/6 | ||||
| No storage | ND | ND | 1.43 ± 0.47 (7) | 2.00 ± 0.19 (7) |
| 4-hour cold ischemia | ND | ND | 2.42 ± 0.52 (6) | 1.93 ± 0.37 (7) |
| bm12 → B/6 | ||||
| No storage | 2.86 ± 0.21 (7) | 3.57 ± 0.17 (7) | ND | ND |
| 4-hour cold ischemia | 3.08 ± 0.54 (6) | 3.25 ± 0.21 (6) | ND | ND |
Values are expressed as mean ± SEM.
PR, parenchymal rejection; GAD, graft coronary artery disease. There was no statistically significant difference in GAD and parenchymal rejection scores between no storage and 4-hour cold ischemia Stanford solution groups in each donor strain. Significantly greater values (p < 0.05) than those of the corresponding saline groups (see Table) are indicated by underlining.
ND, not done.
Expression of Inflammatory Cytokines in Grafts With and Without Prolonged Cold Ischemia
Figure 2 ▶ shows representative RPA gels from mRNA derived from isografts or total mismatched allografts; the associated densitometric analyses of these gels are shown in Figure 3 ▶ . Normal hearts contained minimal levels of mRNAs encoding inflammatory cytokines such as IL-6 and IL-1β. However, expression of the mRNAs for these cytokines increased by 4 hours after transplantation in all grafts. This rise is attributable to the transplantation surgery which entails an obligatory 30 minutes of warm ischemia. Cold ischemia increased expression of IL-6 and IL-1β mRNAs in all donor and recipient strain combinations tested. TGF-β mRNA was expressed weakly in normal hearts, and increased in all grafts within 24 hours of transplantation. Cold ischemia (4 hours) enhanced this increase in TGF-β by 24 hour post-transplantation, reaching statistically significant differences between no-storage group and 4-hour cold ischemia group in TGF-β1, β2 and β3 in isografts, and in TGF-β2 and β3 in allografts. In isografts, TGF-β1 and β2 mRNA decreased by day 7 and there was no significant difference between the no-storage and 4-hour cold ischemia groups. Thus, in isografts, the effects of prolonged cold ischemia (relative to no-storage) on cytokine mRNA expression are evident only at early time points (1 day) after transplant.
Figure 2.

Representative panels of RPA from transplanted hearts. Fifteen micrograms of total RNA from normal non-transplanted hearts, or from hearts explanted at 0 hour, 4 hour, 24 hour, 3 days, or 7 days after transplantation were analyzed using the multiprobe RPA kits mCK-3b (containing DNA templates for TNF-β, LT-β, TNF-α, IL-6, IFN-γ, IFN-β, TGF-β1, TGF-β2, TGF-β3, MIF, L32, and GAPDH) and mCK-2b (containing DNA templates for IL-12p35, IL-12p40, IL-10, IL-1α, IL-1β, IL-1Ra, IL-18, IL-6, IFN-γ, MIF, L32, and GAPDH). The lanes on the far left show the undigested labeled probes that serve as size references, and the lanes on the far right show the results of RPA using a control RNA sample that was supplied by a manufacturer of the kit. Left panel: Representative gel images of B/6 isografts experiment analyzed by mCK-3b. Right panel: Representative gel images of BALB/c to B/6 allografts experiment analyzed by mCK-3b.
Figure 3.
