

Abstract
To elucidate the role of p53/p16INK4a/RB1 pathways in prostate carcinogenesis, we analyzed the p14ARF, p16INK4a, RB1, p21Waf1, p27Kip1, PTEN, p73, p53, and MDM2 gene status of multiple areas within 16 histologically heterogeneous prostate carcinomas using methylation-specific polymerase chain reaction, differential polymerase chain reaction, and immunohistochemistry. All focal areas examined had Gleason scores ranging from 1 to 5. Methylation of either PTEN or p73 was undetected in any sample, whereas expression of MDM2 seemed to be an independent event within small foci of 4 of 16 tumors. Loss of p14ARF, p16INK4a, RB1, and p27Kip1 expression correlated with homozygous deletion or promoter hypermethylation. One carcinoma showed co-deletion of both p14ARF and p16INK4a in two of five areas examined; two areas within another tumor demonstrated concurrent hypermethylation of the promoter regions of the same genes. Focal hypermethylation of RB1, p21Waf1, and p27Kip1 was detected within two, two, and three tumors, respectively. These findings indicate that both genetic and epigenetic events occur independently in intratumor foci and further suggest hypermethylation-induced loss of gene function may be as critical as specific genetic mutations in prostate carcinogenesis.
In recent years, prostate cancer has shown an ∼3% annual increase worldwide. In American men this cancer is clinically diagnosed in 1 of every 11 men; one-third of those diagnosed will develop significant life-threatening disease, making it the second most lethal neoplasia. 1 The prostate cancer mortality rate in Japan has increased more than ten-fold throughout the last 3 decades, making it now the eighth leading cause of male cancer death. Epidemiological studies suggest increasing incidence might coincide with wider adoption of a Western lifestyle. 2
Advances in molecular biology have shown us an accumulation of genetic alterations in the step-wise process of tumorigenesis, but the actual genetic basis of the disease is not fully understood. Indeed, recent data have revealed there is actually little direct proof to support genetic mutation as the primary and/or only cause of prostate cancer. Epigenetic mechanisms, such as hypermethylation, are suspected of being more responsible in tumor progression. 3 For example, the p16INK4a gene was reported to be mutated in one of three prostate cancer cell lines, 4 but methylated in three of five others. 5 Both genetic and epigenetic alterations of the INK4a/ARF locus may provide impairment in both p14ARF/p53 and p16INK4a/RB1 pathways in the development and progression of prostate carcinomas. p14ARF confines MDM2 to a subsection of the nucleus and stabilizes intranuclear p53 protein by preventing its cytoplasmic transport, 6-10 indicating that p14ARF may act as an upstream regulator of p53 function. 11 Although homozygous deletion of p14ARF has been reported in ∼40% of glioblastomas, 12 the human p14ARF promoter has been shown to contain a CpG island that is also aberrantly methylated in gliomas, colorectal adenomas, and carcinomas, 12-14 and in esophageal squamous cell carcinomas. 15 However, there has been no study of silencing of the p14ARF by methylation/deletion specifically in prostate carcinomas. Inactivation of RB1 by mutation, deletion, and/or promoter hypermethylation has been reported as an alternative molecular mechanism to p16INK4a inactivation, CDK4 amplification, or CCND1 amplification/rearrangement in human tumors, including prostate carcinomas. 16
Prostate cancer poses unique problems in terms of treatment and prognosis because of the frequent histological heterogeneity encountered. We previously reported on both known and unknown genetic changes within prostate carcinomas 17 that suggested that multicentric genetic events occur leading to tumor progression. We now additionally investigate the potential role of epigenetic, as well as genetic, mechanisms within the p53/p16INK4a/RB1 pathway in this series of prostate carcinomas.
Materials and Methods
Tumor Samples and Histology
The 16 prostate carcinomas examined in this study were obtained from radical prostatectomies. No initial chemotherapy or hormonal treatments were instituted before tumor excision. A slice of whole prostate was fixed in 10% neutral-buffered formalin and embedded in paraffin, and the remainder of the tumors were frozen at −80°C for later DNA extraction. Consecutive sections were cut at 4 μm and mounted for immunohistochemical analyses and histopathological evaluation using conventional hematoxylin and eosin (H&E) staining; the H&E-stained sections also served as a guide for the immunohistochemical and DNA analyses. Three to five different foci from each tumor were selected based on representative morphology, size, and lack of contamination with normal prostatic tissues and histologically graded according to the Gleason system for prostate carcinoma. 18
Methylation-Specific Polymerase Chain Reaction (PCR)
DNA methylation patterns in the CpG islands of the p14ARF, p16INK4a, RB1, p21Waf1, p27Kip1, PTEN, and p73 genes were determined by methylation-specific PCR (MSP). 19 Sodium bisulfite modification was performed using the CpGenome DNA Modification Kit (Intergen, Oxford, UK) according to the manufacturer’s protocol with minor adjustments. 12,20 The primer sequences for methylated and unmethylated PCR of p14ARF, p16INK4a, RB1, p21Waf1, p27Kip1, and p73 have been previously reported. 19,21-23 The primer sequences for methylated and unmethylated PCR of PTEN are as follows: 5′-TTT TCGT TCG GCG CGG TTT CG-3′ (sense) and 5′-GCC GCG CCG AAA ACC CGA ACG-3′ (anti-sense) for the methylated reaction; 5′-TTG TTT GGT GTG GTT TTG TTT GTT T-3′ (sense) and 5′-ACC ACC ACA CCA AAA ACC CAA ACA-3′ (anti-sense) for the unmethylated reaction. MSP conditions for p14ARF, p16INK4a, RB1, p21Waf1, and p27Kip1 were previously described. 12,20,24 The annealing temperature for both PTEN-methylated and -unmethylated reactions was 64°C, whereas that for both p73-methylated and -unmethylated reactions was 60°C. Amplified products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining.
