

Abstract
Solid-pseudopapillary tumors (SPTs) are unusual pancreatic neoplasms of low malignant potential that most frequently affect young women. Genetic events contributing to the development of SPTs are unknown. Whereas the more common ductal adenocarcinomas of the pancreas essentially never harbor β-catenin or APC gene mutations, we have recently identified alterations of the APC/β-catenin pathway in other nonductal pancreatic neoplasms including pancreatoblastomas and acinar cell carcinomas. We analyzed a series of 20 SPTs for somatic alterations of the APC/β-catenin pathway using immunohistochemistry for β-catenin protein accumulation, direct DNA sequencing of β-catenin exon 3, and direct DNA sequencing of the mutation cluster region in exon 15 of the APC gene in those SPTs that did not harbor β-catenin mutations. Immunohistochemical labeling for cyclin D1 was performed to evaluate the overexpression of this cell-cycle protein as one of the putative downstream effectors of β-catenin dysregulation. In addition, we analyzed the SPTs for genetic alterations commonly found in pancreatic ductal adenocarcinomas, including mutations in the K-ras oncogene and p53 and DPC4 tumor suppressor genes, using direct DNA sequencing of K-ras and immunostaining for p53 and Dpc4. Almost all SPTs harbored alterations in the APC/β-catenin pathway. Nuclear accumulation of β-catenin protein was present in 95% (19 of 20), and activating β-catenin oncogene mutations were identified in 90% (18 of 20) of the SPTs. Seventy-four percent (14 of 19) showed overexpression of cyclin D1, ranging from 10 to 70% of tumor nuclei. In contrast, no K-ras mutations were present in any of the 20 SPTs, and Dpc4 expression was intact in all 16 SPTs for which immunohistochemical labeling was successful. Overexpression of p53 was limited to only 3 of 19 (15.8%) SPTs. These results emphasize the two distinct, divergent genetic pathways of neoplastic progression in pancreatic ductal and nonductal neoplasms.
Solid-pseudopapillary tumors of the pancreas (SPTs) are uncommon tumors, constituting only ∼1% of pancreatic neoplasms. 1 SPTs are histologically, clinically, and prognostically quite distinct from the more common pancreatic ductal adenocarcinomas. In contrast to the infiltrative growth and almost invariably lethal behavior of ductal adenocarcinomas among older individuals, SPTs are neoplasms of young women and are frequently grossly sharply circumscribed and cystic lesions (although their microscopic margins may be indistinct). Most importantly, despite their often large size (mean of >10 cm at presentation in one study), 2 the vast majority of SPTs are indolent neoplasms. They are confined to the pancreas in 85% of patients, and even the 10 to 15% of patients with liver or peritoneal metastases from SPTs commonly enjoy long-term survival. 2-6
The histopathological appearance of SPTs is distinctive among the primary pancreatic neoplasms. 1 The neoplastic epithelial cells are uniform, polygonal, and discohesive in nature. Smaller SPTs may be primarily arranged in solid sheets with a rich microvasculature (Figure 1A) ▶ . Frequent degenerative changes in larger tumors, however, lend a characteristic pseudopapillary pattern because of residual epithelial cells that form perivascular rosettes (Figure 1B) ▶ . SPTs are known to consistently express vimentin, 7-9 α1-anti-trypsin, 7-11 neuron-specific enolase, 8,10,11 progesterone receptors, 10 and more recently, CD10. 9 However, attempts to discern a cell of origin or even a line of cellular differentiation for SPTs based on these results have been unrevealing. 10,12,13
Figure 1.
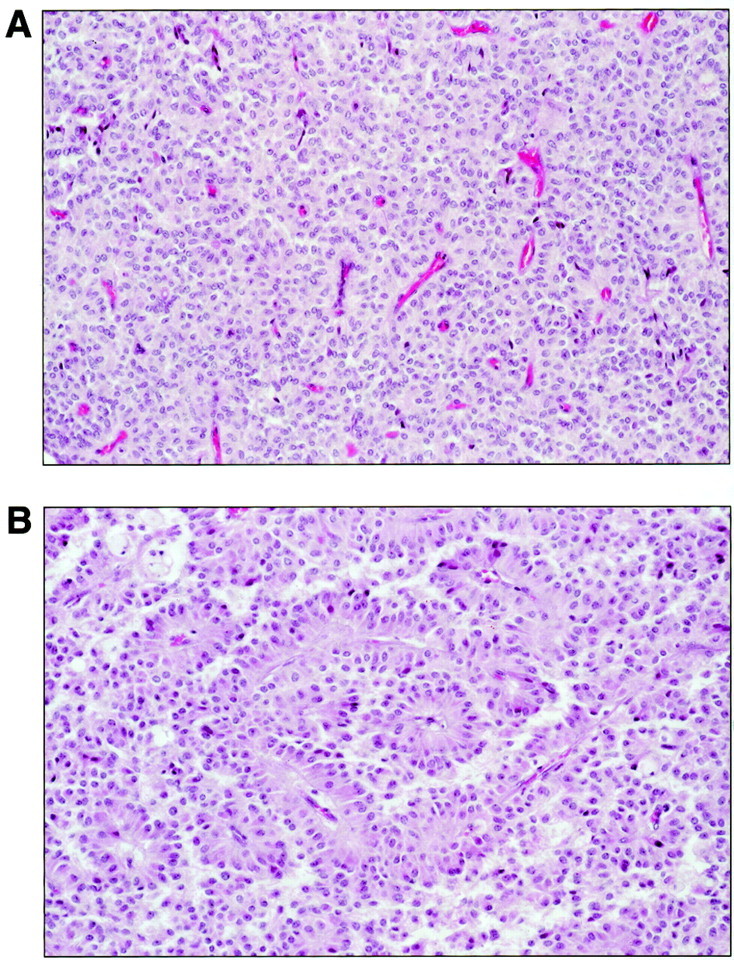
Histological appearance of SPTs. A: In solid areas, patternless sheets of uniform epithelial cells are punctuated by numerous small vascular channels. B: Degenerative changes and the discohesive nature of the epithelial cells give rise to characteristic pseudopapillae in which residual tumor cells appear to rosette around vascular cores.
