

Abstract
Microvascular injury has been proposed to be a main cause of ischemia-reperfusion (I/R) injury. The roles of endothelial nitric oxide synthase (eNOS)-derived NO, a key regulator of vascular function, in I/R injury are incompletely understood. We used transgenic mice overexpressing eNOS in endothelial cells (eNOS-Tg) and their littermates wild-type mice (WT) to investigate the roles of eNOS in I/R injury in skeletal muscle. Superoxide levels in the affected muscles were reduced by approximately 50% in eNOS-Tg compared with WT during reperfusion. In WT, the disassembly of endothelial junctional proteins seen in the early period of reperfusion was recovered in the later phase. These findings were correlated with the increased vascular permeability in vivo. In contrast, eNOS-Tg maintained the endothelial junction assembly as well as vascular permeability during reperfusion. Leukocyte extravasation into tissue and up-regulated expression of adhesion molecules in the reperfused vessels were significantly inhibited in eNOS-Tg. Tissue viability of the affected muscle was decreased in WT time-dependently after reperfusion, whereas eNOS-Tg showed no significant reduction. NOS inhibition completely reversed these protective effects of eNOS overexpression in I/R injury. Thus, eNOS overexpression appears to prevent the I/R injury in skeletal muscle by maintaining vascular integrity.
One of the most serious problems in cardiovascular medicine is ischemia-reperfusion (I/R)-induced tissue injury, in which restoration of blood flow to ischemic tissues or organs causes more deleterious consequences, in various clinical settings such as thrombolytic therapy, coronary angioplasty, cardiopulmonary bypass, and organ transplantation. 1 In the pathological process of I/R injury, various microvascular dysfunctions, including impaired endothelium-dependent vasodilation and promoted oxidant productions, are widely recognized to trigger tissue injury. 1,2 The increased oxidant production activates multiple intracellular signaling and gene expression. In addition, I/R increases vascular permeability in the site of postcapillary venules. 1,3 Vascular permeability is considered to be regulated mainly by cell-cell junctions of endothelial cells consisting of tight junction and vascular endothelial (VE)-cadherin-based adherens junction. 4-6 The disassembly of endothelial cell-cell junctions causes increases in protein leakage, 6 and subsequently various cytotoxic and inflammatory mediators infiltrate into the affected tissues. 1 Further, the disorganization of endothelial junctional proteins is suggested to contribute to leukocyte transmigration. 7,8 Adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) participating in leukocyte-endothelial interactions also play important roles in leukocyte extravasation. 2
Nitric oxide (NO) is a key regulator of vascular functions that play important roles in physiology as well as in pathophysiology of various cardiovascular diseases. There has been a controversy about the generation of NO during I/R. NO production is shown to be increased during ischemia and in the very early periods of reperfusion in rat cerebral or isolated heart I/R model. 9,10 In contrast, other studies suggested that NO generation decreases due to reduced expression and inactivation of endothelial NO synthase (eNOS) in the affected vessels. 11,12 The roles of NO in I/R injury have not been established because of its biphasic aspects, cytoprotective or cytotoxic, in different organs and a variety of experimental conditions. 13-16 L-arginine, a substrate for NOS enzymes, and NO donors have been demonstrated to exert protective effects against I/R injury in rat skeletal muscle and rat mesenteric artery in vivo. 17,18 In contrast, studies using NOS inhibitors have shown either protective or detrimental effects of NO in I/R tissue injury. 16,19 Thus, the roles of endogenous NO, particularly that derived from eNOS, in the pathogenesis of I/R have not been clearly elucidated.
We have previously established transgenic mice in which eNOS is overexpressed mainly in the endothelium, 20 and have presented protective effects of eNOS-derived NO in lung inflammation induced by lipopolysaccharide 21 or various types of vascular remodeling. 22,23 In the present study, we investigated the roles of NO in I/R injury in skeletal muscle of mice hindlimb by the use of eNOS-transgenic (eNOS-Tg) and their littermates wild-type (WT) mice. Particularly, we focused on the vascular integrity such as assembly of endothelial junctions and in vivo vascular permeability of plasma proteins and leukocytes. Also, we examined the effects of eNOS overexpression on oxidants production in the skeletal muscle tissues after I/R.
Materials and Methods
Chemicals and Antibodies
All chemicals were obtained from Sigma Chemical Co. (St. Louis, MO, USA) unless specified. Pentobarbital sodium was purchased from Abbot Laboratories (North Chicago, IL, USA). Evans Blue dye (EBD) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). The following antibodies were used: a rabbit polyclonal anti-eNOS antibody, and a mouse monoclonal anti-β-catenin antibody (Transduction Laboratories, Lexington, KY, USA); a rat monoclonal anti-VCAM-1 antibody (CHEMICON International, Temecula, CA, USA); a goat polyclonal anti-VE-cadherin antibody (Santa Cruz, Santa Cruz, CA, USA); and a mouse monoclonal anti-ICAM-1 antibody (Genzyme/Techne, Cambridge, MA, USA).
I/R Injury Model in Mice
Male eNOS-Tg (n = 166) mice and their littermates WT (n = 100) were derived from the same genetic background (C57BL/6) as previously described. 20 In eNOS-Tg mice, to evaluate the precise effects of eNOS overexpression in the pathological process of I/R, 73 animals were treated with a non-selective NOS inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME), in drinking water (1 mg/ml) for one week before the experiment. Complete inhibition of the endogenous NOS activity was confirmed as described previously. 21,22 Mice were anesthetized with pentobarbital sodium (80 μg/kg) intraperitoneally and placed on a heated table, and the body temperature was maintained at 37°C throughout the experiments. Ischemia was induced by occluding right femoral arteries for 30 minutes with the use of vascular clips (AESCULAP), and the arteries were subsequently reperfused for 60 or 240 minutes by releasing the clips. In the following experimental protocol, all tissue samples were obtained from gastrocnemius muscles. All animal experiments were conducted according to the Guidelines for Animal Experimentation at Kobe University Graduate School of Medicine.