Differential PCR for p14ARF and p16INK4a Deletions
To assess homozygous deletion, we performed differential PCR with primers covering exon 1β of the p14ARF using GAPDH as a reference. Differential PCR for homozygous deletion of p16INK4a exon 1α was performed using the β-actin gene as a reference. The primer sequences and PCR conditions were previously described. 12,15 The PCR product was analyzed on an 8% acrylamide gel. Gels were photographed using a DC290 Zoom digital camera (Eastman Kodak, Rochester, NY), and densitometry of the PCR fragments was performed using Kodak Digital Science ID Image Analysis Software (Version 3.5.2, Eastman-Kodak). Samples presenting <20% of the control signal were considered homozygously deleted. 12,15,24
Loss of Heterozygosity (LOH) Assay for RB1
LOH assays were performed using the Genetic Analyzer 310 (PE Biosystems, Norwalk, CT) capillary electrophoresis system. The marker D13S153 lies in intron 2 of the RB1 gene. 25 PCR was performed for 28 cycles with 58°C annealing temperature. The analysis was performed using the Gene Scan Program (PE Biosystems) following the manufacturer’s protocol.
PCR-Single-Strand Conformation Polymorphism Analyses for p53 Mutations
The p53 mutations in tumors 1 to 9 have been previously reported. 26 For tumors10 to 16, PCR amplification of exons 5 to 9 of p53, single-strand conformation polymorphism analysis, and DNA sequencing was performed according to these published conditions. 26
Immunohistochemistry
The expression of each gene was assessed immunohistochemically using a polyclonal anti-human antibody to p14ARF (FL-132, SC1661; Santa Cruz Biochemicals, Santa Cruz, CA) and monoclonal antibodies to p16INK4a (F-12, SC1661; Santa Cruz Biochemicals), pRB (clone G3-245; Pharmingen, San Diego, CA), p21Waf1 (F-5, SC-6246; Santa Cruz Biochemicals), p27Kip1 (clone 57; Transduction Laboratories, Lexington, KY), and MDM2 (clone IF2; Oncogene Res. Products, Boston, MA). After deparaffinization, sections were heated for 5 minutes in 10 mmol/L of sodium citrate buffer (pH 6.0) in a pressure cooker. The sections were then incubated overnight at 4°C with a 1:500 dilution of antibodies to p14ARF, pRB, and p27Kip1, a dilution of 1:1000 for p16INK4a, and a dilution of 1:100 for p21Waf1 and MDM2. The reactions were visualized using a Histofine SAB-PO kit and diaminobenzidine as the chromogen (Nichirei, Tokyo, Japan) with hematoxylin counterstaining. For immunostaining, the reactivity was recorded as (−) when <5% of cancer cells were positive, according to the previous study. 24
Results
p14ARF and p16INK4a Alterations
As shown in Table 1 ▶ , of the 16 prostate adenocarcinomas evaluated, simultaneous homozygous deletion of the p14ARF and p16INK4a gene was detected by differential PCR in only one case (tumor 3, Figure 1 ▶ ). Similarly, hypermethylation of the promoter regions of both p14ARF and p16INK4a also occurred in a single carcinoma (tumor 8, Figure 2 ▶ ). Concurrent deletion or methylation of p14ARF and p16INK4a was detected in two of five foci within individual tumor (tumors 3 and 8, respectively). The two foci showing deletions were Gleason grades 5 and 4, whereas those with the co-methylation were grades 3 and 4. Hypermethylation of p16INK4a exon 2 was found in all areas examined in each of 11 tumors (69%, Table 1 ▶ ).
Table 1.