Likewise, genetic alterations that characterize SPTs have remained elusive. Numerous flow cytometric evaluations of SPTs have been reported, 7,14-20 with only occasional neoplasms demonstrating aneuploidy 7,14-16 or other structural chromosomal abnormalities. 20-22 Only a few SPTs have been evaluated for molecular alterations, and these studies have been limited to evaluation of the K-ras oncogene and p53, p16, and DPC4 tumor suppressor gene. These genes are frequently involved in the pathogenesis of pancreatic ductal adenocarcinomas. Not surprisingly, given the marked clinicopathological differences between SPTs and ductal adenocarcinomas, alterations in these genes have not been detected in SPTs. 18,19,23-27
In contrast to pancreatic ductal adenocarcinomas in which β-catenin or APC gene mutations are essentially never present, we have recently identified somatic alterations of the APC/β-catenin pathway in other pancreatic nonductal neoplasms including pancreatoblastomas and acinar cell carcinomas. 28,29 We therefore undertook a molecular characterization of a series of 20 SPTs for alterations in the APC/β-catenin pathway and for cyclin D1 protein overexpression, a putative downstream effector of β-catenin dysregulation. 30-33 In addition, we analyzed the SPTs for alterations in the K-ras oncogene and p53 and DPC4 tumor suppressor genes that are known to characterize pancreatic ductal adenocarcinomas.
Materials and Methods
Case Selection
The study population consisted of 20 patients with pancreatic SPTs who either underwent surgical resection or whose surgical material was seen in consultation at The Johns Hopkins Hospital (9 cases) or Memorial Sloan-Kettering Cancer Center (11 cases) between 1989 and 2000. Clinical information including patient age, gender, tumor size, tumor location in the pancreas, and presence or absence of metastatic disease was collected from patient charts or the computerized patient files.
Immunohistochemistry for β-Catenin, p53, and Dpc4
Immunohistochemical labeling using diaminobenzidine as the chromogen was performed on the Techmate 1000 automatic labeling system (BioTek Solutions, Tucson, AZ). Deparaffinized sections of formalin-fixed tissue at 5 μm thickness were labeled with β-catenin antibody (1:500 dilution, mouse monoclonal; Becton Dickinson Transduction Laboratories, Lexington, KY), cyclin D1 antibody (1:50 dilution, rabbit polyclonal; Oncogene Research Products, San Diego, CA), p53 antibody (1:100 dilution, mouse monoclonal clone D07; DAKO, Carpinteria, CA), and Dpc4 antibody (1:100 dilution, monoclonal clone B8; Santa Cruz Biotechnology, Santa Cruz, CA). Heat-induced antigen retrieval using steam for 20 minutes at 80°C was used before incubation with all four antibodies.
For β-catenin, immunohistochemical labeling was evaluated for the presence of nuclear, cytoplasmic, and membranous β-catenin accumulation in the SPTs and surrounding nonneoplastic pancreas, if present. Nuclear and cytoplasmic accumulation of β-catenin in SPTs was graded according to the percentage of neoplastic cells with strong immunolabeling. For cyclin D1, the percentage of moderately or strongly positive tumor nuclei was evaluated. For p53, the percentage of positively labeled nuclei was recorded; we considered strong nuclear labeling in ≥30% of neoplastic cells as the cutoff for positivity. 34 For Dpc4, SPTs were classified as showing intact Dpc4 protein expression if they showed the normal pattern of strong, diffuse cytoplasmic labeling and labeling of scattered nuclei. They were classified as showing loss of normal Dpc4 expression only if they showed a complete loss of cytoplasmic and nuclear Dpc4 labeling. 35
DNA Extraction
Microdissection of SPTs for DNA extraction was performed from formalin-fixed, paraffin-embedded tissues. A 271/2-guage-needle tip was used for microdissection of routinely processed 5-μm hematoxylin and eosin-stained slides under a low-power (×4) objective. Genomic DNA was extracted as described previously. 36 Corresponding normal control DNA was available in 17 cases and was extracted from adjacent nonneoplastic pancreatic acini or stroma in 16 cases and from adjacent duodenum in 1 case.