Protein Analysis for eNOS
The expression of eNOS in hindlimb skeletal muscle was analyzed by immunoblotting, and NOS enzymatic activity was determined by conversion of [3H]-L-arginine to [3H]-L-citrulline as described previously. 20 Enzyme activity was expressed as citrulline production in picomole per milligram of protein per minute.
Measurements of NO Release from Skeletal Muscles
The skeletal muscles in hindlimbs were excised, and incubated in O2-saturated Krebs’ solution at 37°C for 60 minutes. After the supernatants were collected, the muscles were again incubated in O2-saturated Krebs’ solution containing 30 μmol/L acetylcholine at 37°C for 60 minutes. Then, the supernatants collected under these two conditions were subjected to nitrite measurement by using a NOx analyzer (ozone chemiluminescence), as reported previously. 24,25 The difference in NO release, calculated by subtracting NO release in the non-stimulated condition from that in the acetylcholine-stimulated condition, was expressed as picomole per milligram of protein per hour.
Imuunohistochemistry and Immunofluorescence
Immunohistochemical stainings for eNOS, ICAM-1, and VCAM-1 of the vasculatures in gastrocnemius muscles were performed by the labeled streptavidin biotin method as previously described. 20 VE-cadherin and β-catenin were visualized by immunofluorescence microscopy. Briefly, frozen sections were fixed in acetone for 10 minutes, blocked with carrier protein for 60 minutes, and incubated with a primary antibody for 3 hours. In the case of mouse primary antibodies, fixed sections were blocked with blocking solution from the HistoMouse plus kit (Zymed, South San Francisco, CA, USA) following the manufacturer’s instructions. Each section was then incubated with the biotinylated secondary antibody for 30 minutes and subsequently with horseradish peroxidase-labeled streptavidin or FITC-labeled streptavidin for 20 minutes. To detect horseradish peroxidase activity, endogenous peroxides were quenched with 0.03% hydrogen peroxide. Fluorescence was examined by the laser scanning confocal imaging system (MRC-1024, Bio-Rad Laboratories).
Measurements and in Situ Detection of Superoxide
Superoxide levels in muscles were measured by the lucigenin-enhanced chemiluminescence method with the use of 10 μmol/L lucigenin. In situ dihydroethidium fluorescence was performed according to the method described previously. 26 Briefly, the unfixed frozen tissues were cut into 10-μm-thick sections, and incubated with 2 × 10−6 mol/L dihydroethidium at 37°C for 30 minutes in a light-protected humidified chamber. The images were obtained by the laser scanning confocal imaging system (MRC-1024, Bio-Rad).
Evaluation of Vascular Permeability
EBD, which binds to plasma proteins, was used as a tracer for extravascular protein leakage. 27 At 60 or 240 minutes after reperfusion, EBD (30 mg/kg) was injected through the tail vein under anesthesia. Five minutes after EBD administration, the peripheral vascular bed was perfused with 0.9% NaCl containing heparin (10 units/ml) followed by 4% paraformaldehyde in 0.05 mol/ml citrate buffer (pH 3.5). The gastrocnemius muscle was removed, rinsed in ice cold phosphate-buffered solution, gently blotted, and weighed. Half of the tissue was dried by incubation at 60°C for 48 hours, and reweighed. The remaining tissue was incubated in 1 ml of formamide at room temperature for 48 hours to extract EBD. EBD contents in tissues were quantified by measuring the optical density of the formamide extract at the wavelength of 620 nanometer and adjusted with tissue weight. Vascular permeability was expressed as a ratio of EBD extravasation in the ischemic muscle to that in the non-ischemic contralateral muscle.
Myeloperoxidase Assay
Myeloperoxidase (MPO) activity in skeletal muscle was examined on the basis of the method described previously. 21,28 In brief, the excised skeletal muscle was rinsed with phosphate-buffered saline, blotted, and weighed. The muscle was homogenized in 50 mmol/L potassium-phosphate-buffered solution (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (HTAB). After centrifuging at 25,000 × g for 15 minutes at 4°C, the supernatant was collected. The pellet was resuspended in 1 ml of potassium-phosphate buffered solution containing HTAB, and sonicated for 30 seconds. The suspension was centrifuged again at 25,000 × g for 15 minutes at 4°C, and the supernatant was collected. This extraction procedure was repeated three times. The supernatant collected in the above procedure was mixed 1:30 (v/v) with 50 mmol/L potassium-phosphate-buffered solution containing 0.167 mg/ml o-dianisidine dihydrochloride and 0.0005% hydrogen peroxide. Then, the absorbance was measured at 460 nm for 5 minutes. MPO activity, calculated by a change in the absorbance for 5 minutes, was adjusted with the protein content, and shown as delta absorbance (ABS) per minutes per milligram of protein.
Quantitative Analysis of Tissue Viability (WST-1 Assay)
To quantify tissue viability, mitochondrial dehydrogenase activity was evaluated by assaying its cleaving activity of the tetrazolium salt WST-1 (Roche Molecular Biochemicals, Basel, Switzerland), with modifying the method described by Belkin. 29 Briefly, the ischemia-reperfused gastrocnemius muscle and the contralateral non-injured muscle as a control were homogenized in 500 μl of 0.25 mol/L sucrose. After centrifugation at 3,000 × g for 5 minutes, 100 μl of supernatant was mixed with 10 μl of WST-1 reagent in microtiter plates. After shaking thoroughly for 1 minute, the reaction mixture was incubated under a humidified atmosphere (37°C, 5% CO2). The absorbance of the red colored formazan dye cleaved from WST-1 was measured at 450 nm by the spectrophotometer U2000 (Hitachi). Reactions were performed in triplicate. The absorbance was adjusted with the protein content, and the result was expressed as a ratio of the reduced WST-1 activity in the ischemic muscle to that in the contralateral muscle.
Statistical Analysis
Data were expressed as mean ± SEM. Unpaired Student’s t-test was used to compare the protein levels of eNOS, NOS activity, and NO release in the skeletal muscles between WT and eNOS-Tg mice. For other data, two-way analysis of variance was used to compare the differences between two groups with Bonferroni’s test for post hoc analysis. Values of P <0.05 were considered statistically significant.
