

Abstract
Susceptibility to lead toxicity in MT-null mice and cells, lacking the major forms of the metallothionein (MT) gene, was compared to wild-type (WT) mice or cells. Male MT-null and WT mice received lead in the drinking water (0 to 4000 ppm) for 10 to 20 weeks. Lead did not alter body weight in any group. Unlike WT mice, lead-treated MT-null mice showed dose-related nephromegaly. In addition, after lead exposure renal function was significantly diminished in MT-null mice in comparison to WT mice. MT-null mice accumulated less renal lead than WT mice and did not form lead inclusion bodies, which were present in the kidneys of WT mice. In gene array analysis, renal glutathione S-transferases were up-regulated after lead in MT-null mice only. In vitro studies on fibroblast cell lines derived from MT-null and WT mice showed that MT-null cells were much more sensitive to lead cytotoxicity. MT-null cells accumulated less lead and formed no inclusion bodies. The MT-null phenotype seems to preclude lead-induced inclusion body formation and increases lead toxicity at the organ and cellular level despite reducing lead accumulation. This study reveals important roles for MT in chronic lead toxicity, lead accumulation, and inclusion body formation.
Lead is widely recognized as an important environmental toxicant that poses a substantial risk to the human population throughout the world. 1 Toxic effects of lead occur in multiple organ systems but particularly the developing nervous system of infants and children. 2,3 Renal effects are also common in adults with chronic lead exposure. 3 Lead produces renal tumors in rodents, and lead and inorganic lead compounds have been classified as possible human carcinogens. 4 However, the precise mechanisms of lead toxicity or carcinogenicity are incompletely defined.
A remarkable pathogenic feature of lead poisoning is the presence of inclusion bodies composed of lead-protein complex. 5-15 Blackman 6 first reported the formation of lead inclusion bodies in the 1930s in renal epithelial cells of lead-poisoned children. Since then, many investigators have reported inclusion body formation with lead exposure in humans and animals. 5,7,8 Lead-induced inclusion bodies are frequently nuclear, roughly spherical, and typically consist of an electron-dense core with a fibrillary network at the periphery. 2 These inclusion bodies, although common in the kidney, also form in cells of nervous tissue origin such as astrocytes, 9 neuroblastoma cells, 10 and in other cell types such as osteoclasts. 11 Metal analysis shows that lead is highly concentrated within the inclusion bodies. 12 Inclusion bodies may be protective in that, when lead accumulates in the inclusion bodies, it prevents injury to more sensitive cellular targets. 12,13 It is thought that inclusion bodies probably have an important role in the intracellular partitioning and, perhaps, transport and toxicity of lead. 14 Thus, the formation of lead-binding inclusion bodies may function to detoxify lead, 15 although this has yet to be definitively established.
Metallothionein (MT) is a low-molecular-weight metal-binding protein with one-third of its amino acids as cysteine. 16 These cysteinyl sulfhydryls coordinate a variety of metal atoms. 17 Various metals increased the concentration of MT in major organs of rats. 18 MT has been assigned pleiotropic roles from gene regulation to metal homeostasis, transport, and detoxification. 19 For instance, MT has been shown to play a protective role in cadmium-induced hepatotoxicity and nephrotoxicity. 20 Similarly, MT-I/II knock-out (MT-null) mice are more sensitive than wild-type (WT) mice to the nephrotoxicity produced by chronic exposure to cadmium and/or other inorganic metals. 21 MT is highly inducible by many metals, particularly zinc, cadmium, copper, and mercury, and clearly plays a role in mitigating the toxicity of these metals. 17 However, any mitigating role for MT in lead toxicity is still only poorly defined. In this regard, lead has been shown to induce the synthesis of MT in several instances, 19,22-24 which implicates, but does not definitively establish, a role in lead metabolism. On the other hand, this induction seems rather modest compared to many other metals and occurs only in the liver, 18 perhaps indicating stress-mediated induction. Others have found that lead is unable to stimulate the synthesis of MT in human blood lymphocytes. 25 Lead appears to bind to MT or MT-like proteins in human erythrocytes, 26 which suggests sequestration into a nonbioavailable, and thus nontoxic form. The presence of zinc-induced MT will modestly mitigate the toxicity of lead in cultured primary rat hepatocytes 27 and lead can avidly bind to MT ex vivo displacing zinc in the process. 28 Furthermore, the binding of lead to MT seems to reduce lead-induced inhibition of the enzyme δ-aminolevulinic acid dehydratase, at least ex vivo. 29 Although there are indications that MT mitigates lead toxicity, the data are far from convincing and additional work is warranted.
Therefore, the purpose of the present study was to investigate the role of MT in lead toxicity using genetically engineered systems. Initial studies used MT-null mice that are unable to produce the major forms of MT (MT-I and MT-II isoforms) and compared them to WT controls. Despite accumulating less renal lead, MT-null animals were significantly more sensitive than WT mice to the nephrotoxic effects of lead, as assessed by nephromegaly, renal function, and molecular evidence of a toxic response. Surprisingly, MT-null mice did not form inclusion bodies. Additional work in vitro showed MT-null cells similarly accumulated less lead but were still more sensitive to lead-induced cytotoxicity than WT cells. MT-null cells also did not form inclusion bodies after lead exposure, although they were common in WT cells. These data indicate that MT may play a role in lead toxicity and, possibly, in inclusion body formation. In addition, because the inability to produce MT seems to be related to enhanced susceptibility to lead toxicity, individuals that poorly express MT may have increased susceptibility to lead intoxication.
Materials and Methods
Chemicals and Materials
Lead nitrate, lead acetate, and glutamic acid were obtained from Sigma Chemical Company (St. Louis, MO). Nonradioactive cell proliferation assay kit was obtained from Promega (Madison, WI).
Animals and Treatments
Homozygous MT-I/II knock-out mice (129-Mt1tm/Bri, Mt2tm/Bri 129/SvPCJ background) 30 were obtained from Jackson Laboratories (Bar Harbor, ME). The homozygous mutants were mated inter se to maintain the line. Male MT-null mice and the corresponding WT mice were housed in an American Association for Acreditation of Laboratory Animal Care (AAALAC) accredited facility under conditions that met or exceeded recommendations outlined in the Guide for Care and Use of Laboratory Animals (National Institutes of Health Publication no. 86-23, 1985). Mice were provided food (NIH-31 diet; Zeigler Brothers, Gardners, PA) and water ad libitum. At 10 weeks of age, MT-null and WT mice were randomly divided into three treatment groups of 10 mice each and one control group of 20 mice. They were given acidified drinking water containing lead acetate at concentrations of 1000, 2000, or 4000 ppm lead. Control groups of mice received acidified drinking water. Animals were weighed weekly. Mice were killed after 10 weeks of treatment. Their kidneys were removed and weighed individually. For one-half of the controls (n = 10), and the 1000 and 2000 ppm groups, one kidney was fixed in 10% buffered formalin for histopathological analysis and a portion of the contralateral kidney was frozen in liquid nitrogen and used for subsequent lead determination. For the 2000 ppm group and one-half of the controls (n = 10), half of one kidney was frozen in liquid nitrogen for later RNA isolation. Both kidneys in the 4000-ppm group were used for histopathological analysis including quantitation of inclusion bodies. Formalin-fixed kidneys were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) for histological examination.