Heterogeneous Alterations of Multiple Genes in Prostate Cancer
| ID No. | Gleason score | p14ARF | p16INK4a | RB1 | p21Waf1 | p27Kip1 | PTEN | p73 | p53 | MDM2 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IHC* | Methyl or del† | IHC | Methyl or del‡ | Methyl§ | IHC | LOH at D6S153 | Methyl | IHC | Methyl | IHC | Methyl | Methyl | Methyl | IHC | Mutation | IHC | ||||||||||
| 1 | ||||||||||||||||||||||||||
| 1 | 3 | + | − | + | − | Methyl | − | NI¶ | Methyl | − | Methyl | + | − | − | − | + | + | − | ||||||||
| 2 | 4 | + | − | + | − | Methyl | − | NI | Methyl | − | Methyl | + | − | − | − | + | + | − | ||||||||
| 3 | 3 | + | − | + | − | Methyl | − | NI | Methyl | − | − | + | − | − | − | + | + | − | ||||||||
| 4 | 4 | + | − | + | − | Methyl | − | NI | Methyl | − | Methyl | + | − | − | − | + | + | − | ||||||||
| 5 | 3 | + | − | + | − | Methyl | − | NI | − | − | Methyl | + | − | − | − | − | − | − | ||||||||
| 2 | ||||||||||||||||||||||||||
| 1 | 1 | + | − | + | − | Methyl | − | LOH∥ | − | − | − | + | − | − | − | − | − | − | ||||||||
| 2 | 2 | + | − | + | − | Methyl | + | LOH | − | − | − | + | − | − | − | − | − | − | ||||||||
| 3 | 2 | + | − | + | − | Methyl | − | LOH | − | − | − | + | − | − | − | − | − | − | ||||||||
| 4 | 3 | + | − | + | − | Methyl | − | LOH | − | − | − | + | − | − | − | − | − | − | ||||||||
| 5 | 2 | + | − | + | − | Methyl | − | LOH | − | − | − | + | − | − | − | − | − | − | ||||||||
| 3 | ||||||||||||||||||||||||||
| 1 | 5 | − | del | − | del | Methyl | + | RET** | − | + | − | − | Methyl | − | − | − | − | − | ||||||||
| 2 | 4 | − | del | − | del | Methyl | + | RET | − | + | − | − | Methyl | − | − | − | − | − | ||||||||
| 3 | 5 | − | − | − | − | Methyl | + | RET | − | + | − | − | Methyl | − | − | − | − | − | ||||||||
| 4 | 5 | + | − | + | − | Methyl | + | RET | − | + | − | + | Methyl | − | − | − | − | − | ||||||||
| 5 | 4 | − | − | − | − | Methyl | + | RET | − | + | − | − | Methyl | − | − | − | − | + | ||||||||
| 4 | ||||||||||||||||||||||||||
| 1 | 5 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 2 | 4 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 3 | 4 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 4 | 4 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 5 | 3 | + | − | + | − | Methyl | − | n.a.†† | − | + | − | + | − | − | − | − | − | − | ||||||||
| 5 | ||||||||||||||||||||||||||
| 1 | 4 | + | − | + | − | Methyl | − | n.a. | − | − | − | + | − | − | − | − | − | − | ||||||||
| 2 | 4 | + | − | + | − | Methyl | − | n.a. | − | − | − | + | − | − | − | − | − | − | ||||||||
| 3 | 5 | + | − | + | − | Methyl | − | RET | − | − | − | − | − | − | − | − | − | − | ||||||||
| 4 | 5 | + | − | + | − | Methyl | − | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 5 | 5 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | + | ||||||||
| 6 | ||||||||||||||||||||||||||
| 1 | 3 | + | − | + | − | Methyl | + | RET | − | − | − | + | − | − | − | − | − | − | ||||||||
| 2 | 4 | + | − | + | − | Methyl | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 3 | 4 | + | − | + | − | Methyl | + | LOH | − | + | − | + | − | − | − | − | − | − | ||||||||
| 4 | 3 | + | − | + | − | Methyl | − | LOH | − | + | − | − | − | − | − | − | − | − | ||||||||
| 5 | 4 | + | − | + | − | Methyl | + | LOH | − | + | − | + | − | − | − | − | − | − | ||||||||
| 7 | ||||||||||||||||||||||||||
| 1 | 4 | − | − | − | − | − | + | n.a. | − | + | − | + | − | − | − | + | + | − | ||||||||
| 2 | 3 | + | − | − | − | − | + | RET | − | + | − | + | − | − | − | + | + | − | ||||||||
| 3 | 3 | + | − | − | − | − | + | RET | − | + | − | + | − | − | − | + | + | − | ||||||||
| 4 | 3 | + | − | − | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 5 | 3 | + | − | + | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 8 | ||||||||||||||||||||||||||
| 1 | 4 | + | − | + | − | Methyl | + | RET | − | − | Methyl | − | Methyl | − | − | − | − | − | ||||||||
| 2 | 3 | − | Methyl | − | Methyl | Methyl | + | RET | − | − | Methyl | − | − | − | − | − | − | − | ||||||||
| 3 | 4 | − | Methyl | − | Methyl | Methyl | + | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 4 | 5 | + | − | + | − | Methyl | + | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 5 | 4 | + | − | + | − | Methyl | + | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 9 | ||||||||||||||||||||||||||
| 1 | 2 | + | − | + | − | − | − | NI¶ | − | + | − | − | − | − | − | − | − | − | ||||||||
| 2 | 3 | + | − | + | − | − | − | NI | − | + | − | − | − | − | − | − | − | − | ||||||||
| 3 | 4 | + | − | + | − | − | − | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 4 | 4 | + | − | + | − | − | + | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 10 | ||||||||||||||||||||||||||