Results
Expression of eNOS and NOS Activity in Skeletal Muscle
Expression of eNOS in hindlimb skeletal muscle was analyzed by both immunoblotting and immunohistochemistry. As shown in Figure 1A and B ▶ , the protein expression of eNOS in skeletal muscles was significantly increased in eNOS-Tg mice compared to WT mice. The Ca2+-dependent NOS activity in skeletal muscle of eNOS-Tg mice was also 1.5-fold higher than that of WT mice (Figure 1C) ▶ . In addition, the acetylcholine-stimulated NO release in skeletal muscle of eNOS-Tg mice was approximately twofold higher than that of WT mice (24.7 ± 1.5 vs. 12.8 ± 5.7 pmol/mg protein/hour respectively; P <0.05; Figure 1D ▶ ). Immunohistochemistry showed that eNOS expression in endothelial cells of arterioles, capillary microvessels, and postcapillary venules was higher in eNOS-Tg mice than in WT mice (Figure 2) ▶ .
Figure 1.

A: Immunoblotting for eNOS in skeletal muscle. Protein expression of eNOS in skeletal muscle was significantly higher in eNOS-Tg mice than WT mice. B: Immunoblots were analyzed and quantified by densitometry. Open and solid bars indicate the values of WT and eNOS-Tg mice, respectively. C: Ca2+-dependent NOS activity in skeletal muscle. Ca2+-dependent NOS activity in skeletal muscle was also significantly higher in eNOS-Tg mice than WT mice. Enzyme activity was expressed as citrulline production in picomole per milligram of protein per minute. D: NO release from ex vivo skeletal muscle. Acetylcholine-stimulated NO release from ex vivo skeletal muscle was assessed by measuring nitrite production with the use of a NOx analyzer. *P <0.05 vs. the value for WT mice.
Figure 2.
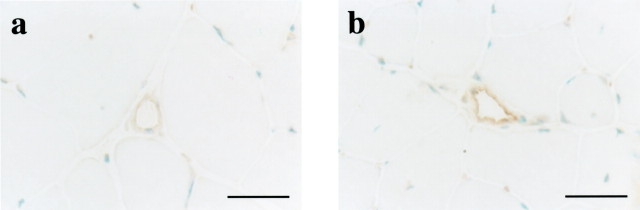
Expression of eNOS in blood vessels of skeletal muscle. Immunohistochemistry using a rabbit polyclonal anti-eNOS antibody revealed higher expression of eNOS in microvessels of skeletal muscle in eNOS-Tg mice (b) than in WT mice (a). Scale bar, 50 μm.
Superoxide Levels in Ischemia-Reperfused Muscles
Reoxygenation by I/R is known to enhance oxidant production and exaggerate reperfusion tissue injury. 2 As shown in Figure 3 ▶ a and c, I/R caused a significant increase in in situ detection of superoxide with dihydroethidium fluorescence in the ischemic limb of WT mice after 60 minutes of reperfusion. This increase was markedly suppressed in the affected limbs of eNOS-Tg mice (Figure 3, b and e) ▶ . In contrast, L-NAME treatment increased the intensity of dihydroethidium fluorescence in eNOS-Tg mice after reperfusion, showing the reversed effect of NOS inhibitor on superoxide production (Figure 3, c and f) ▶ . At 240 minutes after reperfusion, similar results were obtained (data not shown). As revealed also by lucigenin-enhanced chemiluminescence method, superoxide levels in the ischemic muscles of WT mice were increased by twofold of the non-injured control muscles during reperfusion (Figure 4) ▶ . In eNOS-Tg mice, however, the superoxide levels were significantly lower than in WT mice at both reperfusion time points. On the other hand, in L-NAME-treated eNOS-Tg mice, superoxide production in the reperfused muscles were comparable to the levels in WT mice at 60 minutes, and increased at 240 minutes after reperfusion.
Figure 3.
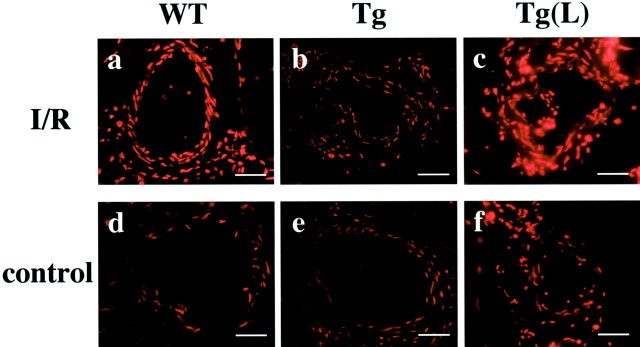
In situ superoxide detection in the reperfused vessels of skeletal muscle. In situ superoxide detection with dihydroethidium fluorescence in blood vessels of ischemia-reperfused (a–c) and non-ischemic control muscle (d–f) from WT (a, d), eNOS-Tg mice (b, e), and eNOS-Tg mice treated with L-NAME (c, f) at 60 minutes after reperfusion. Scale bar, 20 μm.
Figure 4.

Superoxide production in ischemia-reperfused muscle. Superoxide levels in the ischemia-reperfused muscle was measured by the lucigenin-enhanced chemiluminescence method after 60 and 240 minutes of reperfusion following ischemia for 30 minutes. Shown is a ratio of superoxide detected in the affected limb to that in the non-ischemic limb of the same animal in WT (open bar), eNOS-Tg mice (solid bar), and eNOS-Tg mice treated with L-NAME (hatched bar). n = 5 to 6 for each group. *,†P <0.05 vs. the value for WT mice and L-NAME-treated eNOS-Tg mice at 60 or 240 minutes after reperfusion, respectively.