Mutation Analysis of the β-Catenin and APC Genes
Genomic DNA from each SPT and normal tissue (if present) was amplified by polymerase chain reaction (PCR) using the primer pair: 5′-ATGGAACCAGACAGAAAAGC-3′ (sense) and 5′-GCTACTTGTTCTGAGTGAAG-3′ (anti-sense). These amplified a 200-bp fragment of exon 3 of the β-catenin gene that encompasses the region for GSK-3β phosphorylation. PCR reactions were performed under standard conditions in a 50-μl volume containing 38 μl of Platinum PCR SuperMix (Life Technologies, Rockville, MD), 5 μl of both 5′ and 3′ oligonucleotides (final concentration of 1 μmol/L), and 2 μl (∼50 ng) of genomic DNA. PCR conditions consisted of an initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, and a final extension at 72°C for 7 minutes. PCR products were purified with spin columns using QIAquick PCR purification kit (Qiagen, Inc., Valencia, CA) before sequencing. Automated sequencing of purified PCR products was performed on an ABI Prism 3700 DNA Analyzer (Applied Biosystems, Inc., Foster City, CA) using the internal primers: 5′-AAAGCGGCTGTTAGTCACTGG-3′ (sense) and 5′-CCTGTTCCCACTCATACAGG-3′ (anti-sense), and the resulting sequence data were analyzed with the Sequencher analysis program (Gene Codes, Ann Arbor, MI). All mutations were verified in both sense and anti-sense directions on independent PCR products.
Direct sequencing for APC gene mutations was attempted for SPTs that did not contain identifiable β-catenin mutations. Four sets of oligonucleotide primers (A1: 5′-CAGACTTATTGTGTAGAAGA-3′ and A2: 5′-CTCCTGAAGAAAATTCAACA-3′ for codons 1260–1359; B1: 5′-AGGGTTCTAGTTTATCTTCA-3′ and B2: 5′-TCTGCTTGGTGGCATGGTTT-3′ for codons 1339–1436; C1: 5′-GGCATTATAAGCCCCAGTGA-3′ and C2: 5′-AAATGG-CTCATCGAGGCTCA-3′ for codons 1417–1516; D1: 5′-ACTCCAGATGGATTTTCTTG-3′ and D2: 5′-GGCTGGCT-TTTTTGCTTTAC-3′ for codons 1497–1596) were used to amplify the mutation cluster region of the APC gene. 37 PCR reactions were performed in 50-μl volumes using the reaction mixture described above. PCR conditions consisted of an initial denaturation step of 94°C for 3 minutes, 40 cycles (94°C for 1 minute, 55°C for 1 minute, and 68°C for 1.5 minutes for APC-B, APC-C, and APC-D primer pairs and 94°C for 1 minute, 52°C for 1 minute, and 68°C for 1.5 minutes for APC-A), followed by a final extension at 72°C for 7 minutes.
Mutation Analysis of the K-ras Oncogene
The oligonucleotide primers 5′-GAGAATTCATGACTGAATATAAACTTGT-3′ (sense) and 5′-TCGAATTCCTCTATTGTTGGATCATATTCG-3′ (anti-sense) were used to amplify a region in exon 1 of the K-ras gene that includes codons 12 and 13. PCR reactions were performed in 50-μl volumes using the reaction mixture described above, with an initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 50°C for 1 minute, and 72°C for 1 minute, and a final extension at 72°C for 7 minutes. PCR products were purified and sequenced as described above using the internal primer 5′-ATTCGTCCACAAAATGAT-3′.
Results
A summary of the clinicopathological features of the 20 SPTs (designated S1 to S20) is shown in Table 1 ▶ , and the corresponding molecular findings are shown in Table 2 ▶ .
Table 1.
Clinicopathological Features of Pancreatic Solid-Psuedopapillary Tumors
| Case | Age/sex | Tumor size | Location in pancreas | Nuclear pleomorphism | Invasion of adjacent structures | Metastases |
|---|---|---|---|---|---|---|
| S1 | 57/M | 6.0 cm | Tail | − | − | − |
| S2 | 35/F | 3.5 cm | Body | − | − | − |
| S3 | 42/F | 2.0 cm | Head | − | − | − |
| S4 | 25/F | 7.5 cm | Body/tail | − | − | − |
| S5 | 38/F | 4.7 cm | Body/tail | − | − | − |
| S6 | 37/F | 2.5 cm | Body | + (Focal) | + (Peripancreatic fat) | − |
| S7 | 51/F | 3.5 cm | Uncinate | − | − | − |
| S8 | 57/F | 5.2 cm | Head | − | + (Mesocolon; duodenum; peripancreatic fat) | − |
| S9 | 75/F | 5.0 cm | Head | − | − | − |
| S10 | 56/F | 4.0 cm | Distal | − | − | + (Liver)* |
| S11 | 18/F | 7.5 cm | Head | − | − | − |
| S12 | N/A | N/A | N/A | − | N/A | N/A |
| S13 | 13/M | 5.5 cm | N/A | − | − | − |
| S14 | 49/F | 5.5 cm | Distal | − | + (Vascular, perineural) | − |
| S15 | 38/F | 1.0 cm | Distal | − | + (Perineural) | − |
| S16 | 38/M | >2 cm | Tail | − | + (Capsular vessels) | − |
| S17 | 42/F | 2.5 cm | Distal | − | − | − |
| S18 | 26/F | 11.5 cm | Head | − | − | − |
| S19 | 42/F | 3.0 cm | Distal | − | − | − |
| S20 | 27/F | 4.5 cm | Head | − | − | − |
*This patient died of disease 10 years after presentation.