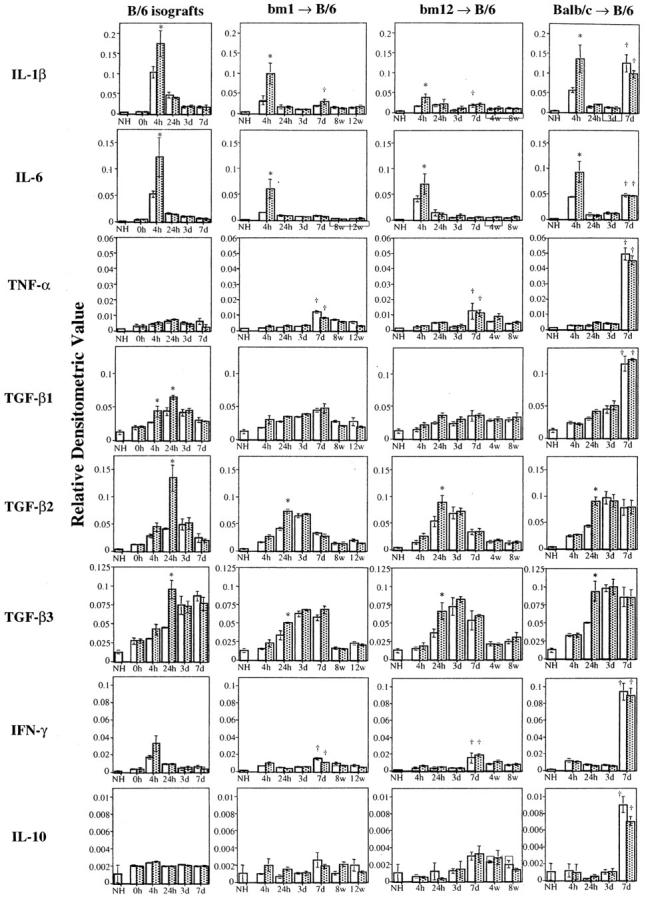
The results of densitometric analysis of cytokine mRNA expression. Volume integrations of the mRNA bands for each cytokine were analyzed using phosphoimaging analyzer and normalized against the bands for GAPDH in the corresponding lane. Graphs showing the relative densitometric values of IL-1β, IL-6, TNF-α, TGF-β1, TGF-β2, TGF- β3, IFN-γ, or IL-10 mRNA expression in isografts and MHC I-disparate (bm1 to B/6), MHC II-disparate (bm12 to B/6), or totally mismatched (B/c to B/6) allografts (from left to right). Three to four animals per each group were analyzed. Each relative densitometric value is expressed as mean ± SEM. *P < 0.05 vs no storage group at the same time point, †P < 0.05 vs each corresponding group at day 3. Open bars, no storage control groups; hatched bars, 4-hour cold ischemia groups.
However, in allografts, TGF-β1 remained elevated at day 7, while TGF-β2 and β3 tended to decrease. Beginning at day 7 post-transplant, mRNAs encoding the T cell cytokine IFN-γ and another proinflammatory cytokine TNF-α were markedly increased in all allografts and, particularly in total allomismatched allografts; transcripts for IL-1β, IL-6, TGF-β1, and IL-10 were also strongly expressed. In contrast to the impact of prolonged cold ischemia on the immediate expression of IL-6, IL-1β, and TGF-β in isografts, there were no differences between the 4-hour cold ischemia group and the control no-storage group in the levels of IL-10 or IFN-γ mRNA at early time points (<3 days). Moreover, there were no significant differences between the 4-hour cold ischemia group and the control no-storage group in the mRNA transcripts of any cytokines measured beginning with the peak of mRNA expression at day 7. The temporal changes of T cell cytokine expression were similar for MHC I, MHC II, or total allogeneic mismatches (B/c hearts into B/6 recipients); the intensity of cytokine expression was greatest in totally mismatched allografts at day 7.
Cell Adhesion Molecules Expression in the Grafts
Normal hearts as well as both isografts and allografts showed no or little staining for P-selectin and E-selectin at any time point examined (Tables 4 and 5) ▶ ▶ . Similarly, the endothelium of large coronary vessels of normal non-transplanted hearts expressed little or no ICAM-1 and VCAM-1. However, the endothelial expression of these molecules increased in all grafts within 24 hours of transplant. Relative to no-storage control hearts, both isografts and allografts in the 4-hour cold ischemia group showed increased expression of ICAM-1 within 4 hours of transplantation. In isografts, the increases in ICAM-1 localized to the endothelium of epicardial and intramyocardial coronary vessels (Figure 4 ▶ , Table 4 ▶ ). ICAM-1 expression on infiltrating leukocytes appeared by day 3, and remained elevated through day 7. Endothelial VCAM-1 was also increased following transplantation, although cold ischemia did not, in general, accelerate expression. (Figure 5 ▶ , Table 5 ▶ ). Thus, 4 hours of cold ischemia resulted in increased endothelial ICAM-1 expression within 4 hours of transplantation, with no significant differences seen for any other adhesion molecules between no-storage and 4-hour cold ischemia groups.
Table 4.
Expression of Cell Adhesion Molecules in Isografts
| Time after transplantation | Normal heart | B/6 isografts | |||
|---|---|---|---|---|---|
| 4 hours | 24 hours | 3 days | 7 days | ||
| ICAM-1 | |||||
| No storage | − | − | + | + | + |
| 4-hour cold ischemia | + | + | + | + | |
| VCAM-1 | |||||
| No storage | − | + | + | − | − |
| 4-hour cold ischemia | + | + | − | − | |
| P-selectin | |||||
| No storage | − | − | − | − | − |
| 4-hour cold ischemia | − | − | − | − | |
| E-selectin | |||||
| No storage | − | − | − | − | − |
| 4-hour cold ischemia | − | − | − | − | |
Immunohistochemical staining of cell adhesion molecules was graded as: −, not present or focal weak staining; +, diffuse or strong staining. Three to four samples were examined for each group. Details are described in Materials and Methods.