| 1 | 2 | + | − | + | − | Methyl | + | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 2 | 3 | + | − | + | − | Methyl | + | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 3 | 3 | + | − | + | − | Methyl | + | n.a.†† | − | + | − | + | − | − | − | − | − | − | ||||||||
| 4 | 4 | + | − | + | − | Methyl | + | n.a. | − | + | − | + | − | − | − | − | − | + | ||||||||
| 5 | 4 | + | − | + | − | Methyl | + | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 11 | ||||||||||||||||||||||||||
| 1 | 5 | + | − | + | − | − | + | RET** | − | + | − | − | − | − | − | − | − | − | ||||||||
| 2 | 5 | + | − | + | − | − | + | RET | − | + | − | − | − | − | − | − | − | − | ||||||||
| 3 | 3 | + | − | + | − | − | + | n.a. | − | − | − | + | − | − | − | − | − | − | ||||||||
| 4 | 4 | + | − | + | − | − | + | LOH∥ | − | − | − | − | − | − | − | − | − | − | ||||||||
| 5 | 4 | + | − | + | − | − | + | LOH | − | − | − | − | − | − | − | − | − | − | ||||||||
| ID No. | Gleason score | p14ARF | p16INK4a | RB1 | p21Waf1 | p27Kip1 | PTEN | p73 | p53 | MDM2 | ||||||||||||||||
| IHC* | Methyl or del† | IHC | Methyl or del‡ | Methyl§ | IHC | LOH at D6S153 | Methyl | IHC | Methyl | IHC | Methyl | Methyl | Methyl | IHC | Mutation | IHC | ||||||||||
| 12 | ||||||||||||||||||||||||||
| 1 | 4 | + | − | + | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 2 | 5 | + | − | + | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 3 | 5 | + | − | + | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 4 | 5 | + | − | + | − | − | + | RET | − | + | − | + | − | − | − | − | − | − | ||||||||
| 13 | ||||||||||||||||||||||||||
| 1 | 3 | + | − | + | − | Methyl | + | RET | − | + | − | − | Methyl | − | − | − | − | − | ||||||||
| 2 | 5 | + | − | + | − | Methyl | − | RET | − | − | − | − | − | − | − | − | − | − | ||||||||
| 3 | 5 | + | − | + | − | Methyl | − | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 4 | 4 | + | − | + | − | Methyl | − | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 5 | 3 | + | − | + | − | Methyl | − | RET | − | − | − | − | Methyl | − | − | − | − | − | ||||||||
| 14 | ||||||||||||||||||||||||||
| 1 | 3 | + | − | + | − | Methyl | + | RET | Methyl | − | − | + | − | − | − | − | − | − | ||||||||
| 2 | 3 | + | − | + | − | Methyl | − | RET | Methyl | − | − | + | − | − | − | − | − | − | ||||||||
| 3 | 4 | + | − | + | − | Methyl | − | RET | Methyl | + | − | + | − | − | − | − | − | − | ||||||||
| 15 | ||||||||||||||||||||||||||
| 1 | 5 | + | − | + | − | Methyl | + | RET | − | − | − | + | − | − | − | − | − | − | ||||||||
| 2 | 4 | + | − | + | − | Methyl | + | RET | − | − | − | − | − | − | − | − | − | − | ||||||||
| 3 | 4 | + | − | + | − | Methyl | + | RET | − | − | − | − | − | − | − | − | − | − | ||||||||
| 4 | 5 | + | − | + | − | Methyl | + | RET | − | − | − | − | − | − | − | − | − | − | ||||||||
| 5 | 4 | + | − | + | − | Methyl | + | RET | − | + | − | − | − | − | − | − | − | − | ||||||||
| 16 | ||||||||||||||||||||||||||
| 1 | 4 | + | − | + | − | − | + | NI | − | + | − | − | − | − | − | − | − | + | ||||||||
| 2 | 4 | + | − | + | − | − | + | NI | − | + | − | + | − | − | − | − | − | − | ||||||||
| 3 | 3 | + | − | + | − | − | + | NI | − | − | − | + | − | − | − | − | − | − | ||||||||
*IHC, immunohistochemistry.
†p14ARF methylation was detected in promoter regions, and deletion in exon 1β.
‡p16INK4a methylation was detected in promoter regions and deletion in exon 1α.
§p16INK4a methylation was detected in exon 2.
¶ NI, noninformative.
∥LOH, loss of heterozygosity.
**RET, retention of heterozygosity.
††n.a., not available.
Figure 1.
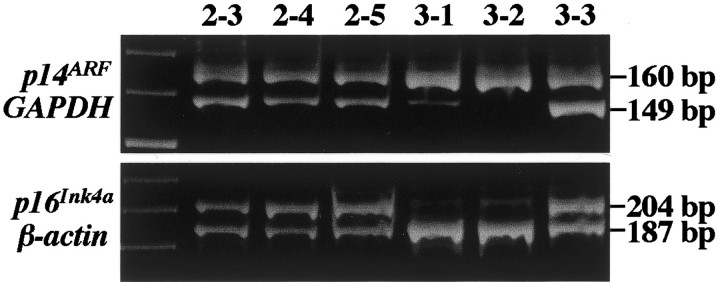
Differential PCR assessing p14ARF and p16INK4a homozygous deletions in prostate carcinomas. Case 2 and foci 3-3 have a normal gene status. Foci 3-1 and 3-2 show p14ARF and p16INK4a co-deletions.
Figure 2.

A: Methylation-specific PCR of CpG islands in the p14ARF and p16INK4a promoters in prostate carcinomas. In foci 7-5, 8-1, and 8-4, only unmethylated DNA (U) is apparent. In case 8, p14ARF and p16INK4a methylation (M) was restricted to foci (8-2 and 8-3) lacking p14ARF and p16INK4a immunoreactivity. N.C., Normal control DNA from a normal blood; P.C., positive control for methylated DNA; U, PCR product amplified by unmethylated-specific primers; M, PCR product amplified by methylated-specific primers. B: Methylation-specific PCR for RB1, p21Waf1, and p27Kip1. Hypermethylation of RB1 and/or p21Waf1 promoters was found in four of five foci in only one tumor (tumor no.1). Hypermethylation of p27Kip1 was demonstrated in four foci in one tumor (tumor no. 8).