Alterations of Vascular Integrity Caused by I/R
It is established that endothelial cells form cell-cell junctions composed of VE-cadherin-based adherens junction and tight junction to maintain vascular integrity, including the regulation of vascular permeability. 6,30 As adherens junction assembly is considered to be essential for the formation of tight junction, 6 we studied the effects of I/R on endothelial adherens junction assembly by immunofluorescence of key components such as VE-cadherin and β-catenin which interacts with cytoplasmic tail of VE-cadherin. 6 After reperfusion for 60 minutes following ischemia for 30 minutes, VE-cadherin and β-catenin were hardly detectable at cell-cell contact sites of endothelial cells in the reperfused vessels in WT mice (Figure 5 ▶ c and i). In contrast, this disappearance of adherens junction components did not occur in eNOS-Tg mice at the same time point (Figure 5, d and j) ▶ . After reperfusion for 240 minutes, VE-cadherin and β-catenin were seen at cell-cell contacts of endothelial cells in the reperfused vessels in both genotypes (Figure 5, e, f, k, and l) ▶ . In eNOS-Tg mice treated with L-NAME, the similar findings seen in WT mice were observed (data not shown). Thus, it seems that the reorganization of endothelial cell-cell junctions occurs at the earlier phase of reperfusion in WT mice, which is prevented by overexpression of eNOS. In the later phase of reperfusion, the assembly of endothelial junctions appears to be maintained in both genotypes.
Figure 5.
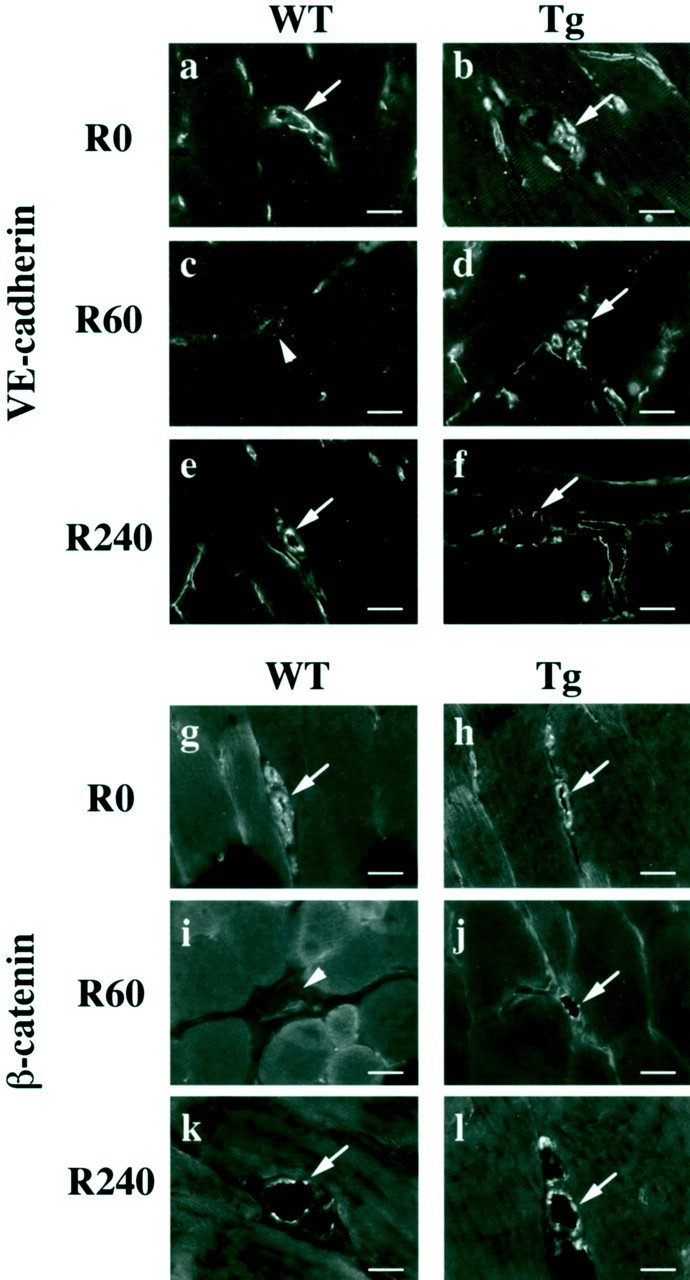
Endothelial adherens junction assembly in reperfused vessels. Immunofluorescence of adherens junction components VE-cadherin (a–f) and β-catenin (g–l) are shown. Immunoreactivities of VE-cadherin and β-catenin were detected using a goat polyclonal anti-VE-cadherin antibody and a mouse monoclonal anti-β-catenin antibody. After reperfusion for 60 minutes, both VE-cadherin and β-catenin disappeared from cell-cell contacts of endothelial cells in reperfused vessels in WT mice (arrowhead, c, i). On the other hand, this disappearance was not seen in eNOS-Tg mice at the same time point (arrow, d, j). After 240 minutes of reperfusion, adherens junction components were localized at cell-cell contacts in endothelial cells in both WT (e, k) and eNOS-Tg mice (f, l). The figures are representatives of WT (n = 6) and eNOS-Tg (n = 6) mice. Similar results were obtained from all of the examined mice of both genotypes. Scale bar, 20 μm.
Next, vascular permeability in vivo was evaluated by measuring extravasation of EBD, which is considered to reflect plasma albumin leakage. 27 EBD extravasation levels in non-ischemic control muscle were similar between WT and eNOS-Tg mice (data not shown). At 60 minutes after reperfusion, the EBD extravasation in the ischemia-reperfused muscle in WT mice was increased by 1.7-fold compared with the non-ischemic contralateral muscle (Figure 6) ▶ . In contrast, the ischemia-reperfused muscle in eNOS-Tg mice showed no significant increase in the EBD extravasation at 60 minutes after reperfusion. In L-NAME-treated eNOS-Tg mice, the inhibitory effect of eNOS overexpression on the EBD extravasation was completely cancelled. In the later phase of reperfusion (at 240 minutes after reperfusion), the EBD extravasation in the ischemia-reperfused muscle in the three groups was not significantly different from that of non-ischemic muscle (Figure 6) ▶ . Thus, in accordance with the reorganization of endothelial cell-cell junctions (Figure 5) ▶ , vascular permeability in vivo appears to increase in the earlier phase of reperfusion, and this increase was attenuated by eNOS overexpression.
Figure 6.