In a separate experiment, urine and orbital blood samples were taken from individual male MT-null and WT mice that were part of an on-going chronic carcinogenesis bioassay and had been exposed to 4000 ppm lead for 20 weeks. Blood urea nitrogen, blood creatinine, and total urinary protein were assessed as biomarkers of renal function and determined through a commercial clinical chemistry laboratory (Ani Lytics, Inc., Gaithersburg, MD).
Renal Lead Accumulation
Kidneys removed from WT and MT-null mice were digested in nitric acid (J.T. Baker, Philipsburg, NJ) overnight at 65°C. These digests were used for determination of the renal lead levels by graphite furnace atomic-absorption spectrophotometry with a Perkin-Elmer Model 5000 spectrophotometer.
Quantitation of Inclusion Bodies
The number of inclusion bodies was counted in three randomly selected H&E-stained kidney sections from each group. In each case a total of 200 randomly selected cells from the inner cortex were scored.
Microarray Analysis
The Atlas Mouse 1.2 cDNA expression microarray (1178 genes) was performed according to the manufacturers’ instructions. Briefly, 10 to 20 μg of total RNA isolated from MT-null control and lead-treated (2000 ppm) mouse kidneys were converted to [α-32P]-dATP-labeled cDNA probe using MMLV reverse transcriptase and Atlas Mouse Stress CDS primer mix (Clontech, Palo Alto, CA). The 32P-labeled cDNA probe was purified using chroma spin-200 columns, denatured in 0.1 mol/L NaOH, 10 mmol/L ethylenediaminetetraacetic acid at 68°C for 20 minutes, followed by neutralization with an equal volume of 1 mol/L NaH2PO4 for 10 minutes. The membrane was prehybridized with Ultrahyb (Ambion, Austin, TX) for 30 to 60 minutes at 42°C, followed by hybridization overnight at 42°C. Arrays were washed two times in 2× standard saline citrate/0.1% sodium dodecyl sulfate, 5 to 10 minutes each, and two times in 0.1× standard saline citrate/0.1% sodium dodecyl sulfate for 15 to 30 minutes. The arrays were then sealed in a plastic bag, and exposed to a phosphoimage screen or X-ray film. The images were analyzed densitometrically using AtlasImage software. The gene expression intensities were normalized with the sum of eight housekeeping genes on the array (40S ribosomal protein S29, 45-kd calcium-binding protein, β-actin, ornithine decarboxylase, myosin 1-α, G3PDH, hypoxanthine-guanine phosphoribosyltransferase, and phospholipase A2) except for ubiquitin (the hybrid intensity of ubiquitin was saturated). Means and SEM of four hybridizations were calculated for this analysis.
Cell Culture and Treatments
A cell line created from the embryonic cells of transgenic mice with a targeted disruption of MT-I/II genes (MT-null cells; also known as MT−/−), along with the corresponding WT control cells (WT; also known as MT+/+) from normal mice, were graciously supplied by Dr. John Lazo, University of Pittsburgh, Pittsburgh, PA. Cells were cultured in Dulbecco’s modified Eagle’s medium media containing 5% fetal bovine serum as described previously. 31 The precipitation of lead in the medium was controlled by complexing lead nitrate with glutamic acid in equimolar amounts, as detailed in a previous report. 32 Thus, cells were exposed to lead nitrate (200 μmol/L) with glutamic acid in equimolar amounts for the time specified throughout this study.
Metabolic Integrity Assay
Promega Cell Titer 96 Nonradioactive Cell Proliferation Assay kits were used to determine acute cytotoxicity of lead in MT-null and WT cells as defined by metabolic integrity. The assay measures the amount of formazan produced by metabolic conversion of Owen’s reagent [(3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)2H-tetrazolium, inner salt; MTS] by dehydrogenase enzymes found in the mitochondria of metabolically active cells. The quantity of formazan product, as measured by absorbance at 490 nm, is directly proportional to the number of living cells. A minimum of 4 replicates of 10,000 cells per well were plated in 96-well plates and allowed to adhere to the plate for 24 hours at which time the media was removed and replaced with media containing various concentrations of lead. Cells were then incubated for an additional 24 hours and cell viability was determined. 33 LC50 values were determined from analysis of the linear portion of the metabolic integrity curves and compared between WT and MT-null cells.
Electron Microscopy
WT and MT-null cells were treated with lead (200 μmol/L) for 48 hours. The cells were harvested by trypsinization and fixed overnight in 3% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.3. After primary fixation, the cells were rinsed in 0.1 mol/L of phosphate buffer for 15 minutes. Postfixation was done in 1% osmium tetroxide in 0.1 mol/L of phosphate buffer, pH 7.3, for 2 hours. The cells were rinsed again in the phosphate buffer for 15 minutes and were followed by treatment with an aqueous solution of 5% uranyl acetate for 2 hours. After dehydration in graded ethanol, the specimens were embedded in PolyBed resin. The resin blocks were cut at ∼90 nm, collected on coated grids, and stained with uranyl acetate and lead citrate. The examination of the grids was done using a Philips 400 electron microscope. 32
Determination of Cellular Lead Accumulation and Efflux
WT and MT-null cells were grown to ∼50% confluence, then the medium was removed and replaced with either fresh control medium or medium containing lead (200 μmol/L). Cells were harvested 24 hours later, counted, and pelleted by centrifugation. The cell pellets were digested overnight in 50% perchloric:nitric acid (2:1). These digests were used for determination of the amount of total lead that had accumulated after 24 hours of exposure. To estimate lead efflux, replicate sets of cells were washed after 24 hours of exposure to lead and allowed to incubate an additional 24 hours in fresh media. These cells were then digested and analyzed for lead. Total cellular lead levels were determined by graphite furnace atomic-absorption spectrophotometry using a Perkin-Elmer Model 5000 spectrophotometer and adjusted to cell numbers. Triplicate determinations were used for each data point.
Determination of MT Levels
WT cells were treated with lead (200 μmol/L) for 48 hours. Cells were harvested by trypsinization and resuspended at a density of 2.5 × 106/ml in 10 mmol/L Tris buffer (pH 7.4) at 4°C. Cells were then lysed by sonication on ice. Complete lysis was confirmed microscopically and cellular debris was removed by centrifugation (15 minutes, 16,000 × g). MT levels were determined in the supernatant using the Cd-hemoglobin method of Onosaka and colleagues 34 as modified by Eaton and Toal. 35
Statistical Analysis
Student’s t-test or analysis of variance with subsequent Dunnett’s test were used as appropriate. All values are expressed as mean ± SEM of three or more replications. Differences were considered significant at level of P < 0.05.