N/A, information was not available.
Table 2.
Genetic Alterations in Pancreatic Solid-Pseudopapillary Tumors
| Case | β-catenin mutation | Nuclear β-catenin, % | Nuclear cyclin D1, % | K-ras mutation | p53 accumulation | Dpc4 loss |
|---|---|---|---|---|---|---|
| S1 | G34V | >90 | 50 | Wild-type | − | Intact |
| S2 | S37F | >90 | 30 | Wild-type | − | Intact |
| S3 | D32Y | >90 | 40 | Wild-type | − | Intact |
| S4 | G34V | >90 | 50 | Wild-type | − | Intact |
| S5 | S37F | >90 | 70 | Wild-type | − | Intact |
| S6 | G34V | >90 | 10 | Wild-type | + (Patchy) | Intact |
| S7 | S33A | >90 | 20 | Wild-type | − | Intact |
| S8 | S37F | >90 | 10 | Wild-type | − | Intact |
| S9 | D32V | >90 | 10 | Wild-type | − | Intact |
| S10 | Wild-type | >90 | − | Wild-type | − | Intact |
| S11 | D32N | 20 | − | Wild-type | − | N/A |
| S12 | S37F | 30 | − | Wild-type | − | Intact |
| S13 | S37F | >90 | 15 | Wild-type | − | Intact |
| S14 | Wild-type | >90 | − | Wild-type | 40% | Intact |
| S15 | G34R | >90 | 10 | Wild-type | − | Intact |
| S16 | S37F | >90 | 30 | Wild-type | − | Intact |
| S17 | D32Y | >90 | 30 | Wild-type | − | N/A |
| S18 | D32N | >90 | − | Wild-type | − | Intact |
| S19 | D32Y | >90 | 10 | Wild-type | + (Patchy) | N/A |
| S20 | G34R | N/A | N/A | Wild-type | N/A | N/A |
Locations of somatic mutations in β-catenin are shown by codon.
Nuclear β-catenin, cyclin D1, and p53 accumulation were evaluated based on the percentage of labeled tumor cell nuclei.
N/A, immunohistochemistry failed.
Clinicopathological Characteristics
Clinical information was available in 19 patients with SPTs. Sixteen (84.2%) patients were female and three (15.8%) were male. They ranged from 13 to 75 years with a mean patient age of 40.3 years. The SPTs were located throughout the pancreas without a particular site predilection. Mean size of the SPTs was 4.6 cm, although there was a wide variation in tumor size, ranging from 1 cm to 11.5 cm. Histopathologically, the 20 SPTs showed uniformly small, regular nuclei with a focal area of nuclear pleomorphism in only one (5%) of the SPTs. Five of the 19 SPTs (26.3%) for which pathological staging was available showed tumor infiltration either into vascular/perineural spaces (three SPTs) or into the adjacent bowel wall and/or peripancreatic fat (two SPTs). However, metastatic disease was present in only one patient (S10, with metastases to the liver), who subsequently died of disease 10 years after initial presentation.
Mutations in the β-Catenin Gene
Activating mutations in exon 3 of the β-catenin oncogene were present in nearly all (18 of 20, 90%) SPTs. All 18 were 1-bp missense mutations that either affected serine residues at GSK-3β phosphorylation sites in codon 33 (one SPT) and codon 37 (six SPTs), or immediately around serine/threonine GSK-3β phosphorylation sites in codon 32 (six SPTs) and codon 34 (five SPTs). All of these mutations were somatic in nature, as evidenced by the lack of mutations in the corresponding normal tissues (available for sequencing in 17 of 20 SPTs overall and 15 of 18 SPTs with β-catenin mutations). All chromatograms showing β-catenin mutations contained an admixture of the mutated and wild-type peaks, consistent with the known dominant-positive nature of β-catenin gene mutations (Figure 2) ▶ .
Figure 2.

β-catenin oncogene mutations in SPTs. Representative DNA sequencing chromatograms demonstrate a GAC (aspartic acid)→AAC (asparagine) mutation in codon 32 of case S11, a GAC (aspartic acid)→GTC (valine) mutation in codon 32 of S9, a GGA (glycine)→AGA (arginine) mutation in codon 34 of S15, a GGA (glycine)→GTA (valine) mutation in codon 34 of S4, and a TCT (serine)→TTT (phenylalanine) mutation in codon 37 of S5. In each case, a mixture of the mutant and wild-type peaks is present because of the dominant-positive nature of β-catenin mutations.