Table 5.
Expression of Cell Adhesion Molecules in Allografts
| Time after transplantation | bm1 → B/6 Allografts | bm12 → B/6 Allografts | B/c → B/6 Allografts | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 hours | 24 hours | 3 days | 7 days | 4 hours | 24 hours | 3 days | 7 days | 4 hours | 24 hours | 3 days | 7 days | |
| ICAM-1 | ||||||||||||
| No storage | − | + | + | + | − | + | + | + | − | + | + | + |
| 4-hour cold ischemia | + | + | + | + | + | + | + | + | + | + | + | + |
| VCAM-1 | ||||||||||||
| No storage | + | + | + | + | − | + | + | + | + | + | + | + |
| 4-hour cold ischemia | + | + | + | + | + | + | + | + | + | + | + | + |
| P-selectin | ||||||||||||
| No storage | − | − | − | − | − | − | − | − | − | − | − | − |
| 4-hour cold ischemia | − | − | − | − | − | − | − | − | − | − | − | − |
| E-selectin | ||||||||||||
| No storage | − | − | − | − | − | − | − | − | − | − | − | − |
| 4-hour cold ischemia | − | − | − | − | − | − | − | − | − | − | − | − |
Immunohistochemical staining of cell adhesion molecules was graded as: −, not present or focal weak staining; +, diffuse or strong staining. Three samples at 4 or 24 hours, or four samples at day 3 or day 7 were examined for each group. Details are described in Materials and Methods.
Figure 4.
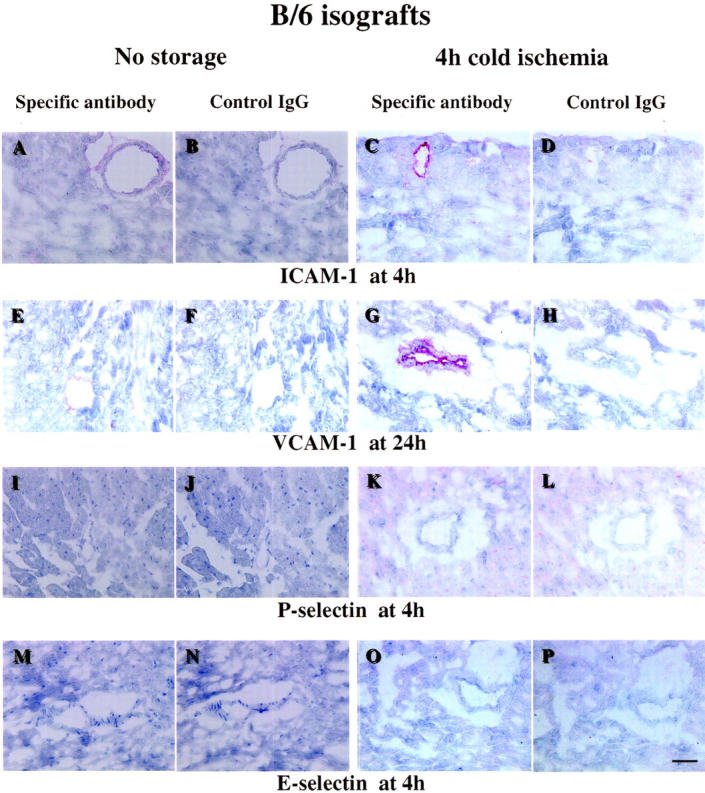
Immunohistochemical staining of representative sections from B/6 isografts. Scale bar, 50 μm. A: ICAM-1 staining of a section from a B/6 isograft in no storage group, at 4 hours after transplantation. ICAM-1 is weakly expressed in the endothelium of coronary vessels. B: Negative control staining for A using isotype-matched hamster IgG instead of the anti-ICAM-1 antibody. C: ICAM-1 staining of a section from a B/6 isograft in the cold ischemia group, 4 hours after transplantation. ICAM-1 was strongly expressed in isografts following 4-hour cold ischemia. D: Negative control staining for C. E: VCAM-1 staining of a section from a B/6 isograft in no storage group, 24 hours after transplantation. Endothelial VCAM-1 is increased by the transplant procedure, compared with normal heart. F: Negative control staining for E using isotype-matched rat IgG. G: VCAM-1 staining of a section from a B/6 isograft in 4-hour cold ischemia group, 24 hours after transplantation. Positive staining for VCAM-1 expression was seen in the majority of the vessels in isografts following 4-hour cold ischemia. H: Negative control staining for G. I: P-selectin staining of a section from a B/6 isograft in no storage group, 4 hours after transplantation. J: Negative control staining for I using isotype-matched rat. K: P-selectin staining of a section from a B/6 isograft in 4-hour cold ischemia group, 4 hours after transplantation. L: Negative control staining for K. M: E-selectin staining of a section from a B/6 isograft in no storage group, 4 hours after transplantation. N: Negative control staining for M. O: E-selectin staining of a section from a B/6 isograft in 4-hour cold ischemia group, 4 hours after transplantation. P: Negative control staining for O. P-selectin and E-selectin are sparsely expressed in isografts at any time point.