Vascular permeability in ischemia-reperfused skeletal muscle. Vascular permeability in vivo was evaluated by measuring extravasation of tracer EBD. Shown is a ratio of EBD extravasation in ischemic limb to that in non-ischemic limb in WT (open bar), eNOS-Tg mice (solid bar), and L-NAME-treated eNOS-Tg mice (hatched bar). n = 5 to 9 for each group. *P <0.05 vs. WT and L-NAME-treated eNOS-Tg mice at 60 minutes after reperfusion.
Expression of Adhesion Molecules in Endothelial Cells and Leukocyte Infiltration after I/R
There is accumulating evidence that exaggerated trafficking of leukocytes in postischemic venules is one of important pathogenic factors in I/R tissue injury. 1,2 Leukocyte extravasation is mediated by leukocyte-endothelial interactions via adhesion molecules expressed on endothelial cells such as ICAM-1 and VCAM-1. 2 Therefore, the expression of ICAM-1 and VCAM-1 in postischemic microvessels was examined by immunohistochemistry. In non-ischemic control muscle of both genotypes, ICAM-1 and VCAM-1 were hardly detectable in microvessels (Figure 7 ▶ b, d, f, and h). After 240 minutes, but not 60 minutes, of reperfusion, the immunoreactivities of both ICAM-1 and VCAM-1 were increased in the reperfused microvessels of WT mice (Figure 7, a and e) ▶ , whereas this increase of ICAM-1 or VCAM-1 expression did not occur in eNOS-Tg mice (Figure 7, c and g) ▶ . On the other hand, the up-regulated expression of these adhesion molecules was observed in L-NAME-treated eNOS-Tg mice (data not shown).
Figure 7.
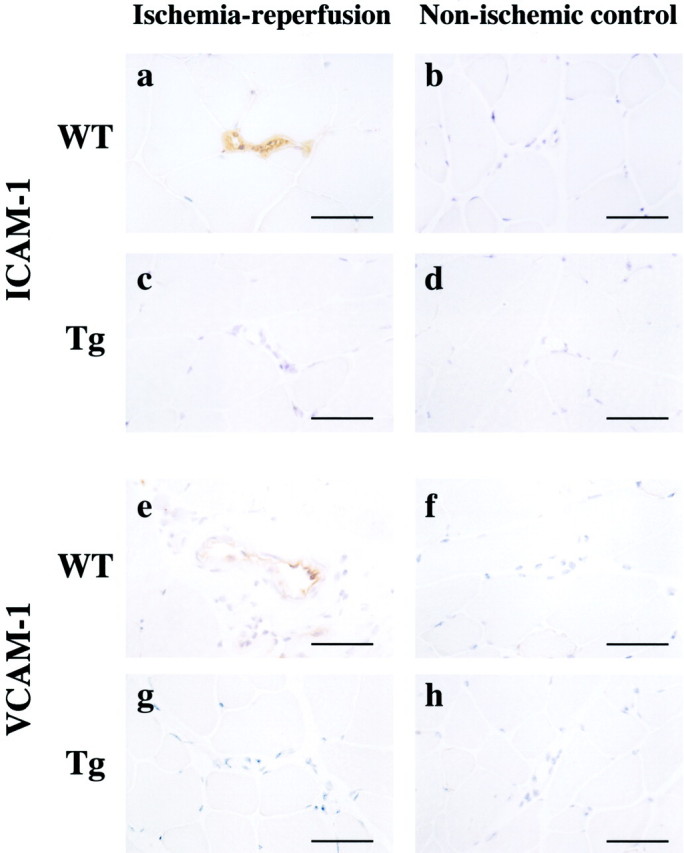
ICAM-1 and VCAM-1 expression in reperfused muscle. After 240 minutes of reperfusion, affected tissues were subject to immunohistochemistry of ICAM-1 (a–d) and VCAM-1 (e–h) using a mouse monoclonal anti-ICAM-1 antibody and a rat monoclonal anti-VCAM-1 antibody. The figures are representatives of WT (n = 6) and eNOS-Tg mice (n = 6). ICAM-1 and VCAM-1 were hardly detectable in vessels of non-injured control skeletal muscles (b, d, f, and h). After 240 minutes of reperfusion, expression of both ICAM-1 and VCAM-1 in microvessels were increased in WT mice (a, e), but attenuated by eNOS overexpression (c, g). Similar results were obtained from all of the examined mice of both genotypes. Scale bar, 50 μm.
Leukocyte infiltration into the reperfused tissues was assessed by quantifying MPO activity in skeletal muscle. After 60 minutes of reperfusion, MPO activities in ischemia-reperfused muscle were similar between ischemic and non-ischemic limbs of all three groups (Figure 8) ▶ . However, at 240 minutes after reperfusion, MPO activity in the affected limb of WT mice was remarkably increased, when compared to that in the non-ischemic contralateral muscle (Figure 8) ▶ . In contrast, the increase in MPO activity was significantly repressed in eNOS-Tg mice. L-NAME treatment for eNOS-Tg mice increased MPO activity to the level comparable to that in WT mice. These results indicate that eNOS overexpression suppresses expression of ICAM-1 and VCAM-1 leading to leukocyte infiltration in the later phase of reperfusion.
Figure 8.

MPO activity in reperfused tissues. MPO activity was assayed in both ischemia-reperfused (I) and non-ischemic contralateral muscle (NI) in WT (n = 5), eNOS-Tg mice (n = 5), and L-NAME-treated eNOS-Tg mice (n = 5). The increases in MPO activities in the reperfused muscles detected in WT and L-NAME-treated eNOS-Tg mice were significantly inhibited in eNOS-Tg mice. *P <0.001 vs. the value for WT mice and L-NAME-treated eNOS-Tg mice at 240 minutes after reperfusion.
Attenuated Tissue Injury after Reperfusion in eNOS-Tg Mice
Tissue injury triggers disruption of mitochondria and subsequent depletion of essential reduction-oxidation coenzymes like mitochondrial dehydrogenase. 31 To evaluate the effects of I/R on tissue viability, mitochondrial dehydrogenase activity was quantified by spectrophotometrically measuring its cleaving activity of tetrazolium salt WST-1 to formazan. After 30 minutes of ischemia, mitochondrial dehydrogenase activity of limb muscle was declined to about 80% of non-ischemic control limb muscle in all groups (Figure 9) ▶ . This observation indicates that ischemia for 30 minutes produced tissue injury to similar extents among three groups and that eNOS overexpression did not affect the ischemia-induced tissue injury.