Results
Pathological and Functional Analysis of Kidneys from Lead-Exposed MT-Null and WT Mice
Male MT-null and WT mice received lead in drinking water (0 to 4000 ppm; 10 to 20 weeks) and renal pathology and function were assessed. Lead did not alter body weight in either MT-null or WT mice throughout the exposure period (data not shown). MT-null mice showed a dose-related nephromegaly indicative of renal toxicity, although the kidneys of WT mice were unaffected by lead (Figure 1) ▶ .
Figure 1.

Kidney weight analysis. MT-null and WT mice were given drinking water containing lead acetate at concentrations of 1000, 2000, or 4000 ppm for 10 weeks. Their kidneys were removed and weighed individually. Results are presented as the mean ± SEM (n = 10); a indicates a significant (P < 0.05) difference from appropriate dosage-matched WT; b indicates a significant (P < 0.05) difference from appropriate untreated control.
Although, no microscopically obvious pathological lesions occurred in lead-exposed kidneys, after chronic exposure to 4000 ppm lead in the drinking water MT-null mice showed evidence of diminished renal function when compared to WT mice. Specifically, there were significant (P ≤ 0.05) increases in blood creatinine (0.53 ± 0.03 mg/dl; mean ± SEM, n = 3 to 4) and total urinary protein (206 ± 14.1 mg/dl) in lead-treated MT-null mice when compared to similarly treated WT mice (blood creatinine = 0.33 ± 0.03 mg/dl; total urinary protein = 158 ± 1.76 mg/dl). Additionally, increases in blood urea nitrogen occurred in lead-exposed MT-null mice (30.3 ± 0.32 mg/dl) that approached significance (P = 0.062) when compared to WT mice (26.7 ± 1.86 mg/dl). This pattern of increases in blood urea nitrogen, blood creatinine, and total urinary protein is typically considered functional evidence of nephrotoxicity, and is consistent with reports on lead-induced nephrotoxicity.
Surprisingly, MT-null mice did not form renal lead-containing inclusion bodies, whereas inclusion bodies were common at all doses in WT mice (Figure 2) ▶ . These inclusion bodies were primarily nuclear. Quantitative analysis of cells from the inner cortex of lead-treated and control sections of kidneys showed that inclusion bodies were increased in a dose-dependent manner in WT mice, but, again were completely absent from MT-null animals (Table 1) ▶ .
Figure 2.
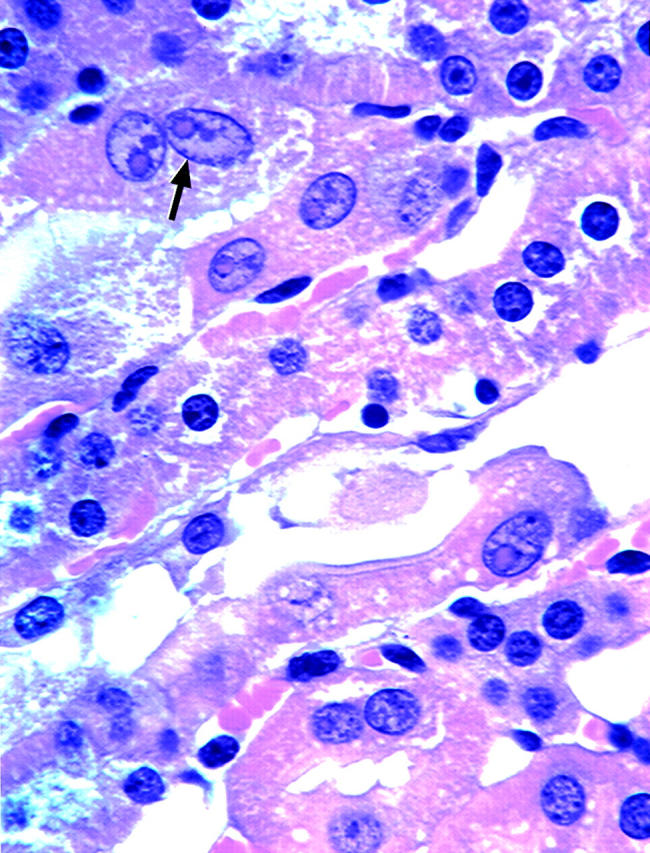
Lead-induced inclusion body formation in kidneys from WT mice. MT-null and WT mice were treated with lead as described in the legend to Figure 1 ▶ . Portions of kidneys were fixed in 10% buffered formalin. Acid-fast stain was used to determine the presence of lead-induced inclusion body in renal tubular cells. Arrows indicate typical karyomegaly of P3 proximal tubular cell in WT mice given lead (lead, 1000 ppm; H&E: original magnification, ×300).
Table 1.
Quantitation of Lead-Induced Inclusion Body Formation in Kidney From WT Mice
| Mouse strain | Lead dose (ppm, p.o.) | |||
|---|---|---|---|---|
| 0 | 1000 | 2000 | 4000 | |
| WT | N.D. | 10± 1 | 16± 1 | 21± 1 |
| MT-null | N.D. | N.D. | N.D. | N.D. |
WT and MT-null mice were given lead p.o. at 0, 1000, 2000, or 4000 ppm for 10 weeks and renal inclusion body formation was assessed. In each case 200 nuclei selected from random fields of the inner cortex of lead-treated and control sections of kidneys were scored. Data are given as the mean ± SEM (n = 3).
N.D., not detected.
Lead-treated WT kidneys were analyzed immunohistochemically for MT localization to see if MT played a direct role in lead-induced inclusion body formation. MT in lead-treated kidneys from WT mice was primarily cytosolic with minimal nuclear staining and no apparent association with inclusion bodies (data not shown).
Renal Lead Accumulation in MT-Null and WT Mice
After 10 weeks of exposure to 0, 1000, or 2000 ppm lead in drinking water, renal lead levels were determined in MT-null and WT mice. Surprisingly, MT-null mice accumulated significantly less renal lead than WT mice at all doses tested (Table 2) ▶ .
Table 2.
Lead Accumulation in Kidney from WT and MT-Null Mice (μg/g Wet Weight)
| Mouse strain | Lead dose (ppm, p.o.) | ||
|---|---|---|---|
| 0 | 1000 | 2000 | |
| WT | N.D. | 10.9 ± 0.3 | 14.4 ± 0.3 |
| MT-null | N.D. | 8.9 ± 0.2* | 11.0 ± 0.7* |
WT and MT-null mice were given lead p.o. at 0, 1000, or 2000 ppm for 10 weeks, renal lead levels were measured by AAS. Data given as the mean ± SEM (n = 10).
*Significant (P < 0.05) difference from appropriate dose-matched WT mice.
N.D., not detectable.