Figure 5.

Immunohistochemical staining for adhesion molecules. Representative sections from bm1, bm12, or B/c to B/6 allografts 7 days after transplant, without 4-hour cold storage. Scale bar, 50 μm. A: ICAM-1 staining of a section from a bm1 to B/6 allograft. ICAM-1 expression is increased on the endothelium and on graft infiltrating mononuclear leukocytes. B: Negative control staining for A using isotype-matched hamster IgG. C: VCAM-1 staining of a section from a bm1 to B/6 allograft. VCAM-1 expression is increased on the endothelium and on some of graft-infiltrating mononuclear leukocytes. D: Negative control staining for C using isotype-matched rat IgG. E: ICAM-1 staining of a section from a bm12 to B/6 allograft. F: Negative control staining for E. G: VCAM-1 staining of a section from a bm12 to B/6 allograft. H: Negative control staining for G. I: ICAM-1 staining of a section from a B/c to B/6 allograft. J: Negative control staining for I. K: VCAM-1 staining of a section from a B/c to B/6 allograft. L: Negative control staining for K. Strong expression of both ICAM-1 and VCAM-1 was observed at day 7 for all allografts. Four-hour cold ischemia did not have any significant effect on the expression of these cell adhesion molecules at day 7 post-transplant. Expression of both ICAM-1 and VCAM-1 was most pronounced in B/c to B/6 allografts, reflecting the most severe acute rejection.
Discussion
Prolonged cold ischemia before heart transplantation is one plausible non-immunological risk factor 6,7 for GAD, accounting for selective involvement of the graft vessels, with sparing of the native donor arteries. Despite many clinical studies, however, it remains controversial whether prolonged cold ischemia can trigger or exacerbate GAD. 19 A few experimental studies 9,13 using heart transplantation in rats have also reported conflicting results regarding the exacerbating effects of prolonged cold ischemia on the development of GAD in allografts. However, the mechanisms underlying the induction or accentuation of GAD by cold ischemia have not been clarified.
The availability of congenic strains in mice permit isograft controls that rigorously eliminate immunological contributions. Our results indicate that cold ischemia (4 hours) alone is sufficient to initiate the development of mild GAD in isografts within 8 to 12 weeks of transplantation. Molecular and cellular pathways potentially involved in the pathogenesis of GAD include transiently augmented expression of inflammatory cytokines such as IL-6, IL-1β, and TGF-β, as well as increased ICAM-1 adhesion molecule expression. Notably, the increased cytokine and ICAM-1 expression seen in isografts were comparable to those seen in all allograft combinations from 4 hours to 3 days post-transplantation, a time when the transient effects of perioperative ischemia will predominate. Moreover, cold ischemia in isografts had comparable effects to cold ischemia in all allograft combinations in the same time period. By day 7 post-transplant, when the allo-responses predominate, isografts show diminished cytokines and adhesion molecules relative to allografts. In addition, 4 hours of cold ischemia did not augment the severity of parenchymal rejection or GAD in non-immunosuppressed MHC I- or MHC II-mismatched allografts. Regardless of the degree of alloantigen disparity (MHC I, MHC II, or total allomismatches), all cytokines examined (IFN-γ, IL-10, TGF-β, IL-1β, IL-6, and TNF) showed comparable tempo and level of mRNA expression in the cold ischemia and control groups in allografts beginning 3 days after transplantation. These results show that cold ischemia (4 hours) did not significantly affect either net helper T cell-mediated alloresponses (IFN-γ, IL-10) or antigen non-specific innate responses (IL-1β, IL-6, TGF-β, TNF). Abundant ICAM-1 and VCAM-1 expression occurred in the arterial walls and parenchyma in all allografts at day 7 after transplantation with no difference between the cold ischemia and control groups, while levels of P-selectin and E-selectin were low, even in allografts.