Figure 9.

Tissue viability after ischemia-reperfusion. To evaluate tissue viability, mitochondrial dehydrogenase activity was determined in hindlimb skeletal muscle after 60 or 240 minutes of reperfusion following 30 minutes of ischemia in WT (open bar), eNOS-Tg mice (solid bar), and L-NAME-treated eNOS-Tg mice (hatched bar). y axis shows a ratio of mitochondrial dehydrogenase activity in the affected limb to that in the contralateral control limb, at 0, 60, and 240 minutes after reperfusion in three groups. Data were obtained from 5 to 8 animals per each group. *P <0.05 vs. the value for WT before reperfusion, †P <0.05 vs. the value for L-NAME-treated eNOS-Tg mice before reperfusion, ‡,§P <0.05 vs. the value for WT mice or L-NAME-treated eNOS-Tg mice at 240 minutes after reperfusion, respectively.
In the reperfusion phase, mitochondrial dehydrogenase activity of the affected limb muscle of WT mice was further decreased in a time-dependent manner (Figure 9) ▶ . In eNOS-Tg mice, however, the decrease in mitochondrial dehydrogenase activity in the affected limb muscle was significantly attenuated and hardly detectable (Figure 9) ▶ . In L-NAME-treated eNOS-Tg mice, as in WT mice, the mitochondrial dehydrogenase activity progressively declined after reperfusion. These results suggest that eNOS overexpression is protective against I/R-induced tissue injury in mice skeletal muscle.
Discussion
In the present study, we demonstrated with the use of eNOS-Tg mice that overproduction of NO derived from eNOS protected against I/R injury in the hindlimb skeletal muscle by reducing superoxide levels, preventing protein leakage from vessels, and abrogating leukocyte extravasation. I/R is known to trigger microvascular response to injury that exhibits various endothelial cell dysfunctions. These include enhanced oxidants production as well as increased vascular permeability to plasma protein and leukocytes in postischemic vessels. 1,2 Oxidants induce endothelial cytotoxity through oxidation of cellular lipids, protein, and nucleic acids. 2 In addition, oxidants promote the production of inflammatory cytokines and the expression of adhesion molecules in endothelial cells, and inactivate endothelial cell-derived NO. 2 In the process of I/R, both overproduced superoxide and reduced NO synthase activity appear to decrease NO availability within endothelial cells. 2,11 In this study, superoxide levels in the postischemic muscles of WT mice, measured by the lucigenin-enhanced chemiluminescence method, were elevated in the reperfusion phase (Figure 4) ▶ . In eNOS-Tg mice, but not L-NAME-treated eNOS-Tg mice, however, superoxide levels in the postischemic muscles were not enhanced in the reperfusion phase. In situ detection of superoxide supports these observations (Figure 3) ▶ . The superoxide levels detected by the method used in this study may not provide the accurate quantity of endogenous superoxide generation because of the methodological limitation as previously pointed out. 32,33 However, the decreased levels of superoxide detected in the affected tissues of eNOS-Tg mice were completely reversed by L-NAME treatment (Figure 4) ▶ . Therefore, our present findings reveal that eNOS overexpression decreases superoxide levels in the reperfused skeletal muscles.
Vascular permeability, a key component of vascular integrity, is regulated mainly by endothelial cell-cell adhesion maintained by cell-cell junctions. 4-6 We found transient and reversible disassembly of endothelial junctions in the earlier phase of reperfusion in our I/R model, which showed a good correlation with an increase in vascular plasma leakage observed in a similar time course (Figures 5 and 6) ▶ ▶ . eNOS overexpression seems to prevent the reorganization of endothelial junctions leading to an increase in vascular plasma leakage. Our findings demonstrate a novel function of NO in the maintenance of endothelial cell-cell adhesion in vivo. On the other hand, NO is shown to mediate increases in vascular permeability, particularly in vascular endothelial growth factor-induced vascular permeability. 34,35 In some animal models of I/R, NO is also shown to promote vascular leakage after I/R. 36,37 The discrepant results may be related to the differences in disease models, including duration and severity of ischemia, targeted organs examined, or animal species. However, most of those studies were performed by using NO donors or NOS inhibitors, which did not allow the direct assessment of NO derived from eNOS on vascular permeability. In the present study, we examined, for the first time, the role of eNOS-derived NO on vascular leakage elicited by I/R in skeletal muscle with the use of eNOS gene-engineered mice. Our study may serve to elucidate the discrepancy on the role of NO on I/R-induced vascular leakage.
I/R-induced increases in vascular permeability have been linked to pathological events such as oxidant stress 38 and interaction between leukocytes and endothelial cells. 7,8 Activated leukocytes and injured endothelial cells generate enormous amounts of reactive oxygen species, and adhesion of leukocyte to the vessel wall is proposed to trigger the intracellular signals that disorganize VE-cadherin/β-catenin complex in endothelial cells and increase leukocyte extravasation. 7 In eNOS-Tg mice, superoxide levels in the postischemic tissues were consistently reduced throughout the reperfusion periods, and eNOS overexpression inhibited both the expression of adhesion molecules and the concomitant leukocyte infiltration in the reperfused tissues (Figures 7 and 8) ▶ ▶ . These results suggest that the overproduced NO derived from eNOS maintains the microvascular integrity by decomposing superoxide as well as abrogating leukocyte adhesion to the endothelial cells through inhibition of adhesion molecule expression.
A possibility that the reduced perfusion pressure in postcapillary vessels, due to systemic hypotension in eNOS-Tg mice, 20 contributes to the attenuation of vascular protein leakage is to be argued. EBD extravasation levels in non-ischemic muscles were, however, similar between WT and eNOS-Tg mice. Also, in L-NAME-treated eNOS-Tg mice, where systemic blood pressure is increased by 10 mmHg compared to WT mice, 20 EBD extravasation levels in the reperfused tissues were similar to WT mice. Therefore, the reduced perfusion pressure is unlikely to be responsible for the attenuated EBD extravasation.