We attempted to sequence the two SPTs without β-catenin mutations for mutations in the mutation cluster region of the APC gene, but the PCR amplification failed in both cases.
Supposed Downstream Effects of β-Catenin Mutation
The location of the identified β-catenin mutations either at or around GSK-3β phosphorylation sites would be expected to interfere with normal phosphorylation by GSK-3β and subsequent ubiquitin-mediated degradation of β-catenin protein. Consistent with this effect, strong nuclear and cytoplasmic accumulation of β-catenin protein was present in nearly all SPTs. In 17 of 20 cases (85%), the immunolabeling was strong and diffuse, present throughout the tumor in >90% of neoplastic epithelial cells (Figure 3A) ▶ . In an additional 2 of 20 cases (10%, both outside material with unknown tissue fixation parameters) nuclear and cytoplasmic accumulation was patchy, detected in 30% of the neoplastic epithelial cells in one SPT, and 20% of neoplastic epithelial nuclei with only faint cytoplasmic staining in the other SPT. In the one remaining SPT (also outside material), all immunolabeling reactions failed, with no normal membranous pattern of β-catenin staining in the adjacent nonneoplastic tissue. In all other SPTs that contained adjacent nonneoplastic tissues, only the normal membranous pattern of β-catenin labeling was observed.
Figure 3.
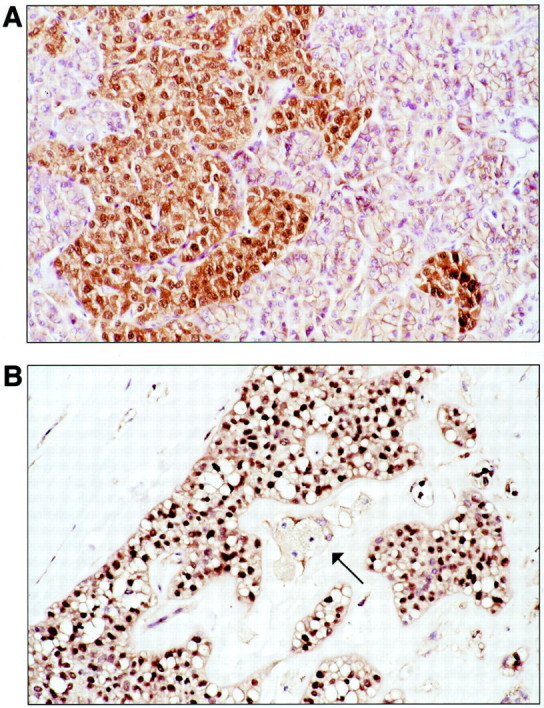
Downstream effects of β-catenin dysregulation in SPTs. A: Nuclear translocation and accumulation of β-catenin protein is seen in neoplastic epithelial cells that have infiltrated into normal pancreas at the edge of the tumor. Nonneoplastic acini show only normal membranous β-catenin labeling, but are negative for nuclear accumulation. B: Overexpression of cyclin D1, a downstream transcriptional target of nuclear β-catenin-Tcf/Lef complex. Cyclin D1 labeling is seen in the nuclei of neoplastic epithelial cells of this SPT, but not in the foamy histiocytes at center (arrow).
There was no correlation between poor immunolabeling for β-catenin and lack of detectable β-catenin mutations. Each of the three SPTs that either showed only patchy or negative β-catenin protein accumulation contained β-catenin gene mutations on sequencing, and the two SPTs that did not contain β-catenin gene mutations showed strong and diffuse nuclear β-catenin protein accumulation. This finding suggests that β-catenin was dysregulated in all 20 SPTs examined in this study.
A majority of SPTs also demonstrated overexpression of cyclin D1, a downstream target of nuclear β-catenin. Seventy-four percent of SPTs (14 of 19 cases in which immunostaining was successful) contained moderately or strongly labeled nuclei that ranged from 10 to 70% of neoplastic epithelial cells (mean, 27.5%) (Figure 3B) ▶ . We were better able to detect cyclin D1 overexpression among the 9 in-house SPTs with relatively uniform fixation conditions (9 of 9 cases positive, with a mean nuclear labeling of 32.2%) than among the 11 outside SPTs (5 of 11 cases positive, mean nuclear labeling of 19% in positive cases), suggesting that the true frequency of cyclin D1 activation may be higher than we were able to confirm in this study. No nuclear cyclin D1 was present in normal pancreatic acini or ductal epithelial cells; strong cytoplasmic and nuclear labeling of occasional islet cells was present, of uncertain etiology.
Alterations in K-ras, Dpc4, and p53
All 20 SPTs (100%) contained only wild-type K-ras gene sequences around codons 12 and 13 (Figure 4A) ▶ . Normal Dpc4 protein expression was preserved in all 16 SPTs (100%) for which the immunolabeling was successful (Figure 4B) ▶ . Only 3 of 19 (15.8%) SPTs demonstrated nuclear p53 overexpression. In one case, ∼40% of neoplastic epithelial cell nuclei were positive for p53 in a diffuse pattern throughout the tumor. In the other two cases, intense nuclear labeling for p53 was present but was confined to distinct patchy foci that overall occupied <30% of the tumor volume (Figure 4C) ▶ .