A number of studies focusing on warm ischemia/reperfusion and myocardial tissue injury suggest important roles for expression of inflammatory cytokines and adhesion molecules in vascular cells and cardiac myocytes. 10-12 Some cytokines and adhesion molecules will promote inflammation and tissue destruction, 11,12,20 while others protect against tissue injury, including myocardial necrosis. 21,22 For example, IL-1β or IL-6 stimulate ICAM-1, VCAM-1 and E-selectin expression on cultured human umbilical vein endothelial cells. 23,24 Sources of IL-6 in the heart grafts include cardiac myocytes themselves as well as infiltrating inflammatory cells. 25,26 Therefore, endothelial activation by inflammatory cytokines can increase adhesion molecule expression and stimulate leukocyte adhesion to coronary endothelial cells, potentially augmenting coronary arterial injury, and triggering a response to injury cascade. 27 As observed in this study, enhanced expression of IL-1β and IL-6 after prolonged cold ischemia may transiently increase adhesion molecule expression, suggesting a mechanism for the development of mild GAD in isografts after prolonged cold ischemia.
Contrary to the proinflammatory cytokines such as IL-1β and IL-6, TGF-β may have a protective effect in the setting of ischemia and reperfusion. 28 TGF-β can also protect the endothelium and myocardium indirectly by decreasing the release of TNF-α and reactive oxygen species. In addition, TGF-β1, the dominant member of TGF-β family produced by leukocytes, suppresses alloimmune responses. For instance, TGF-β1 inhibits IL-2-dependent T cell proliferation, 29 IFN-γ production, 30 cytotoxic T cell generation, 31 and responsiveness of antigen presenting cells to IFN-γ, 32 in vitro. During rejection, rat cardiac allografts have elevated levels of TGF-β, 33 and moreover, systemic administration of recombinant TGF-β1 prolonged cardiac allograft survival in rats. 34 Because frequent episodes of acute rejection and immunological injury to vascular cells can contribute to early development of GAD, 4 immunosuppression via TGF-β1 could mitigate GAD.
Conversely, other biological activities of TGF-β might exacerbate the development of GAD. The mature intimal lesion of GAD consists of accumulated extracellular matrix and vascular smooth muscle cells (SMC), along with infiltrating macrophages and T lymphocytes. 35-37 TGF-β increases extracellular matrix production such as proteoglycans and collagens by SMC. 36,38 Although the mitogenic effects of TGF-β on SMC vary depending on the conditions, 39,40 local expression of TGF-β1 gene in porcine arteries in vivo resulted in the development of fibrocellular intimal hyperplasia. 41 This observation suggests that, in the absence of other major factors causing arterial injury, TGF-β could induce vascular sclerotic lesion formation. These effects of TGF-β on fibrocellular proliferation could partially explain the development of relatively mild intimal thickening at 8 to 12 weeks post-transplant in isografts from the 4-hour cold ischemia group, where there was early elevated TGF-β expression relative to no-storage controls.
In MHC I- and MHC II-mismatched allografts, we found no differences between the no-storage control group and the 4-hour cold ischemia group in either parenchymal rejection or GAD grades at 4 to 12 weeks post-transplantation. This observation agrees with the results from RNase protection assays demonstrating that prolonged cold ischemia did not augment the already significant helper T cell-mediated alloimmune responses (as assessed by the induction of IFN-γ and IL-10 genes). Thus, prior enhanced inflammation due to prolonged cold ischemia does not significantly affect vascular injury due to alloimmune responses.
Histological examination of bm1 allografts subjected to 4-hour cold storage showed a GAD score at 12 weeks that was significantly decreased relative to the 8 week GAD score (P = 0.046). Because GAD scores were not significantly different between 8 weeks and 12 weeks in no-storage bm1 allografts, it cannot be concluded that GAD regressed in bm1 allografts. Rather, the maturation of GAD lesions from a highly cellular to a more fibrotic intima 15 with vascular remodeling will likely be associated with an apparent diminution in the severity of GAD. Our results showing the decreased GAD scores for MHC 1-disparate allografts at 12 weeks relative to 8 weeks agree with the report by Russell et al, 42 in an MHC I-disparate transplant model.
We also evaluated the effects of the Stanford preservation solution on development of GAD and parenchymal rejection, comparing a no-storage control group with a 4-hour cold ischemia group. We used the Stanford preservation solution because a previous clinical study suggested that its use reduced the incidence of GAD, compared with the University of Wisconsin high potassium solution. 16 The Stanford solution experiments were performed as a distinct cohort from saline experiments, at a later time, and with their own set of internal controls. The histological examination showed no reduction in the development of GAD in isografts following prolonged cold ischemia in the Stanford solution. Similarly, GAD and parenchymal rejection did not differ between the no-storage group and the 4-hour cold ischemia group in allografts from the Stanford solution experiments. In this study, the use of the Stanford solution did result in apparently greater parenchymal rejection and GAD than that seen in saline incubated hearts. However, we cannot draw any conclusions about the relative efficacy of saline or Stanford solution preservation because the two groups were transplanted as distinct cohorts at separate times, and with different baseline levels of rejection and GAD for the control hearts not exposed to prolonged cold ischemia. Nevertheless, the results do not indicate any unique beneficial effects of Stanford cardioplegic preservation solution for the prevention of GAD.