Subsequently, we analyzed viability of ischemia-reperfused muscle to evaluate the effects of NO on I/R injury. The extent of tissue injury induced by short-time ischemia was similar between WT and eNOS-Tg mice, however the reperfusion-induced aggravation of tissue injury was prevented by eNOS overexpression (Figure 9) ▶ . Because of the lack of definitive evidence on roles of endogenous NO from eNOS in I/R injury, genetically engineered eNOS-Tg mice are useful to study in vivo pathophysiological roles of NO. In this study, the relative short duration of ischemia was chosen because mice are shown to be more susceptible to I/R injury compared with other species such as rats, rabbits, and humans. 16 The differences in susceptibility to tissue injury among species are an important issue for data interpretation. To apply the data obtained from our I/R model to clinical situations, further studies are required.
In contrast to our study, there are reports implicating NO as a mediator of reperfusion injury in various tissues. In a variety of I/R model, NO derived from inducible NOS (iNOS) is shown to react with superoxide and generates peroxynitrite, resulting in the reperfusion injury. 15,16 Considerable amounts of NO derived from neuronal NOS (nNOS) are also suggested to contribute to I/R injury in brain via peroxynitrite formation or induction of intracellular Ca2+ overload in neuronal cells. 39 In these studies, reperfusion injury was protected by NOS inhibitors via inactivation of iNOS or nNOS, which decreases NO production and inhibits peroxynitrite generation. On the other hand, the effects of NOS inhibitors on I/R injury are suggested to diversify depending on the doses and timing of drug administration. 40 We found that L-NAME treatment cancelled the beneficial effects of eNOS overexpression and rather worsened the reperfusion injury. In the present experimental model, the detrimental effects of L-NAME support the idea that eNOS overexpression serves to prevent I/R injury.
Our study demonstrates that overexpression of eNOS in the endothelium could attenuate reperfusion injury after short-time ischemia in mice skeletal muscle by decreasing superoxide production, preventing protein leakage, and abrogating leukocyte-endothelial interactions. Up-regulation of eNOS or augmentation of eNOS-derived NO production is indicated to be beneficial in the treatment of I/R injury. The present findings that eNOS overexpression maintains vascular integrity in I/R could contribute to identify a novel therapeutic approach for I/R injury.
Acknowledgments
We thank Kiyoko Matsui for her skillful technical assistance.
Footnotes
Address reprint requests to Seinosuke Kawashima, Division of Cardiovascular and Respiratory Medicine, Department of Internal Medicine, Kobe University Graduate School of Medicine, 7–5-1, Kusunoki-cho, Chuo-ku, Kobe, 650-0017, Japan. E-mail: kawashim@med.kobe-u.ac.jp.
Supported by the Ministry of Education, Science, Sports and Culture of Japan (10470165) and the Japan Heart Foundation/Pfizer Grant for Research on Hypertension and Vascular Metabolism.
References
- 1.Carden DL, Granger DN: Pathophysiology of ischaemia: reperfusion injury. J Pathol 2000, 190:255-266 [DOI] [PubMed] [Google Scholar]
- 2.Grisham MB, Granger DN, Lefer DJ: Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radic Biol Med 1998, 25:404-433 [DOI] [PubMed] [Google Scholar]
- 3.Harris NR, Granger DN: Neutrophil enhancement of reperfusion-induced capillary fluid filtration associated with hypercholesterolemia. Am J Physiol 1996, 271:H1755-H1761 [DOI] [PubMed] [Google Scholar]
- 4.Alexander JS, Blaschuk OW, Haselton FR: An N-cadherin-like protein contributes to solute barrier maintenance in cultured endothelium. J Cell Physiol 1993, 156:610-618 [DOI] [PubMed] [Google Scholar]
- 5.Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC, Roos D: Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J Cell Sci 1999, 112:1915-1923 [DOI] [PubMed] [Google Scholar]
- 6.Dejana E, Bazzoni G, Lampugnani MG: Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp Cell Res 1999, 252:13-19 [DOI] [PubMed] [Google Scholar]
- 7.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E: Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol 1996, 135:497-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY: Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem 1999, 274:24930-24934 [DOI] [PubMed] [Google Scholar]
- 9.Sato S, Tominaga T, Ohnishi T, Ohnishi ST: Role of nitric oxide in brain ischemia. Ann NY Acad Sci 1994, 738:369-373 [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Zweier JL: Measurement of nitric oxide and peroxynitrite generation in the postischemic heart: evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem 1996, 271:29223-29230 [DOI] [PubMed] [Google Scholar]
- 11.Ma XL, Weyrich AS, Lefer DJ, Lefer AM: Diminished basal nitric oxide release after myocardial ischemia and reperfusion promotes neutrophil adherence to coronary endothelium. Circ Res 1993, 72:403-412 [DOI] [PubMed] [Google Scholar]
- 12.Messina A, Knight KR, Dowsing BJ, Zhang B, Phan LH, Hurley JV, Morrison WA, Stewart AG: Localization of inducible nitric oxide synthase to mast cells during ischemia/reperfusion injury of skeletal muscle. Lab Invest 2000, 80:423-431 [DOI] [PubMed] [Google Scholar]
- 13.Johnson G, Tsao PS, Lefer AM: Cardioprotective effects of authentic nitric oxide in myocardial ischemia with reperfusion. Crit Care Med 1991, 19:244-252 [DOI] [PubMed] [Google Scholar]