Figure 4.
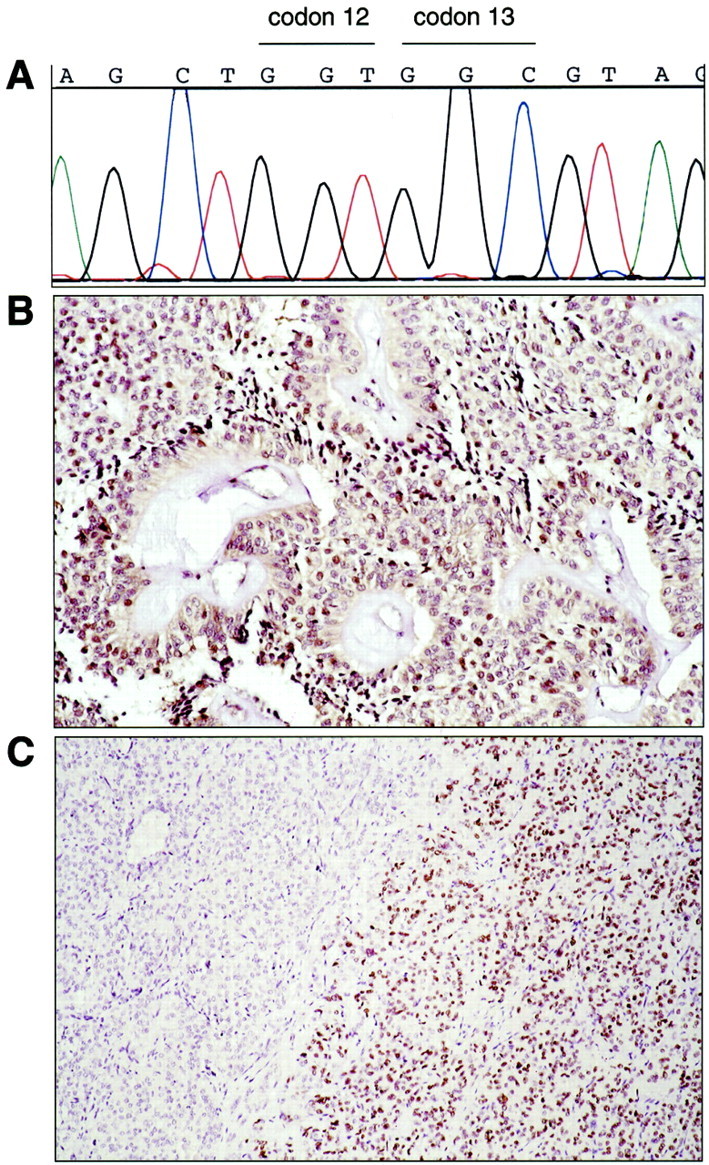
SPTs rarely harbor alterations in the genes commonly involved in ductal adenocarcinomas. A: Only wild-type K-ras sequences were present at codons 12 and 13 in all SPTs. B: Intact Dpc4 protein expression was present in all SPTs. C: Only 3 of 19 (15.9%) SPTs showed p53 protein overexpression, as in this case (S6) that contained a distinct patch of neoplastic epithelial cells with nuclear labeling.
Discussion
We have shown that nearly all SPTs (18 of 20, 90%) harbor β-catenin gene mutations. β-catenin protein functions both as a submembranous component of the cadherin-mediated cell adhesion system and as a downstream transcriptional activator in the Wnt signaling pathway. 38,39 Normal β-catenin degradation is promoted by the APC tumor suppressor protein, which in conjunction with glycogen synthase kinase-3β (GSK-3β) and AXIN, promotes phosphorylation of β-catenin on serine/threonine residues that are encoded in exon 3 of the β-catenin gene. 38,40,41 Normal levels of β-catenin protein are maintained by this phosphorylation, which targets β-catenin for subsequent ubiquitin-mediated degradation. 42,43 Abnormal accumulation of β-catenin and loss of β-catenin regulatory activity can result from stabilizing β-catenin gene mutations, truncating APC tumor suppressor gene mutations, or AXIN mutations. 41,44,45 For example, most colorectal adenomas and carcinomas can be demonstrated to contain either bi-allelic inactivation of the APC gene or (less commonly) activating β-catenin oncogene mutations. 46,47 All β-catenin mutations detected in SPTs in this study were 1-bp missense mutations that affected either serine phosphorylation residues in codons 33 and 37 or residues immediately surrounding phosphorylation sites in codons 32 and 34. These mutations would therefore be predicted to interfere with normal phosphorylation and degradation of β-catenin protein.
In addition to the high frequency of β-catenin gene mutations in SPTs, the results of this study demonstrate in part the downstream effects of β-catenin dysregulation. The consequences of cytosolic β-catenin accumulation because of mutations in the APC/β-catenin pathway include its interaction with T-cell transcription factor (Tcf)/lymphoid enhancer-binding factor (Lef) and translocation of the β-catenin-Tcf/Lef complex to the nucleus. 30-33 Consistent with this, we observed abnormal nuclear and cytoplasmic β-catenin accumulation in nearly all SPTs, including the two SPTs that did not contain identifiable β-catenin or APC gene mutations. In fact, all 20 SPTs in this study showed evidence for a dysregulated APC/β-catenin pathway in the form of nuclear β-catenin accumulation or activating β-catenin mutation on sequencing.