The murine heterotopic heart transplantation model used here has certain limitations. First, heterotopic transplantation causes load reduction of the implanted hearts, particularly in the left ventricle, while clinical orthotopic transplantation does not. These differences could affect the metabolic demand of myocardium or coronary vessel tissues under ischemic condition, causing modification of the effects of cold ischemia on the development of GAD. To our knowledge, however, there are no applicable data for the assessment of the metabolic demand of ischemic mouse hearts or relative susceptibility to ischemic injury between human and mice. Secondly, the donor mice, regardless of strain, had normal arteries pre-transplantation. In contrast, the arteries of human donor hearts frequently have intimal thickening and not infrequently pre-existing atheroma. Hence, the arterial substrates in human heart transplantation may vary from the simple, reproducible, and well-controlled situation in the mouse. Finally, although we did examine a variety of MHC-mismatched strain combinations, we did not use any immunosuppression in this study. Thus, it is possible that the effects of cold ischemia might be accentuated under conditions where the alloimmune response is tempered. It is important to note, however, that the changes in cytokine and adhesion molecule expression due either to surgery or to cold ischemia were comparable between isografts and all allograft combinations, up to 3 days post-transplant, when effects of ischemia will predominate. Moreover, subsequent cytokine and adhesion molecule expression, parenchymal rejection and GAD were all unaffected by the presence or absence of cold ischemia. Thus, given that cold ischemia induced only minimal GAD in isografts, it seems unlikely that cold ischemia alone materially accentuates the GAD induced by alloresponses. Although immunosuppression with the mouse allograft model may be achieved with anti-CD4 and anti-CD8 antibody administration, 15,42 we elected not to use these treatments because they also dramatically affect macrophage recruitment and activation during the early time periods when ischemic effects will be greatest. Cyclosporine immunosuppression is also not as effective in mice as in humans. 43
The present experimental results indicate that an ischemic insult similar to that allowed maximally in human heart transplantation causes only slight arteriopathy, and does not exacerbate the much more profound intimal response due to allogeneic stimulation. Our results indicate distinct molecular mechanisms underlying the mild and limited intimal thickening in transiently ischemic isografts versus the severe arteriopathy induced by allo- responses. We conclude that cold ischemic injury can induce GAD in the absence of alloimmune responses, but does not accentuate antigen-dependent mechanisms for the development of GAD in transplanted mouse hearts.
Acknowledgments
We thank Ms. Eugenia Shvartz, Mr. Gregory Russo, Ms. Krista Condon, Ms. Sarah E. Cole, Ms. Elissa Simon-Morrissey, Ms. Karen E. Williams, and Dr. Jun-Ichi Suzuki, Brigham and Women’s Hospital, for their skillful assistance.
Footnotes
Address reprint requests to Dr. Richard N. Mitchell, M.D., Ph.D., Immunology Research Division, Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, 221 Longwood Avenue, Boston, MA 02115. E-mail: rmitchell@rics.bwh.harvard.edu.
Supported by National Institutes of Health grant RO1 HL 43364–07 (to P.L. and R.N.M.). Yutaka Furukawa is a recipient of Research Fellowship Grant from Japan Heart Foundation.