- 14.Kurose I, Wolf R, Grisham MB, Granger DN: Modulation of ischemia/reperfusion-induced microvascular dysfunction by nitric oxide. Circ Res 1994, 74:376-382 [DOI] [PubMed] [Google Scholar]
- 15.Liu P, Hock CE, Nagele R, Wong PY-K: Formation of nitric oxide, superoxide, and peroxynitrite in myocardial ischemia-reperfusion injury in rats. Am J Physiol 1997, 272:H2327-H2336 [DOI] [PubMed] [Google Scholar]
- 16.Knight KR, Zhang B, Morrison WA, Stewart AG: Ischaemia-reperfusion injury in mouse skeletal muscle is reduced by Nω-nitro-L-arginine methyl ester and dexamethasone. Eur J Pharmacol 1997, 332:273-278 [DOI] [PubMed] [Google Scholar]
- 17.Huk I, Nanobashvili J, Neumayer C, Punz A, Mueller M, Afkhampour K, Mittlboeck M, Losert U, Polterauer P, Roth E, Patton S, Malinski T: L-arginine treatment alters the kinetics of nitric oxide and superoxide release and reduces ischemia/reperfusion injury in skeletal muscle. Circulation 1997, 96:667-675 [DOI] [PubMed] [Google Scholar]
- 18.Meldrum DG, Stephenson LL, Zamboni WA: Effects of L-NAME and L-arginine on ischemia-reperfusion injury in rat skeletal muscle. Plast Reconstr Surg 1999, 103:935-940 [DOI] [PubMed] [Google Scholar]
- 19.Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL, Billiar TR: Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation 2000, 101:2742-2748 [DOI] [PubMed] [Google Scholar]
- 20.Ohashi Y, Kawashima S, Hirata K, Yamashita T, Ishida T, Inoue N, Sakoda T, Kurihara H, Yazaki Y, Yokoyama M: Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest 1998, 102:2061-2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita T, Kawashima S, Ohashi Y, Ozaki M, Ueyama T, Ishida T, Inoue N, Hirata K, Akita H, Yokoyama M: Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation 2000, 101:931-937 [DOI] [PubMed] [Google Scholar]
- 22.Ozaki M, Kawashima S, Yamashita T, Ohashi Y, Rikitake Y, Inoue N, Hirata K, Hayashi Y, Itoh H, Yokoyama M: Reduced hypoxic pulmonary vascular remodeling by nitric oxide from the endothelium. Hypertension 2001, 37:322-327 [DOI] [PubMed] [Google Scholar]
- 23.Kawashima S, Yamashita T, Ozaki M, Ohashi Y, Azumi H, Inoue N, Hirata K, Hayashi Y, Itoh H, Yokoyama M: Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol 2001, 21:201-207 [DOI] [PubMed] [Google Scholar]
- 24.Mügge A, Peterson T, Harrison DG: Release of nitrogen oxides from cultured bovine aortic endothelial cells is not impaired by calcium channel antagonists. Circulation 1991, 83:1404-1409 [DOI] [PubMed] [Google Scholar]
- 25.Hirata K, Kuroda R, Sakoda T, Katayama M, Inoue N, Suematsu M, Kawashima S, Yokoyama M: Inhibition of endothelial nitric oxide synthase activity by protein kinase C. Hypertension 1995, 25:180-185 [DOI] [PubMed] [Google Scholar]
- 26.Miller FJ, Gutterman DD, Rios CD, Heistad DD, Davidson BL: Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res 1998, 82:1298-1305 [DOI] [PubMed] [Google Scholar]
- 27.Dallal MM, Chang SW: Evans blue dye in the assessment of permeability-surface area product in perfused rat lungs. J Appl Physiol 1994, 77:1030-1035 [DOI] [PubMed] [Google Scholar]
- 28.Bradley PP, Christensen RD, Rothstein G: Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 1982, 60:618-622 [PubMed] [Google Scholar]
- 29.Belkin M, Brown RD, Wright JG, LaMorte WW, Hobson RW: A new quantitative spectrophotometric assay of ischemia-reperfusion injury in skeletal muscle. Am J Surg 1988, 156:83-86 [DOI] [PubMed] [Google Scholar]
- 30.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM, Ward PA, Dejana E: Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA 1999, 96:9815-9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein HH, Puschmann S, Schaper J, Schaper W: The mechanism of the tetrazolium reaction in idenfying experimental myocardial infarction. Virchows Arch 1981, 393:287-297 [Google Scholar]
- 32.Liochev SI, Fridovich I: Lucigenin as mediator of superoxide production: revisited. Free Radic Biol Med 1998, 25:926-928 [DOI] [PubMed] [Google Scholar]
- 33.Vásquez-Vivar J, Hogg N, Pritchard KA, Martasek P, Kalyanaraman B: Superoxide anion formation from lucigenin: an electron spin resonance spin-trapping study. FEBS Lett 1997, 403:127-130 [DOI] [PubMed] [Google Scholar]
- 34.Wu HM, Huang Q, Yuan Y, Granger HJ: VEGF induces NO-dependent hyperpermeability in coronary venules. Am J Physiol 1996, 271:H2735-H2739 [DOI] [PubMed] [Google Scholar]
- 35.Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner JM: Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97:99-107 [DOI] [PubMed] [Google Scholar]
- 36.Gursoy-Ozdemir Y, Bolay H, Saribas O, Dalkara T: Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 2000, 31:1974-1980 [DOI] [PubMed] [Google Scholar]
- 37.Schutte H, Mayer K, Burger H, Witzenrath M, Gessler T, Seeger W, Grimminger F: Endogenous nitric oxide synthesis and vascular leakage in ischemic-reperfused rabbit lungs. Am J Respir Crit Care Med 2001, 164:412-428 [DOI] [PubMed] [Google Scholar]
- 38.Kadambi A, Skalak TC: Role of leukocytes and tissue-derived oxidants in short-term skeletal muscle ischemia-reperfusion injury. Am J Physiol 2000, 278:H435-H443 [DOI] [PubMed] [Google Scholar]
- 39.Segawa D, Hatori N, Yoshizu H, Uriuda Y, Shimizu M, Tanaka S: The effects of nitric oxide synthase inhibitor on reperfusion injury of the brain under hypothermic circulatory arrest. J Thorac Cardiovasc Surg 1998, 115:925-930 [DOI] [PubMed] [Google Scholar]
- 40.Spinnewyn B, Cornet S, Auguet M, Chabier PE: Synergistic protective effects of antioxidant and nitric oxide inhibitor in transient focal ischemia. J Cereb Blood Flow Metab 1999, 19:139-143 [DOI] [PubMed] [Google Scholar]