Nuclear translocation of the β-catenin-Tcf/Lef complex, in turn, has been shown in studies of colon carcinoma cells to stimulate transcription of target genes involved in cell cycle regulation, including c-myc and cyclin D1. 30,48 The β-catenin complex activates transcription of cyclin D1 via binding to consensus sequences on the cyclin D1 promoter; high levels of cyclin D1 mRNA and constitutive production of cyclin D1 protein have previously been demonstrated in colon carcinoma cells harboring β-catenin mutations. 30 Consistent with this, we found overexpression of cyclin D1 by immunohistochemical labeling in 74% of SPTs, ranging from 10 to 70% of neoplastic epithelial cell nuclei. We observed a greater frequency of cyclin D1 labeling, as well as a higher proportion of labeled nuclei, in our in-house SPTs in which tissue fixation and processing were more uniform (9 of 9 in-house cases, mean nuclear labeling of 32.2% versus 5 of 11 outside cases, mean nuclear labeling of 18%), suggesting that the true rate of cyclin D1 overexpression in SPTs may be higher than what was demonstrable in this study. Muller-Hocker and colleagues 25 have also recently shown overexpression of cyclin D1 (as well as the cell-cycle protein cyclin D3) in 50% and 30% of tumor nuclei in two SPTs, respectively, although analysis of β-catenin was not performed.
In contrast to the nearly universal presence of an abnormal APC/β-catenin pathway in SPTs, we found little evidence for involvement of genes known to be important in the stepwise molecular pathogenesis of the more common pancreatic ductal adenocarcinomas, including K-ras, DPC4, and p53. K-ras oncogene mutations occur early in the development of nearly all in situ ductal neoplasms, 49,50 DPC4 tumor suppressor gene inactivation later in the progression of slightly more than 50% of cases, 51,52 and p53 inactivation in up to 70% of invasive pancreatic adenocarcinomas. 27,49,53,54 In contrast, alterations of the APC/β-catenin pathway are essentially never observed in ductal adenocarcinomas. Only a few SPTs have previously been investigated for similar molecular genetic alterations. A total of 16 SPTs in four previous studies have been shown to harbor only wild-type K-ras gene sequences, 19,23,24,27 and 18 SPTs from four other studies have been shown to be negative for p53 alterations by immunohistochemistry 18,25,26 or gene sequencing. 27 In addition, four SPTs analyzed by Moore and colleagues 27 were negative for p16 and DPC4 gene mutations. We found only wild-type K-ras gene sequences in codons 12 and 13 among our 20 SPTs, intact Dpc4 protein expression in all 16 stainable SPTs, and low-level and/or patchy abnormal p53 accumulation in only 3 of 19 (15.8%) SPTs. Notably, immunohistochemical labeling for Dpc4 has been shown to accurately mirror the status of DPC4 gene inactivation. 35 Although there is imperfect correlation between immunohistochemical labeling for p53 and p53 gene mutations (p53 may be overexpressed for reasons other than gene mutation, and conversely, truncating p53 mutations may be present that do not result in p53 protein accumulation by immunohistochemistry), the results of this study and the previous investigations of p53 immunohistochemistry and gene sequencing in SPTs suggest that p53 alterations are at least uncommon in SPTs.
These marked molecular differences between SPTs and pancreatic ductal adenocarcinomas in this study and previous studies are not surprising given the sharp clinicopathological and prognostic distinction between the two types of neoplasms. In contrast to ductal adenocarcinomas, SPTs are characteristically tumors of young women, although cases in males, 55-58 children, 3,4,58,59 and in elderly patients 55 have been described. Furthermore, SPTs are typically indolent, with examples of long-term survival even among the 10 to 15% of patients who have liver or peritoneal metastases. 2-6 The results of this study do not explain the distinctive histopathological appearance of SPTs, nor identify the cell of origin/cellular differentiation of the neoplastic epithelial cells of SPTs, concepts that have remained elusive despite numerous studies detailing their immunophenotype. It is also conceptually difficult to establish a connection between a disrupted APC/β-catenin pathway and the peculiar tendency for SPTs to occur primarily in young females. However, we have shown other examples of lesions with frequent APC/β-catenin alterations including breast fibromatoses and juvenile nasopharyngeal angiofibromas that also show a distinct sex predilection. 60,61
We have recently studied the genetic alterations present in two other clinicopathologically distinct nonductal pancreatic neoplasms, pancreatoblastomas and acinar cell carcinomas. 28,29 Like SPTs, pancreatoblastomas and acinar cell carcinomas contrast with ductal adenocarcinomas in their distinctive histopathological features, characteristically young age at presentation (in the case of pancreatoblastomas), and somewhat better prognosis than the usual ductal adenocarcinoma. 62,63 Also like SPTs, these neoplasms only rarely harbor the alterations in K-ras, Dpc4, and p53 that are common to ductal adenocarcinomas, but 67% of pancreatoblastomas and 23.5% of acinar cell carcinomas had either activating β-catenin mutations or inactivating APC mutations. 28,29,64-66 The results of this study suggest that pancreatic ductal and nonductal neoplasms progress genetically through two primarily dichotomous pathways: K-ras, p16, DPC4, and p53 gene alterations in the case of ductal neoplasms, and varying rates of APC/β-catenin alterations in nonductal neoplasms, including virtually all SPTs.