Microarray Analysis of Lead-Treated Kidneys from MT-Null and WT Mice
To help define more subtle differential toxicity after lead exposure, gene expression array studies were performed with RNA isolated from the kidneys of lead-treated (2000 ppm for 10 weeks) WT and MT-null mice. Lead exposure altered the expression of a variety of genes, and such alterations were generally much more common in MT-null mice. Details of gene expression changes are given in Table 3 ▶ . Among the 1178 genes investigated, more than 60 genes (5.0%) were aberrantly expressed in MT-null mice after lead exposure whereas only 35 genes (2.9%) were aberrantly expressed in WT mice. Specifically, various oxidative stress and cellular defense-related genes were up-regulated in MT-null mice because of lead treatment, indicative of a molecular response to a toxic insult. Notably, the expression of the genes encoding for glutathione S-transferase-5 (GST-μ), glutathione S-transferase θ 1 (GST-theta), and glutathione S-transferase π (GST-π) were increased ∼2.5-fold to threefold in MT-null mice treated with lead as compared with untreated MT-null mice. However, expression of GST-μ, GST-θ, and GST-π, was not altered in WT mice. Thus, although lead does not induce overt pathology at the microscopic level in MT-null mice, it did induce gross pathological changes (nephromegaly), as well as diminished renal function, and clearly caused more subtle lesions leading to altered gene expression much more commonly in MT-null mice.
Table 3.
Differentially Expressed Genes in MT-Null and WT Mice Treated with Lead*
| Gene | MT-null control | MT-null lead 4000 ppm | MT-null lead/ control | WT control | WT lead 4000 ppm | WT lead/ control | |
|---|---|---|---|---|---|---|---|
| Stress-response proteins (first group) | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM | |||
| Heat shock protein HSP27 | 1915 ± 2 | 4181 ± 891 | 2.18 | 3411 ± 364 | 3302 ± 934 | 0.97 | |
| Heat shock protein HSP84 | 10222 ± 4143 | 14301 ± 640 | 1.40 | 14991 ± 811 | 12755 ± 925 | 0.85 | |
| Microsomal GST (MGST1) | 4784 ± 596 | 8721 ± 306 | 1.82 | 6681 ± 27 | 7029 ± 1134 | 1.05 | |
| Glutathione S-transferase-5 (GST mu) | 14369 ± 1999 | 35330 ± 2752 | 2.46 | 33379 ± 6472 | 35428 ± 5084 | 1.06 | |
| Glutathione S-transferase theta 1 | 1409 ± 33 | 4213 ± 898 | 2.99 | 3216 ± 78 | 2909 ± 601 | 0.91 | |
| Glutathione S-transferase Pi | 7920 ± 1609 | 20892 ± 4302 | 2.64 | 17357 ± 7238 | 17715 ± 4877 | 1.02 | |
| Oxidative stress protein A170 | 2477 ± 67 | 4526 ± 419 | 1.83 | 2534 ± 211 | 2571 ± 154 | 1.26 | |
| Cell signaling and transducers (second group) | |||||||
| Insulin-like growth factor IGFB6 | 4675 ± 3115 | 10823 ± 2335 | 2.32 | 10567 ± 5353 | 11596 ± 3393 | 1.10 | |
| Insulin-like growth factor II IGF-II | 1792 ± 215 | 4275 ± 2500 | 2.39 | 3519 ± 3216 | 4701 ± 3062 | 1.34 | |
| Wingless MMTV integration WNT 3 | 3218 ± 1088 | 6683 ± 1962 | 2.07 | N.D. | N.D. | ||
| WNT5B | 2424 ± 260 | 4972 ± 655 | 2.05 | N.D. | N.D. | ||
| Tumor necrosis factor-beta | 744 ± 214 | 2744 ± 1913 | 3.68 | 524 ± 139 | 298 ± 156 | 0.57 | |
| Cytokine inducible CISH7 | 6492 ± 2264 | 10727 ± 2423 | 1.65 | 12660 ± 1620 | 13995 ± 3714 | 1.11 | |
| Nuclear factor-kappa B (IkB-beta) | 1626 ± 88 | 8941 ± 5194 | 5.45 | 1162 ± 9 | 1272 ± 418 | 1.09 | |
| DNA synthesis, repair (third group) | |||||||
| DNA excision repair protein ERCC1 | 711 ± 266 | 1840 ± 779 | 2.59 | N.D. | N.D. | ||
| DNA repair protein XPBC | 3170 ± 740 | 5819 ± 1410 | 1.84 | 2767 ± 201 | 5141 ± 1569 | 1.86 | |
| UV excision repair protein RAD23 | 4186 ± 1417 | 7080 ± 878 | 1.69 | 508 ± 389 | 1716 ± 1028 | 3.42 | |
| DNA polymerase delta POLD1 | 1324 ± 373 | 3903 ± 2341 | 2.95 | 1826 ± 763 | 1454 ± 173 | 0.80 | |
| DNAse 1 | 33264 ± 9000 | 21764 ± 4110 | 0.65 | 29969 ± 2862 | 28308 ± 3807 | 0.95 | |
| Apoptosis-associated proteins (fourth group) | |||||||
| Tumor necrosis factor receptor 1 | 1121 ± 155 | 4601 ± 1409 | 4.11 | 2043 ± 2007 | 1619 ± 1450 | 0.79 | |
| Tumor necrosis factor receptor 2 | 1018 ± 81 | 13492 ± 5031 | 13.3 | 3062 ± 1100 | 2382 ± 1060 | 0.78 | |
| Caspase-7 | 2858 ± 2734 | 245 ± 138 | 0.09 | 200 ± 61 | 75 ± 40 | 0.37 | |
| MCL-1 | 5178 ± 1850 | 2221 ± 540 | 0.43 | 2296 ± 270 | 3236 ± 509 | 1.40 | |
| Sentrin; ubiquitin-like protein | 7783 ± 1849 | 3532 ± 606 | 0.45 | 1484 ± 817 | 1654 ± 1454 | 1.11 | |
| Inhibitor of neuronal NO synthetase | 12867 ± 2830 | 8963 ± 1697 | 0.71 | 12010 ± 753 | 10204 ± 408 | 0.85 | |
| Cell surface antigens (fifth group) | |||||||
| Connexin 26 | 10096 ± 2018 | 4518 ± 1057 | 0.46 | 5944 ± 1841 | 7544 ± 2178 | 1.27 | |
| Connexin 40 | 2835 ± 210 | 4737 ± 712 | 1.67 | 2942 ± 531 | 7067 ± 2054 | 2.40 | |
| m-numb (m-NB) | 3592 ± 453 | 1642 ± 193 | 0.46 | 1688 ± 285 | 1464 ± 768 | 0.87 | |
| Glutamate receptor subunit | 2864 ± 1204 | 696 ± 493 | 0.24 | 1189 ± 519 | 1222 ± 513 | 1.03 | |
| Transcription factors (sixth group) | |||||||
| Msx-interacting Zn finger protein | 5069 ± 1976 | 1675 ± 842 | 0.33 | 2537 ± 944 | 3565 ± 1390 | 1.40 | |