Methylation of RB1, p21Waf1, p27Kip1, PTEN, and p73
Hypermethylation of the RB1, p21Waf1, or p27Kip1 promoter was detected in two, two, and three tumors, respectively (Table 1 ▶ , Figure 2 ▶ ). In general, methylation occurred in most, but not all, large foci within these few tumors and seemed to be independent events, ie, methylation of both the RB1 and p21Waf1 promoters was detected in three of five foci in tumor 1 and concurrent methylation of p21Waf1 and p27Kip1 was detected in one of five foci in tumor 8. Methylated and unmethylated control DNAs showed the expected fragment sizes of 163 bp for RB1, 89 bp and 118 bp for p21Waf1, and 195 bp and 212 bp for p27Kip1 (Figure 2) ▶ . Methylation of the PTEN and p73 promoters was not detected in any tumor examined. For RB1, 3 of 12 informative cases had allelic loss in more than two foci. In case 2, LOH was detected in all foci within the tumor.
Immunohistochemistry and Correlation with Genetic and Epigenetic Changes
All 13 tumors with p14ARF expression showed a normal p14ARF gene status. Three tumors showed loss of p14ARF and p16INK4a expression (Table 1) ▶ . Loss of p14ARF/p16INK4a expression seemed to correlate to homozygous co-deletion or promoter co-methylation. In one case (tumor 8), three foci showed expression of both p14ARF and p16INK4a, but the neoplastic cells within two other areas lacked expression of p14ARF/p16INK4a while showing promoter hypermethylation in both (Table 1 ▶ , Figure 3 ▶ ); exon 2 of p16INK4a was also methylated in these two immunonegative foci. Two foci (3-3 and 3-5) in tumor 3 and focus 7-1 in tumor 7 showed loss of both p14ARF and p16INK4a expression without simultaneous deletion or methylation in the genes. Another anomaly of tumor 7 was that four of the five individual foci examined were immunonegative for expression of p16INK4a without further alteration in that gene. Nuclear immunoreactivity to p14ARF and p16INK4a was observed in normal prostate tissues.
Figure 3.

Loss of expression of p14ARF and p16INK4a was associated with promoter methylation. A: p14ARF immunohistochemistry showing nuclear immunoreactivity in the majority of tumor cells within a focus (case 8-1). B: A separate focus within the same tumor (case 8-2) without p14ARF expression. C: Case 8-1 exhibits a focus with extensive marked immunoreactivity for p16INK4a. D: Within the same tumor, another area of tumor cells, 8-2, shows no p16INK4a immunoreactivity. Original magnifications, ×300; hematoxylin counterstain.
Negative immunoreactivity to pRB was evident in a majority of areas within six tumors and five tumors were primarily negative to p27Kip1. Of these samples, tumor 13 showed loss of expression of both genes, whereas no correlation was seen in the remainder of cases. Within this subset, two lesions (tumors 1 and 14) showed promoter hypermethylation of RB1 concurrent with immunonegativity and three tumors (tumors 3, 8, and 13) showed the same phenomenon with p27Kip1. Loss of protein expression tended to correlate with RB1 methylation in six of eight foci (75%) examined within tumors 1 and 14. Thirty-four of 42 foci without LOH showed RB1 expression, but six foci without RB1 methylation showed loss of the expression. In the three lesions with overall loss of p27Kip1 expression, methylation was similarly detected in 12 of 15 foci (80%). However, as can be seen from Table 1 ▶ , focal loss of protein expression without any other detected aberrations in the promoter regions of the genes discussed occurred far more frequently.
A large fraction of prostate carcinomas (10 of 16 cases, 33 of 46 foci) revealed some loss of p21Waf1 expression, but p21Waf1 concurrent hypermethylation was detected in only six immunonegative foci among two tumors (tumors 1 and 8). Five tumors showed collective negative immunoreactivity to both pRB and p21Waf1, and four tumors expressed neither p21Waf1 nor p27Kip1. MDM2 protein was overexpressed in only one focus in each of four tumors and did not seem to correlate with any other gene alterations.
p53 Gene Mutations and p14ARF/MDM2
In our previous examination of these prostate tumors, we detected p53 mutations in seven foci between two tumors; 26 these seven foci also showed expression of p53 protein with immunostaining. In this study, we confirmed the previous results and, additionally, found no significant association between p53 status and alterations in p14ARF or overexpression of MDM2.
Discussion
Within the multistep process of carcinogenesis, clonal populations arising within tumors may undergo separate individual genetic and epigenetic changes leading to aggressive growth advantages. The 16 prostate carcinomas analyzed in the current study demonstrated multiple genetic/epigenetic patterns; several foci showed promoter methylation or deletions in promoter regions that were not detected in other areas within the same tumor.