References
- 1.Uretsky BF, Murali S, Reddy PS, Rabin B, Lee A, Griffith BP, Hardesty RL, Trento A, Bahnson HT: Development of coronary artery disease in cardiac transplant patients receiving immunosuppressive therapy with cyclosporine and predonisone. Circulation 1987, 76:827-834 [DOI] [PubMed] [Google Scholar]
- 2.Paul LC, Fellstrom B: Chronic vascular rejection of the heart and the kidney; have rational treatment options emerged? Transplantation 1992, 53:1169-1179 [DOI] [PubMed] [Google Scholar]
- 3.Gao SZ, Hunt SA, Schroeder JS, Alderman EL, Hill IR, Stinson EB: Early development of accelerated graft coronary artery disease: risk factors and course. J Am Coll Cardiol 1996, 28:673-679 [DOI] [PubMed] [Google Scholar]
- 4.Hornick P, Smith J, Pomerance A, Mitchell A, Banner N, Rose M, Yacoub M: Influence of acute rejection episodes, HLA matching, and donor/recipient phenotype on the development of “early” transplant-associated coronary artery disease. Circulation 1997, 96(Suppl II):148-153 [PubMed] [Google Scholar]
- 5.Weis M, von Scheidt W: Cardiac allograft vasculopathy: a review. Circulation 1997, 96:2069-2077 [DOI] [PubMed] [Google Scholar]
- 6.Johnson MR: Transplant coronary disease: nonimmunologic risk factors. J Heart Lung Transplant 1992, 11:S124-S132 [PubMed] [Google Scholar]
- 7.Day JD, Rayburn BK, Gaudin PB, Baldwin WM 3rd, Lowenstein CJ, Kasper EK, Baughman KL, Baumgartner WA, Hutchins GM, Hruban RH: Cardiac allograft vasculopathy: the central pathogenetic role of ischemia-induced endothelial cell injury. J Heart Lung Transplant 1995, 14:S142–S149 [PubMed]
- 8.Gaudin PB, Rayburn BK, Hutchins GM, Kasper EK, Baughman KL, Goodman SN, Lecks LE, Baumgartner WA, Hruban RH: Peritransplant injury to the myocardium associated with the development of accelerated arteriosclerosis in heart transplant recipients. Am J Surg Pathol 1994, 18:338-346 [DOI] [PubMed] [Google Scholar]
- 9.Schmid C, Heemann U, Tilney NL: Factors contributing to the development of chronic rejection in heterotopic rat heart transplantation. Transplantation 1997, 64:222-228 [DOI] [PubMed] [Google Scholar]
- 10.Kukielka GL, Hawkins HK, Michael L, Manning AM, Youker K, Lane C, Entman ML, Smith CW, Anderson DC: Regulation of intercellular adhesion molecule-1 (ICAM-1) in ischemic and reperfused canine myocardium. J Clin Invest 1993, 92:1504-1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wyble CW, Desai TR, Clark ET, Hynes KL, Gewertz BL: Physiologic concentrations of TNF-α and IL-1β released from reperfused human intestine up-regulate E-selectin and ICAM-1. J Surg Res 1996, 63:333-338 [DOI] [PubMed] [Google Scholar]
- 12.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA: Role of tumor necrosis factor-α in the pathophysiologic alterations after hepatic ischemia-reperfusion injury in the rat. J Clin Invest 1990, 85:1936-1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight RJ, Dikman S, Liu H, Martinelli GP: Cold ischemic injury accelerates the progression to chronic rejection in a rat cardiac allograft model. Transplantation 1997, 64:1102-1107 [DOI] [PubMed] [Google Scholar]
- 14.Corry RJ, Winn HJ, Russell PS: Primary vascularized allografts of hearts in mice. Transplantation 1973, 16:344-350 [DOI] [PubMed] [Google Scholar]
- 15.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P: Interferon-γ deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest 1997, 100:550-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drinkwater DC, Rudis E, Laks H, Ziv E, Marino J, Stein D, Ardehali A, Aharon A, Moriguchi J, Kobashigawa J: University of Wisconsin solution versus Stanford cardioplegic solution and the development of cardiac allograft vasculopathy. J Heart Lung Transplant 1995, 14:891-896 [PubMed] [Google Scholar]
- 17.Billingham ME, Cary NR, Hammond ME, Kemnitz J, Marboe C, McCallister HA, Snovar DC, Winters GL, Zerbe A: A working formation for the standardization of nomenclature in the diagnosis of heart and lung rejection: heart rejection study group. J Heart Lung Transplant 1990, 9:587-593 [PubMed] [Google Scholar]
- 18.Nagano H, Libby P, Taylor MK, Hasegawa S, Stinn JL, Becker G, Tilney NL, Mitchell RN: Coronary arteriosclerosis after T-cell-mediated injury in transplanted mouse hearts: role of interferon-γ. Am J Pathol 1998, 152:1187-1197 [PMC free article] [PubMed] [Google Scholar]
- 19.Escobar A, Ventura HO, Stapleton DD, Mehra MR, Ramee SR, Collins TJ, Jain SP, Smart FW, White CJ: Cardiac allograft vasculopathy assessed by intravascular ultrasonography and nonimmunologic risk factors. Am J Cardiol 1994, 74:1042-1046 [DOI] [PubMed] [Google Scholar]
- 20.Eppihimer MJ, Granger DN: Ischemia/reperfusion-induced leukocyte-endothelial interactions in post-capillary venules. Shock 1997, 8:16-25 [DOI] [PubMed] [Google Scholar]