| Brain-specific homebox (BRN-1) | 7308 ± 1780 | 3992 ± 727 | 0.55 | 4347 ± 931 | 3855 ± 1216 | 0.91 | |
| TBX2 protein | 4752 ± 1154 | 1980 ± 483 | 0.43 | 10279 ± 2927 | 8546 ± 4260 | 0.83 | |
| Homoboxcux2 (CUTL2) | 2918 ± 235 | 8661 ± 3018 | 2.97 | 3233 ± 1825 | 2496 ± 404 | 0.78 | |
| Eyes absent homolog2 | 4352 ± 2058 | 10837 ± 4030 | 2.49 | 12694 ± 1847 | 11597 ± 5639 | 0.91 | |
| Transcriptional coactivator of AML-1 | 5533 ± 1372 | 2119 ± 1126 | 0.38 | 11081 ± 1791 | 7959 ± 3366 | 0.72 | |
| Transcription factor 3B | 6794 ± 202 | 3160 ± 596 | 0.47 | 6085 ± 1097 | 4219 ± 1330 | 0.69 | |
| Ret finger protein | 599 ± 15 | 1842 ± 638 | 3.10 | 906 ± 371 | 1565 ± 899 | 1.73 | |
| Trans-acting transcription factor 3 | 4190 ± 670 | 1545 ± 794 | 0.37 | 1724 ± 805 | 569 ± 184 | 0.33 | |
| A T motif-binding factor (ATBF1) | 14324 ± 2979 | 25596 ± 6946 | 1.79 | 23293 ± 3372 | 21088 ± 76826 | 0.91 | |
| D-binding protein (DBP) | 10812 ± 2609 | 169 ± 316 | 0.29 | 2604 ± 41 | 3512 ± 1418 | 1.34 | |
| Cell cycle Regulators (seventh group) | |||||||
| Protein kinase-β cAMP dependent | 6192 ± 1787 | 3016 ± 464 | 0.49 | 2421 ± 1045 | 2558 ± 1303 | 1.06 | |
| G2/M-specific cyclin G | 12799 ± 3658 | 8715 ± 3360 | 0.68 | 9080 ± 1522 | 10282 ± 4090 | 1.13 | |
| Extracellular signal regulator ERK1 | 5473 ± 380 | 2126 ± 663 | 0.39 | 1981 ± 765 | 2312 ± 1011 | 1.16 | |
| T ob antiproliferative factor | 11180 ± 938 | 7322 ± 1260 | 0.66 | 2039 ± 77 | 2793 ± 969 | 1.36 | |
| Cell adhesion receptors and proteins (eighth group) | |||||||
| α-E-catenin | 9458 ± 2098 | 5941 ± 1800 | 0.63 | 8413 ± 1211 | 8876 ± 852 | 1.05 | |
| Cell surface glycoprotein MAC-1 | 1840 ± 228 | 5140 ± 1516 | 2.79 | 3498 ± 444 | 6519 ± 2306 | 1.86 | |
| Integrin alpha 6 | 3516 ± 844 | 2268 ± 601 | 0.65 | 3188 ± 390 | 2943 ± 570 | 0.92 | |
| Oncogenes and tumor suppressors (nineth group) | |||||||
| Transcription termination factor TTF1 | 11446 ± 1103 | 5614 ± 2085 | 0.49 | 521 ± 515 | 779 ± 308 | 1.49 | |
| Tumor susceptibility protein TSG101 | 3440 ± 840 | 1844 ± 380 | 0.54 | 3836 ± 836 | 2879 ± 1610 | 0.75 | |
| c-myc oncogene | 464 ± 105 | 1145 ± 110 | 2.47 | 537 ± 299 | 774 ± 176 | 1.44 | |
| Erin; Villin 2 | 9229 ± 1202 | 5792 ± 1039 | 0.63 | 7936 ± 1436 | 6553 ± 1582 | 0.83 | |
| c-met oncogene | 10622 ± 3924 | 6705 ± 1681 | 0.63 | 8104 ± 945 | 8506 ± 1464 | 1.05 | |
| Retinoblastoma-like protein RBL2 | 1263 ± 149 | 2851 ± 1053 | 2.26 | 1330 ± 525 | 2125 ± 855 | 1.60 | |
| Ion channels and transporters (tenth group) | |||||||
| Glutamate receptor, ionotropic NMD2A | 618 ± 77 | 4697 ± 1691 | 6.31 | 2533 ± 1059 | 1636 ± 721 | 0.65 | |
| P-glycoprotein (MDR1) | 1237 ± 50 | 3277 ± 952 | 2.65 | 3052 ± 674 | 2055 ± 844 | 0.67 | |
| Receptors (eleventh group) | |||||||
| IL-6 receptor alpha | 553 ± 141 | 4217 ± 1750 | 7.62 | 1891 ± 393 | 1841 ± 120 | 0.99 | |
| 5-hydroxytryptamine receptor | 1066 ± 824 | 11149 ± 9102 | 10.5 | N.D. | N.D. | ||
| Serine protease inhibitor 1-2 | 4486 ± 610 | 17023 ± 1151 | 3.79 | 10910 ± 5711 | 13300 ± 5171 | 1.22 | |
*The data are mean ± SEM of four separate experiments from MT-null and WT mice treated with lead.
N.D., not detected.
Lead Toxicity in MT-Null and WT Cells in Vitro
To help further define sensitivity to lead and propensity to form inclusion bodies, an additional study was conducted in vitro using cell lines derived from MT-null and WT mice. 31 MT-null and WT cells were treated with lead for 24 hours and cytotoxicity was measured as metabolic integrity (Figure 3) ▶ . MT-null cells were much more sensitive than WT cells to lead cytotoxicity. The LC50 value for lead in WT cells was 645 ± 26 μmol/L as compared to 230 ± 17 μmol/L in MT-null cells, which constitutes a 2.8-fold difference in sensitivity to the metal.
Figure 3.

Acute cytotoxicity of lead in MT-null and WT cells. MT-null and WT cells were treated with the lead glutamate at the concentration indicated for 24 hours. Cytotoxicity was measured by the MTS assay. Data are expressed as percentage of untreated control that is set at 100%. Results are presented as the mean ± SEM (n = 4).
Formation of Inclusion Bodies in WT Cells But Not in MT-Null Cells
WT and MT-null cells were exposed to lead and the formation of lead inclusion bodies was examined by electron microscopy (Figure 4) ▶ . Both WT and MT-null cells appeared to have normal ultrastructural features regardless of lead exposure. However, groups of irregularly shaped inclusion bodies were observed around the nuclear membrane in the cytoplasm only in lead-treated WT cells. No inclusion bodies were observed in MT-null cells.
Figure 4.
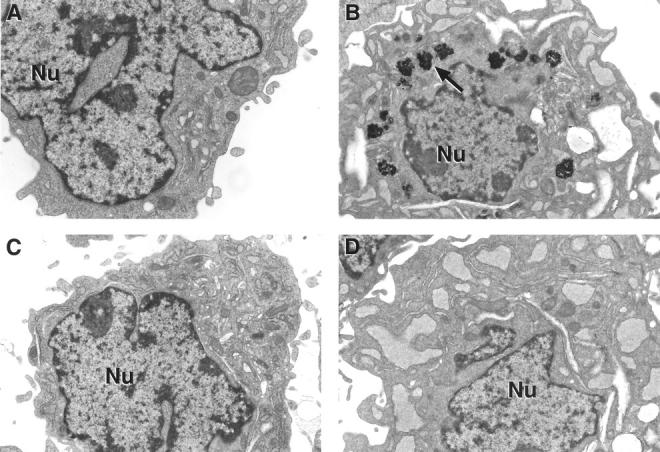
Lead-induced inclusion bodies as detected by EM. MT-null and WT cells were treated with lead for 48 hours; after fixation and staining they were viewed by EM (original magnification, ×15,000; scale bar, 1 μm). A: Control WT cells. B: Lead-treated WT cells with groups of irregularly shaped inclusion bodies around the nucleus present in the cytoplasm. C: Control MT-null cells. D: Lead-treated MT-null cells (note absence of inclusion bodies).