Methylation of DNA is important in the genetic regulation of mammalian cells. CpG islands are GC-rich areas of the genome corresponding to the promoter regions of genes and are associated with transcriptional activity. The methylation status of these islands has been shown to be involved with inactivation of tumor suppressor genes. Recent available evidence would suggest that changes in gene expression through epigenetic mechanisms may be responsible for tumor progression in prostate cancer. 3 Methylation silencing of p14ARF and p16INK4a is frequent in some types of tumors, including gliomas, colon, and esophageal carcinomas; 12,15,21 however, methylation silencing of p14ARF is extremely rare in lymphomas 13,14,24 and in pancreatic and hepatic carcinomas. 13,14 This indicates that methylation silencing of p16INK4a, rather than of p14ARF, may be the predominant event in the INK4a/ARF (p14ARF/p16INK4a) locus on 9p21 in human cancers.
The p16INK4a and p14ARF genes are also frequently co-deleted in human neoplasms. 27 In this study, we detected co-deletion of p16INK4a and p14ARF genes in two foci within a single tumor. In addition, co-methylation of both genes was found in another two foci of a separate lesion. Either event, ie, co-deletion or co-methylation of p16INK4a and p14ARF, might be involved in deregulating the RB1 or p53 pathway. p14ARF plays a major role in the p53 pathway by binding specifically to MDM2 and thus stabilizing both p53 and MDM2. 8 Recently, Esteller and colleagues 13 reported that p14ARF silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Mutations in p53 may thus occur more rarely in tumors with inactivation of this locus than in tumors with wild-type INK4a/ARF genes. 7 In contrast, the reverse correlation between p14ARFand p53 has not been detected in some human tumors analyzed, including leukemia-lymphoma cell lines and large B-cell lymphomas. 28-30 With regards to our particular study, it is possible that should alterations in p14ARF occur early in the development of prostate carcinomas, the tumors may retain wild-type p53.
In foci 3-1 and 3-2, both p16INK4a homozygous deletion and methylation were observed, which seems to be unusual because the exon 1α and exon 2 are closely located. Similar situation also occurs between exon 1α of p16INK4a and exon 1β of p14ARF, which are encoded, by the same exon 2. A somewhat higher frequency of p14ARF than p16INK4a has been reported in other human neoplasms, including esophageal carcinomas, gliomas, and intracranial malignant lymphomas. 15 12,24 Although the mechanism is currently unknown, the specific deletion of p16INK4a exon 1α may occur in some prostate cancers. Nguyen and colleagues 31 reported that exon 2 of p16INK4a was methylated in both the tumor and normal adjacent prostates, and the normal tissues from prostate cancer patients showed increased exon 2 methylation levels compared to normal prostate from patients without prostate cancer. Therefore, the possibility of normal cell contamination could not be excluded.
In the majority of human neoplasms, a clear correlation has been reported between promoter deletion/methylation and loss of gene expression as detected by immunohistochemistry. 12,20,22,24 The present study also revealed a close correlation between gene inactivation and loss of expression. This was also corroborated by the finding that, in at least one case, promoter hypermethylation was detected only in foci lacking p14ARF and p16INK4a immunoreactivity, although there were foci showing loss of expression without concurrent detectable alterations in the promoter regions. We suggest that mutations other than deletion or methylation may occur in these cases.
Exon 2 in p16INK4a was frequently methylated in the prostate carcinomas examined in this study. Nguyen and colleagues 31 reported a high frequency of p16INK4a exon 2 methylation in human prostate cancers (73%, 8 of 11), which correlated with upregulated p16INK4a transcripts. Methylation patterns of p16INK4a exon 2 in our study were different from those of the other genes investigated in that it seemed to be an all-or-nothing event. The underlying mechanism(s), however, are not clear because hypermethylation of p16INK4a exon 2 was not correlated with p16INK4a expression as demonstrated by immunohistochemistry. This might relate to a complex function for DNA methyltransferase, with possible transcriptional inactivation being responsible for this phenomenon.
Similar situations occurred in between abnormalities in RB1 and protein expression. In brain tumor, 20,22,24 a clear correlation has been reported between RB1 homozygous deletion and/or promoter hypermethylation and loss of pRB expression as detected by immunohistochemistry, yet in our study, some nonmethylated foci were immunohistochemically negative for RB1. We found the correlation between pRB expression and LOH on RB1 locus. pRB expression was detected in a majority of foci showing retention of both alleles. We have previously surveyed all 27 exons of RB1 in a series of prostate carcinomas. Five of 32 cancers (16%) had mutations, but only one exonic mutation occurred. 16 We therefore are led to conclude that RB1 alterations may occur in either introns or exons and may have no effect on amino acid composition. On the other hand, allelic loss of the RB1 gene has been extensively investigated and has been found in 27 to 67% of informative prostate cancers, 1 suggesting that loss of RB1 expression is more likely because of allelic loss of the gene. However, in a few cases, the mutation might be considered one of the mechanisms of inactivation of the RB1 gene, although we did not examine all exons of RB1 in this study, and could not exclude the possibility of normal tissue contamination.
p21Waf1 point mutations seem to be very rare in human tumors. 32 A recent investigation reported significant expression of p21 in prostate carcinoma, but not in normal prostate tissue, and expression did not correlate with p53 status. 33 On the other hand, the loss of expression of both p21Waf1 and p27Kip1 was associated specifically with a reduction in metastases-free, recurrent prostate carcinoma. 34 We found that immunohistochemical expression and methylation occurred independently in both genes and, with the exception of one tumor, did not correlate to p53 status. p27Kip1 is an important cyclin-dependent kinase inhibitor and the mechanism by which p27Kip1 expression is reduced or lost during tumorigenesis remains unclear. Specific alterations of the p27Kip1 gene, including mutations and homozygous deletions, are exceedingly rare in human cancers. 35,36 Worm and colleagues 37 indicated that p27Kip1 methylation might be a cause of monoallelic p27Kip1 silencing in malignant melanoma. Regulation of p27Kip1 expression occurs at various stages. Some cancers, such as osteosarcomas and breast and colon cancers, may express p27Kip1 mRNA, but p27Kip1 protein is not detectable because of rapid proteosome-mediated degradation in the tumor cells. 38 These findings suggest that aberrant p27Kip1 methylation is not the only mechanism causing reduced levels of p27Kip1expression and further imply that epigenetic changes in this gene may also play a role in prostate carcinogenesis.