- 21.Lefer AM, Tsao P, Aoki N, Palladino MA, Jr: Mediation of cardioprotection by transforming growth factor-beta. Science 1990, 249:61-64 [DOI] [PubMed] [Google Scholar]
- 22.Lefer AM, Ma XL, Weyrich AS, Scalia R: Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proc Natl Acad Sci USA 1993, 90:1018-1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weill D, Wautier JL, Dosquet C, Wautier MP, Carreno MP, Boral B: Monocyte modulation of endothelial leukocyte adhesion molecules. J Lab Clin Med 1995, 125:768-774 [PubMed] [Google Scholar]
- 24.Watson C, Whittaker S, Smith N, Vora AJ, Dumonde DC, Brown KA: IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin Exp Immunol 1996, 105:112-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML: Induction of interleukin-6 synthesis in the myocardium: potential role in postreperfusion inflammatory injury. Circulation 1995, 92:1866-1875 [DOI] [PubMed] [Google Scholar]
- 26.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML: Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99:546-551 [DOI] [PubMed] [Google Scholar]
- 27.Ross R: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362:801-809 [DOI] [PubMed] [Google Scholar]
- 28.Nose PS: Cytokines and reperfusion injury. J Card Surg 1993, 8(Suppl):305-308 [DOI] [PubMed] [Google Scholar]
- 29.Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS: The production of TGF-β by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med 1986, 163:1037-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Espevik T, Figari IS, Shalaby MR, Lackides GA, Lewis GD, Shepard HM, Palladino MA, Jr: Inhibition of cytokine production by cyclosporin A and transforming growth factor beta. J Exp Med 1987, 166:571-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ranges GE, Figari IS, Espevik T, Palladino MA, Jr: Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med 1987, 166:991-998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czarniecki CW, Chiu HH, Wong GHW, McCabe SM, Palladino M: Transforming growth factor-β1 modulates the expression of class II histocompatibility antigen on human cells. J Immunol 1988, 140:4217-4223 [PubMed] [Google Scholar]
- 33.Waltenberger J, Wanders A, Fellström B, Miyazono K, Heldin C-H, Funa K: Induction of transforming growth factor-β during cardiac allograft rejection. J Immunol 1993, 151:1147-1157 [PubMed] [Google Scholar]
- 34.Raju GP, Belland SE, Eisen HJ: Prolongation of cardiac allograft survival with transforming growth factor-β1 in rats. Transplantation 1994, 58:392-395 [PubMed] [Google Scholar]
- 35.Libby P, Tanaka H: The pathogenesis of coronary arteriosclerosis (“chronic rejection”) in transplanted hearts. Clin Transplant 1994, 8:313-318 [PubMed] [Google Scholar]
- 36.Roberts AB, Sporn MB: The transforming growth factor-βs. Sporn MB Roberts AB eds. Peptide Growth Factors and Their Receptors. 1990, :pp 421-472 Germany, Springer, Heidelberg [Google Scholar]
- 37.Salomon RN, Hughes CC, Schoen FJ, Payne DD, Pober JS, Libby P: Human coronary transplantation-associated arteriosclerosis: evidence for a chronic immune reaction to activated graft endothelial cells. Am J Pathol 1991, 138:791-798 [PMC free article] [PubMed] [Google Scholar]
- 38.Amento EP, Ehsani N, Palmer H, Libby P: Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb 1991, 11:1223-1230 [DOI] [PubMed] [Google Scholar]
- 39.Stouffer GA, Owens GK: TGF-β promotes proliferation of cultured SMC via both PDGF-AA-dependent and PDGF-AA-independent mechanisms. J Clin Invest 1994, 93:2048-2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grainger DJ, Kemp PR, Witchell CM, Weissberg PL, Metcalfe JC: Transforming factor β decreases the rate of proliferation of rat vascular smooth muscle cells by extending the G2 phase of the cell cycle and delays the rise in cyclic AMP before entry into M phase. Biochem J 1994, 299:227-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nabel EG, Shum L, Pompili VJ, Yang ZY, San H, Shu HB, Liptay S, Gold L, Gordon D, Derynck R, Nabel GJ: Direct transfer of transforming growth factor β1 gene into arteries stimulates fibrocellular hyperplasia. Proc Natl Acad Sci USA 1993, 90:10759-10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell PS, Chase CM, Winn HJ, Colvin RB: Coronary atherosclerosis in transplanted mouse heart: I. time course and immunogenetic and immunopathological considerations. Am J Pathol 1994, 144:260-274 [PMC free article] [PubMed] [Google Scholar]
- 43.Ochiai T, Nakajima K, Sakamoto K, Nagata M, Gunji Y, Asano T, Isono K, Sakamaki T, Hamaguchi K: Comparative studies on the immunosuppressive activity of FK506, 15-deoxyspergualin, and cyclosporine. Transplant Proc 1989, 21:829-832 [PubMed] [Google Scholar]