Lead Accumulation in MT-Null and WT Cells
To determine whether cellular lead disposition could play a role in the lack of inclusion body formation in MT-null cells, WT and MT-null cells were exposed to lead and cellular lead levels were measured (Table 4) ▶ . MT-null cells accumulated significantly less lead in comparison to the WT cells. As an indication of lead efflux, lead-loaded cells were placed in lead-free medium for an additional 24 hours and remaining cellular lead was measured. The amount of lead effluxed during this period was not significantly different between MT-null and WT cells (data not shown).
Table 4.
Lead Uptake in WT and MT-Null Cells
| Cell type | Lead uptake |
|---|---|
| WT | 8.7 ± 1.1 |
| MT-null | 4.9 ± 1.2* |
WT and MT-null cells were treated with lead for 48 hours, and intracellular lead levels (μg lead/106 cells) were measured by AAS. Data given as the mean ± SEM (n = 3).
*Significant (P < 0.05) difference from WT cells.
Effect of Lead on MT Levels
To detect the effects of lead on cellular MT levels, WT cells were exposed to lead for 48 hours and MT levels were measured (Table 5) ▶ . Lead caused a significant dose-dependent increase in MT levels in WT cells.
Table 5.
The Levels of MT in the WT Cells Treated with Lead
| Lead exposure concentration, μmol/L | |||
|---|---|---|---|
| 0 | 100 | 200 | |
| Metallothionein (μg/106 cells) | 10.9± 1.1 | 86.1± 8.2* | 112.5± 12.2* |
WT cells were treated with lead for 48 hours. MT was measured by the Cd-hemoglobin method. Data are given as the mean ± SEM (n = 3).
*Significant (P < 0.05) difference from untreated cells.
Discussion
Lead is one of the most important environmental toxicants in the United States and throughout the world because of its ubiquitous nature and the spectrum of toxicological effects it induces, potentially including carcinogenicity. 36 Chronic nephropathy from lead exposure often shows interstitial fibrosis and cystic hyperplasia in humans and animals. 2,9,37,38 Lead is also a renal carcinogen in animals 14,39 and possibly in humans. 1 The profound nephrotoxicity induced by lead is characterized by cellular inclusion bodies and renal tubular dysfunction. 14 These inclusion bodies are a diagnostic feature of lead poisoning and are particularly common in the kidney epithelial cells, although they can occur in the brain and elsewhere. 14 Several studies have shown that inclusion bodies are composed of a lead-protein complex, and it has been hypothesized that inclusion bodies play an important role in the mitigation of lead toxicity. 14 The present results indicate that the inability to produce MT predisposes animals or cells to lead toxicity and is associated with an inability to produce lead inclusion bodies. In this regard, MT-deficient mice were more sensitive than WT mice to lead-induced nephromegaly, depression of renal function, and molecular lesions resulting in aberrant gene expression. In vitro, cells incapable of MT production were clearly hypersensitive to lead-induced cytotoxicity. MT deficiency was associated with the inability to produce lead inclusion bodies in both the in vivo and in vitro system. Thus, it seems that MT is somehow required for inclusion body production because the MT-null genotype does not allow for the formation of inclusion bodies after lead exposure. Precisely how MT may facilitate inclusion body formation is not known, but because MT deficiency enhances lead toxicity as well as preventing inclusion body formation, assuming there is a connection between these events, these results seem to support the concept that inclusion bodies may mitigate lead toxicity. 7,12 What is perhaps more important is that this study predicts that individuals less able to produce MT may be hypersensitive to lead intoxication, indicating a potential genetic basis for lead sensitivity. It is clear that genetic polymorphisms exist in the human MT genes, 40 although how these polymorphisms might affect lead toxicity is unknown. In addition, based on the current results, we would predict that MT-null animals would be more sensitive to lead-induced neurotoxicity. Subtle neurotoxicity of lead is a major issue in child health in the United States and identifying sensitive subpopulations would be a considerable advance in this important public health issue.
Exactly how the inability to produce MT may enhance lead toxicity is unclear. MTs contain numerous thiol groups because of their very high cysteine content, which provides the basis for high-affinity binding of many metals. 16-18 It is likely that a major purpose of these proteins is detoxification of metals. 16 The mitigation of the adverse effects of many toxic metals, including cadmium and mercury, by MT is quite well established and probably occurs through sequestration of the toxic metal in a nonbioavailable, and thus, toxicologically inert form. 16 However, the role of MT in lead toxicity has been only poorly defined. Lead can stimulate MT production in vitro and in vivo in some cases 18,19,23 but not in others. 25 Our results indicate that in vitro lead exposure induces MT in the WT cells used in the present study. The finding that lead induces MT does not, in and of itself, establish that it plays a role in reduction of lead toxicity. However, the results of the present study clearly show that the ability to express the major forms of MT reduces the toxic impact of lead in vivo and in vitro. It is important to note that MT-null animals or cells were more sensitive to lead despite accumulating significantly less of the metal. Even in the face of favorable biokinetics, the MT-null genotype is more sensitive to lead toxicity, as manifested in vivo as nephromegaly, impaired renal function, and aberrant gene expression and in vitro as acute cytotoxicity. Therefore, the present results implicate MT as an important factor in lead toxicity, through an as yet undefined mechanism.
It is also seems from the present results that MT may play a role in lead-induced inclusion body formation. In fact, there was a total absence of inclusion body formation in the MT-null genotype both in vitro and in vivo, even at toxic levels of lead. Exactly how MT may facilitate inclusion body formation is, at present, unknown. Perhaps the simplest explanation for the absence of inclusion body formation with the MT-null phenotype would be biokinetic in nature. In essence, the hypothesis here would be that, because MT-null animals or cells accumulate less lead, the levels of the metal in MT-null mice would be below that needed to stimulate inclusion body formation. However, an examination of the present data (Table 2) ▶ shows that renal lead levels in MT-null animals given 2000 ppm lead (11.0 μg/g wet weight), where no inclusion bodies were found (Table 1) ▶ , actually exceed lead levels in the kidneys of WT mice given 1000 ppm lead (10.9 μg/g wet weight), where inclusion bodies were quite common. On this basis, it would seem that levels of lead sufficient to stimulate inclusion body formation in WT mice did in fact reach the kidneys in MT-null animals without producing any inclusion bodies. This does not entirely exclude biokinetics as an aspect of the inability to form lead inclusion bodies associated with the MT-null genotype. For instance, it is possible that MT may act as a temporary intracellular transport biocomplex with lead to facilitate localization of the metal to the appropriate cellular point for production of inclusion bodies. A variety of high-affinity renal-binding proteins have come to light. 41 Chemical analysis of inclusion bodies indicates a relatively constant protein-to-lead ratio, suggesting an orderly process, 7 which would be consistent with a facilitory role for MT in this process. Defining the exact nature of inclusion bodies has been problematic, 42,43 but it is clear they contain both lead and protein. 7,42,43 Immunohistochemical analysis clearly showed MT was not prominently associated with inclusion bodies, but this is only after formation of the inclusion bodies. MT could still possibly be within inclusion bodies, but in an immunologically changed form that would not be detected by the antibody used in this study. Further research will be required to more fully define the role of MT in lead-induced inclusion body formation, but the present results indicate MT is required for such formation, perhaps in a facilitative or temporary transport role.