With regards to PTEN and p73, we could not detect hypermethylation in either gene in the prostate carcinomas examined. Previous studies have reported frequent loss of PTEN expression in localized prostate cancer with poor prognosis. 39,40 However, there is no direct proof for loss of PTEN expression by methylation in the promoter and enhancer regions in PTEN gene. The present results indicate that loss of PTEN expression might be because of the genetic alterations, not hypermethylation, in prostate carcinoma. Although silencing of p73 in acute lymphatic leukemia and in Burkitt’s lymphoma seems to occur through methylation of the untranslated exon 1 of the gene, p73 methylation was not observed in a survey of tumor cell lines nor in a study of breast, renal, and colon cancers. 23
We previously demonstrated that carcinoma of the prostate is genetically multicentric as well as histologically heterogeneous and multifocal. 17,26 Although prostate carcinoma has been recognized as a multifocal disease, it is possible that these areas with alterations might be multifocal smaller foci within a large tumor. The data obtained in this study indicates that prostate carcinoma is not only histologically heterogeneous and multifocal but also genetically multicentric. We could not detect any specific combination of methylation patterns or epigenetic changes occurring in representative focal areas within prostate tumors. However, the overall frequency of independent methylation events uncovered points to a role for methylation, possibly in progression, in prostate carcinogenesis.
Footnotes
Address reprint requests to Noboru Konishi, M.D., Second Department of Pathology, Nara Medical University, 840 Shijo-cho, Kashihara, Nara, 634-8521, Japan. E-mail: nkonishi@naramed-u.ac.jp.
Supported in part by a grant-in-aid for Scientific Research (no. 12670171) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
References
- 1.Konishi N, Cho M, Yamamoto K, Hiasa Y: Genetic changes in prostate cancer. Pathol Int 1997, 47:735-747 [DOI] [PubMed] [Google Scholar]
- 2.Watanabe M, Nakayama T, Shiraishi T, Stemmermann GN, Yatani R: Comparative studies of prostate cancer in Japan versus the United States. A review. Urol Oncol 2000, 5:274–283 [DOI] [PubMed]
- 3.Rennie PS, Nelson CC: Epigenetic mechanisms for progression of prostate cancer. Cancer Metastasis Rev 1998, 17:401-409 [DOI] [PubMed] [Google Scholar]
- 4.Liu Q, Neuhausen S, McClure M, Frye C, Weaver-Feldhaus J, Gruis NA, Eddington K, Allalunis-Turner MJ, Skolnick MH, Fujimura FK, Kamb A: CDKN2 (MTS1) tumor suppressor gene mutations in human tumor cell lines. Oncogene 1995, 10:1061-1067 [PubMed] [Google Scholar]
- 5.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB: Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 55:4525-4530 [PubMed] [Google Scholar]
- 6.Zhang Y, Xiong Y, Yarbrough WG: ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92:725-734 [DOI] [PubMed] [Google Scholar]
- 7.Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA: The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92:713-723 [DOI] [PubMed] [Google Scholar]
- 8.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ: Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA 1998, 95:8292-8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G: The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 1998, 17:5001-5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA: Cancer. Death and methylation. Nature 2001, 409:143-144 [DOI] [PubMed] [Google Scholar]
- 11.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ: Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91:649-659 [DOI] [PubMed] [Google Scholar]
- 12.Nakamura M, Watanabe T, Klangby U, Asker C, Wiman K, Yonekawa Y, Kleihues P, Ohgaki H: p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol 2001, 11:159-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esteller M, Cordon-Cardo C, Corn PG, Meltzer SJ, Pohar KS, Watkins DN, Capella G, Peinado MA, Matias-Guiu X, Prat J, Baylin SB, Herman JG: p14ARF silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Cancer Res 2001, 61:2816-2821 [PubMed] [Google Scholar]
- 14.Esteller M, Corn PG, Baylin SB, Herman JG: A gene hypermethylation profile of human cancer. Cancer Res 2001, 61:3225-3229 [PubMed] [Google Scholar]
- 15.Xing EP, Nie Y, Song Y, Yang GY, Cai YC, Wang LD, Yang CS: Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin Cancer Res 1999, 5:2704-2713 [PubMed] [Google Scholar]
- 16.Konishi NHY, Tsuzuki T, Matsuda H, Tao M, Nakamura M, Naitoh H, Kitahori Y, Shiraishi T, Yatani R, Shimazaki J, Lin JC: Detection of pRB, p16/CDKN2 and p15INK4B gene alterations with immunohistochemical studies in human prostate carcinomas. Int J Oncol 1996, 8:107-112 [DOI] [PubMed] [Google Scholar]
- 17.Konishi N, Hiasa Y, Nakamura M, Kitahori Y, Matsubara K, Nagai H: Different patterns of DNA alterations detected by restriction landmark genomic scanning in heterogeneous prostate carcinomas. Am J Pathol 1997, 150:305-314 [PMC free article] [PubMed] [Google Scholar]
- 18.Gleason DF: Histologic grading of prostate cancer: a perspective. Hum Pathol 1992, 23:273-279 [DOI] [PubMed] [Google Scholar]
- 19.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H: Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 2001, 81:77-82 [DOI] [PubMed] [Google Scholar]
- 21.Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB, Herman JG: Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res 2000, 60:129-133 [PubMed] [Google Scholar]
- 22.Simpson DJ, Hibberts NA, McNicol AM, Clayton RN, Farrell WE: Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res 2000, 60:1211-1216 [PubMed] [Google Scholar]
- 23.Corn PG, Kuerbitz SJ, van Noesel MM, Esteller M, Compitello N, Baylin SB, Herman JG: Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt’s lymphoma is associated with 5′ CpG island methylation. Cancer Res 1999, 59:3352-3356 [PubMed] [Google Scholar]
- 24.Nakamura M, Sakaki T, Hashimoto H, Nakase H, Ishida E, Shimada K, Konishi N: Frequent alterations of the p14ARF and p16INK4a genes in primary central nervous system lymphomas. Cancer Res 2001, 61:6335-6339 [PubMed] [Google Scholar]
- 25.Toguchida J, McGee TL, Paterson JC, Eagle JR, Tucker S, Yandell DW, Dryja TP: Complete genomic sequence of the human retinoblastoma susceptibility gene. Genomics 1993, 17:535-543 [DOI] [PubMed] [Google Scholar]
- 26.Konishi N, Hiasa Y, Matsuda H, Tao M, Tsuzuki T, Hayashi I, Kitahori Y, Shiraishi T, Yatani R, Shimazaki J, Lin JC: Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol 1995, 147:1112-1122 [PMC free article] [PubMed] [Google Scholar]
- 27.Orlow I, LaRue H, Osman I, Lacombe L, Moore L, Rabbani F, Meyer F, Fradet Y, Cordon-Cardo C: Deletions of the INK4A gene in superficial bladder tumors. Association with recurrence. Am J Pathol 1999, 155:105-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharpless NE, DePinho RA: The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev 1999, 9:22-30 [DOI] [PubMed] [Google Scholar]
- 29.Moller MB, Ino Y, Gerdes AM, Skjodt K, Louis DN, Pedersen NT: Aberrations of the p53 pathway components p53, MDM2 and CDKN2A appear independent in diffuse large B cell lymphoma. Leukemia 1999, 13:453-459 [DOI] [PubMed] [Google Scholar]
- 30.Drexler HG: Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998, 12:845-859 [DOI] [PubMed] [Google Scholar]
- 31.Nguyen TT, Nguyen CT, Gonzales FA, Nichols PW, Yu MC, Jones PA: Analysis of cyclin-dependent kinase inhibitor expression and methylation patterns in human prostate cancers. Prostate 2000, 43:233-242 [DOI] [PubMed] [Google Scholar]
- 32.Shiohara M, el-Deiry WS, Wada M, Nakamaki T, Takeuchi S, Yang R, Chen DL, Vogelstein B, Koeffler HP: Absence of WAF1 mutations in a variety of human malignancies. Blood 1994, 84:3781-3784 [PubMed] [Google Scholar]
- 33.Aaltomaa S, Lipponen P, Eskelinen M, Ala-Opas M, Kosma VM: Prognostic value and expression of p 21waf1/cip1 protein in prostate cancer. Prostate 1999, 39:8-15 [DOI] [PubMed] [Google Scholar]
- 34.Cheng L, Lloyd RV, Weaver AL, Pisansky TM, Cheville JC, Ramnani DM, Leibovich BC, Blute ML, Zincke H, Bostwick DG: The cell cycle inhibitors p21WAF1 and p27KIP1 are associated with survival in patients treated by salvage prostatectomy after radiation therapy. Clin Cancer Res 2000, 6:1896-1899 [PubMed] [Google Scholar]
- 35.Morosetti R, Kawamata N, Gombart AF, Miller CW, Hatta Y, Hirama T, Said JW, Tomonaga M, Koeffler HP: Alterations of the p27KIP1 gene in non-Hodgkin’s lymphomas and adult T-cell leukemia/lymphoma. Blood 1995, 86:1924-1930 [PubMed] [Google Scholar]
- 36.Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP: p27/Kip1 mutation found in breast cancer. Cancer Res 1996, 56:2400-2404 [PubMed] [Google Scholar]
- 37.Worm J, Bartkova J, Kirkin AF, Straten P, Zeuthen J, Bartek J, Guldberg P: Aberrant p27Kip1 promoter methylation in malignant melanoma. Oncogene 2000, 19:5111-5115 [DOI] [PubMed] [Google Scholar]
- 38.Hengst L, Reed SI: Translational control of p27Kip1 accumulation during the cell cycle. Science 1996, 271:1861-1864 [DOI] [PubMed] [Google Scholar]
- 39.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D: Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res 1997, 57:4997-5000 [PubMed] [Google Scholar]
- 40.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR: Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res 1999, 59:4291-4296 [PubMed] [Google Scholar]