GSTs are a family of phase II detoxification enzymes involved in the conjugation of a diverse group of electrophilic substrates with glutathione followed by excretion of the conjugate. 44 Wright and colleagues 44 first reported that increases in GSTs are closely linked to tissue damage resulting from lead exposure. These data suggest that increases in GST precede cellular and physical changes induced by lead, and thereby provide an extremely sensitive tissue biomarker of lead exposure. 44 Moser and colleagues 45 and Oberley and colleagues 46 have also reported that acute or chronic inorganic lead exposure during development produces cell-type-specific increases in GST expression in the rat kidney. However, whether these increases in GSTs are a result of lead-induced injury or serve as a protective adaptation is not clear. 44 Regardless of whether this is a toxic response or an adaptive response to intoxication, the present study demonstrates that the expression of genes encoding for GSTs (including μ, θ, π) were significantly increased in MT-null mice by lead treatment but not in WT mice. This indicates that the molecular responses to lead-induced toxic insult are exaggerated by the inability to produce MT. It is thought that MT may also serve as a scavenger for reactive oxygen species, 47 although this is not clearly established. Thus, the up-regulation of defense-related genes, such as those encoding GSTs, because of lead exposure in MT-null mice, may act as a cellular adaptive mechanism in the absence of MT. Therefore, it appears in the present study that up-regulation of GST may serve as a subtle indicator of lead toxicity as previously suggested. 44 The exact role of the alterations in defense-related genes, as well as the relationship of these genes to lead toxicity, are worth further investigation. Furthermore, how overexpression of heat shock proteins HSP27 and HSP84, as well as cell signaling and transducers in MT-null mice, might contribute to lead toxicity requires additional study.
In summary, the MT-null phenotype does not allow inclusion body formation after lead exposure and predisposes to lead toxicity both in vivo and in vitro despite reducing accumulation of the metal. Thus, MT seems to play an important role in lead toxicity and in inclusion body formation. These results indirectly support a role for inclusion bodies as a potential element in cellular lead tolerance. From these results it is possible to conclude that individual variation in the ability to express MT may dictate sensitivity to lead toxicity in exposed populations, which may have important public health implications.
Acknowledgments
We thank Drs. Larry K. Keefer and William E. Achanzar for critical review of this manuscript, Dr. Jerrold Ward for assistance with photomicrography, and Dan Logsdon for expert technical assistance.
Footnotes
Address reprint requests to Dr. Michael P. Waalkes, Chief, Inorganic Carcinogenesis Section, NCI at NIEHS, PO Box 12233, Mail Drop F0-09, 111 Alexander Dr., Research Triangle Park, NC 27709. E-mail: waalkes@niehs.nih.gov.
Supported in part by the National Cancer Institute (contract N01-CO-56000).
References
- 1.International Agency for Research on Cancer (IARC): IARC Monograph on the Evaluation of Carcinogenic Risks to Humans: Overall Evaluations of Carcinogenicity. An Update of IARC. Lyon, IARC, Monographs 1 to 42, suppl 7, 1987, pp 230–232
- 2.Nolan CV, Shaikh ZA: Lead nephrotoxicity and associated disorders: biochemical mechanisms. Toxicology 1992, 73:127-146 [DOI] [PubMed] [Google Scholar]
- 3.Goyer RA: Lead. Bingham E Cohrssen B Powell CH eds. Patty’s Toxicology. 2001, :pp 611-675 John Wiley & Sons, New York [Google Scholar]
- 4.International Agency for Research on Cancer (IARC): IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans: Some Metals and Metallic Compounds. Lyon, IARC, 1980, pp 325–415
- 5.Choie DD, Ricchter GW: Lead poisoning: rapid formation of intranuclear inclusion. Science 1972, 177:1194-1195 [DOI] [PubMed] [Google Scholar]
- 6.Blackman SS: Intranuclear inclusion-bodies in the kidney and liver caused by lead poisoning. Bull John Hopkins Hosp 1936, 58:584 [Google Scholar]
- 7.Goyer RA, May P, Cates M, Krigman MR: Lead and protein content of isolated intranuclear inclusion bodies from kidneys of lead-poisoned rats. Lab Invest 1970, 22:245-251 [PubMed] [Google Scholar]
- 8.Richter GW: Evolution of cytoplasmic fibrillar bodies induced by lead in rat and mouse kidneys. Am J Pathol 1976, 83:135-148 [PMC free article] [PubMed] [Google Scholar]
- 9.Goyer RA, Rhyne BC: Pathological effects of lead. Int Rev Pathol 1973, 12:1-77 [PubMed] [Google Scholar]
- 10.Klann E, Shelton KR: The effect of lead on the metabolism of a nuclear matrix protein which becomes prominent in lead-induced intranuclear inclusion bodies. J Biol Chem 1989, 264:16969-16972 [PubMed] [Google Scholar]
- 11.Van Mullen PJ, Stadhouders AM: Bone marking and lead-intoxication: early pathological changes in osteoclasts. Virchows Arch B Cell Pathol 1974, 15:345-350 [DOI] [PubMed] [Google Scholar]
- 12.Goyer RA, Leonard JF, Rhyne B, Krigman MR: Lead dosage and the role of the intranuclear inclusion body. Arch Environ Health 1970, 20:705-711 [DOI] [PubMed] [Google Scholar]
- 13.Goyer RA: Lead toxicity: a problem in environmental pathology. Am J Pathol 1971, 64:167-179 [PMC free article] [PubMed] [Google Scholar]
- 14.Goyer RA, Wilson MH: Lead-induced inclusion bodies. Lab Invest 1975, 32:149-156 [PubMed] [Google Scholar]
- 15.Cherian MG, Nordberg M: Cellular adaptation in metal toxicology and metallothionein. Toxicology 1983, 28:1-15 [DOI] [PubMed] [Google Scholar]
- 16.Waalkes MP, Perez-Olle R: Role of metallothionein in metabolism, transport, and toxicity of metals. Molecular Biology and Toxicology of Metals. 2000, R Zalups. London, Taylor & Francis, Edited by DJ Koropatnick
- 17.Cherian MG, Howell SB, Imura N, Klaassen CD, Koropatnick J, Lazo JS, Waalkes MP: Role of metallothionein in carcinogenesis. Toxicol Appl Pharmacol 1994, 126:1-5 [DOI] [PubMed] [Google Scholar]
- 18.Waalkes MP, Klaassen CD: Concentration of metallothionein in major organs of rats after administration of various metals. Fund Appl Toxicol 1985, 5:473-477 [DOI] [PubMed] [Google Scholar]
- 19.Rhee SJ, Huang PC: Metallothionein accumulation in CHO Cdr cells in response to lead treatment. Chem-Biol Interact 1989, 72:347–361 [DOI] [PubMed]
- 20.Klaassen CD, Liu J: Role of metallothionein in cadmium-induced hepatotoxicity and nephrotoxicity. Drug Metab Rev 1997, 29:79-102 [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Liu Y, Habeebu SM, Waalkes MP, Klaassen CD: Chronic combined exposure to cadmium and arsenic exacerbates nephrotoxicity, particularly in metallothionein-I/II null mice. Toxicology 2000, 147:157-166 [DOI] [PubMed] [Google Scholar]
- 22.Maitani T, Watahiki A, Suzuki KT: Induction of metallothionein after lead administration by three injection routes in mice. Toxicol Appl Pharmacol 1986, 83:211-217 [DOI] [PubMed] [Google Scholar]
- 23.Ikebuchi H, Teshima R, Suzuki K, Terao T, Yamane Y: Simultaneous induction of lead-metallothionein-like protein and zinc-thionein in the liver of rats given lead acetate. Biochem J 1986, 233:541-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikebuchi H, Teshima R, Suzuki K, Sawada JI, Terao T, Yamane Y: An immunological study of a lead-thionein-like protein in rat liver. Biochem Biophys Res Commun 1986, 136:535-541 [DOI] [PubMed] [Google Scholar]
- 25.Yamada H, Koizumi S: Metallothionein induction in human peripheral blood lymphocytes by heavy metals. Chem-Biol Interact 1991, 78:347–354 [DOI] [PubMed]
- 26.Church HJ, Day JP, Braithwaite RA, Brown SS: Binding of lead to a metallothionein-like protein in human erythrocytes. J Inorg Biochem 1993, 49:55-68 [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Kershaw WC, Klaassen CD: The protective effect of metallothionein on the toxicity of various metals in rat primary hepatocyte culture. Toxicol Appl Pharmacol 1991, 107:27-34 [DOI] [PubMed] [Google Scholar]
- 28.Waalkes MP, Harvey MJ, Klaassen CD: Relative in vitro affinity of hepatic metallothionein for metals. Toxicol Lett 1984, 20:33-39 [DOI] [PubMed] [Google Scholar]
- 29.Goering PL, Fowler BA: Kidney zinc-thionein regulation of delta-aminolevulinic acid dehydratase inhibition by lead. Arch Biochem Biophys 1987, 253:48-55 [DOI] [PubMed] [Google Scholar]
- 30.Masters BA, Kelly EJ, Quaife CJ, Brinster RL, Palmiter RD: Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc Natl Acad Sci USA 1994, 91:584-588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazo JS, Kondo Y, Dellapiazza D, Michalska AE, Choo KH, Pitt BR: Enhanced sensitivity to oxidative stress in cultured embryonic cells from transgenic mice deficient in metallothionein I and II genes. J Biol Chem 1995, 270:5506-5510 [DOI] [PubMed] [Google Scholar]
- 32.McLachlin JR, Goyer RA, Cherian MG: Formation of lead-induced inclusion bodies in primary rat kidney epithelial cell cultures: effect of actinomycin D and cycloheximide. Toxicol Appl Pharmacol 1980, 56:418-431 [DOI] [PubMed] [Google Scholar]
- 33.Romach EH, Zhao CQ, Del Razo LM, Cebrian ME, Waalkes MP: Studies on the mechanisms of arsenic-induced self tolerance developed in liver epithelial cells through continuous low-level arsenite exposure. Toxicol Sci 2000, 54:500-508 [DOI] [PubMed] [Google Scholar]
- 34.Onosaka S, Tanaka K, Doi M, Okahara K: A simplified procedure for determination of metallothionein in animal tissues. Eisei Kagaku 1978, 24:128-131 [Google Scholar]
- 35.Eaton DL, Toal BF: Evaluation of the Cd/hemoglobin affinity assay for the rapid determination of metallothionein in biological tissues. Toxicol Appl Pharmacol 1982, 66:134-142 [DOI] [PubMed] [Google Scholar]
- 36.: World Health Organization (WHO): Inorganic lead. Geneva, IPCS. Environmental Health Criteria 1995, 165:222-224 [Google Scholar]
- 37.Beck BD: An update on exposure and effects of lead. Fundam Appl Toxicol 1992, 18:1-16 [DOI] [PubMed] [Google Scholar]
- 38.Goyer RA: Lead toxicity: current concerns. Environ Health Perspect 1993, 100:177-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waalkes MP, Diwan BA, Ward JM, Devor DE, Goyer RA: Renal tubular tumors and atypical hyperplasias in B6C3F1 mice exposed to lead acetate during gestation and lactation occur with minimal chronic nephropathy. Cancer Res 1995, 55:5265-5271 [PubMed] [Google Scholar]
- 40.Wu MT, Demple B, Bennett RAO, Christiani DC, Fan R, Hu H: Individual variability in the zinc inducibility of metallothionein-IIA mRNA in human lymphocytes. J Toxicol Environ Health 2000, 61:553-567 [DOI] [PubMed] [Google Scholar]
- 41.Smith DR, Kahng MW, Quintanilla-Vega B, Fowler BA: High-affinity renal lead-binding proteins in environmentally-exposed humans. Chem-Biol Interact 1998, 115:39–52 [DOI] [PubMed]
- 42.Moore JF, Goyer RA, Wilson M: Lead-induced inclusion-bodies: solubility, amino-acid content and relationship to residual acidic nuclear proteins. Lab Invest 1973, 29:488-495 [PubMed] [Google Scholar]
- 43.Moore JF, Goyer RA: Lead-induced inclusion-bodies: composition and probable role in lead metabolism. Environ Health Perspect 1974, 7:121-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright LS, Kornguth SE, Oberley TD, Siegel FL: Effects of lead on glutathione S-transferase expression in rat kidney: a dose-response study. Toxicol Sci 1998, 46:254-259 [DOI] [PubMed] [Google Scholar]
- 45.Moser KE, Oberley TD, Daggett DA, Friedman AL, Johnson JA, Siegel FL: Effects of lead on developing rat kidney. I. Glutathione S-transferase isoenzymes. Toxicol Appl Pharmacol 1995, 131:85-93 [DOI] [PubMed] [Google Scholar]
- 46.Oberley TD, Friedman AL, Moser R, Siegel FL: Effects of lead on developing rat kidney. II. Functional, morphologic and immunohistochemical studies. Toxicol Appl Pharmacol 1995, 131:94-107 [DOI] [PubMed] [Google Scholar]
- 47.Gobel H, van der Wal AC, Teeling P, van der Loos CM, Becker AE: Metallothionein in human atherosclerotic lesions: a scavenger mechanism for reactive oxygen species in the plaque? Virchows Arch 2000, 437:528-533 [DOI] [PubMed] [Google Scholar]