Note added in proof
Subsequent to submission of this manuscript, similar molecular findings were reported in solid-pseudopapillary tumors in a study by Tanaka et al. 67
Footnotes
Address reprint requests to Susan Abraham, M.D., Division of GI/Liver Pathology, Department of Pathology, Ross Building, Room 632, The Johns Hopkins University School of Medicine, 720 Rutland Ave., Baltimore, MD 21205-2196. E-mail: sabraha1@jhmi.edu.
Supported by a National Cancer Institute SPORE grant (P50-CA62924) in gastrointestinal cancer.
References
- 1.Klimstra DS, Wenig BM, Heffess CS: Solid-pseudopapillary tumor of the pancreas: a typically cystic carcinoma of low malignant potential. Semin Diagn Pathol 2000, 17:66-80 [PubMed] [Google Scholar]
- 2.Mao C, Guvendi M, Domenico DR, Kim K, Thomford NR, Howard JM: Papillary cystic and solid tumors of the pancreas: a pancreatic embryonic tumor? Studies of three cases and cumulative review of the world’s literature. Surgery 1995, 118:821-828 [DOI] [PubMed] [Google Scholar]
- 3.Horisawa M, Niinomi N, Sato T, Yokoi S, Oda K, Ichikawa M, Hayakawa S: Frantz’s tumor (solid and cystic tumor of the pancreas) with liver metastasis: successful treatment and long-term follow-up. J Pediatr Surg 1995, 30:724-726 [DOI] [PubMed] [Google Scholar]
- 4.Rebhandl W, Felberbauer FX, Puig S, Paya K, Hochschorner S, Barlan M, Horcher E: Solid-pseudopapillary tumor of the pancreas (Frantz tumor) in children: report of four cases and review of the literature. J Surg Oncol 2001, 76:289-296 [DOI] [PubMed] [Google Scholar]
- 5.Ogawa T, Isaji S, Okamura K, Noguchi T, Mizumoto R, Ishihara A: A case of radical resection for solid-cystic tumor of the pancreas with widespread metastases in the liver and greater omentum. Am J Gastroenterol 1993, 88:1436-1439 [PubMed] [Google Scholar]
- 6.Saiura A, Umekita N, Matsui Y, Maeshiro T, Miyamoto S, Kitamura M, Wakikawa A: Successful surgical resection of solid cystic tumor of the pancreas with multiple liver metastases and a tumor thrombus in the portal vein. Hepatogastroenterology 2000, 47:887-889 [PubMed] [Google Scholar]
- 7.Pettinato G, Manivel JC, Ravetto C, Terracciano LM, Gould EW, di Tuoro A, Jaszcz W, Albores-Saavedra J: Papillary cystic tumor of the pancreas. A clinicopathologic study of 20 cases with cytologic, immunohistochemical, ultrastructural, and flow cytometric observations, and a review of the literature. Am J Clin Pathol 1992, 98:478-488 [DOI] [PubMed] [Google Scholar]
- 8.Lompo O, Hofman V, Soler C, Valla JS, Michiels JF, Bedossa P, Hofman P: Solid and pseudopapillary tumor of the pancreas: immunohistochemical and ultrastructural study of 2 pediatric cases. Ann Pathol 2000, 20:221-224 [PubMed] [Google Scholar]
- 9.Notohara K, Hamazaki S, Tsukayama C, Nakamoto S, Kawabata K, Mizobuchi K, Sakamoto K, Okada S: Solid-pseudopapillary tumor of the pancreas: immunohistochemical localization of neuroendocrine markers and CD10. Am J Surg Pathol 2000, 24:1361-1371 [DOI] [PubMed] [Google Scholar]
- 10.Kosmahl M, Seada LS, Janig U, Harms D, Kloppel G: Solid-pseudopapillary tumor of the pancreas: its origin revisited. Virchows Arch 2000, 436:473-480 [DOI] [PubMed] [Google Scholar]
- 11.Stommer P, Kraus J, Stolte M, Giedl J: Solid and cystic pancreatic tumors. Clinical, histochemical, and electron microscopic features in ten cases. Cancer 1991, 67:1635-1641 [DOI] [PubMed] [Google Scholar]
- 12.Balercia G, Zamboni G, Bogina G, Mariuzzi GM: Solid-cystic tumor of the pancreas. An extensive ultrastructural study of fourteen cases. J Submicrosc Cytol Pathol 1995, 27:331-340 [PubMed] [Google Scholar]
- 13.Kissane JM: Pancreatoblastoma and solid and cystic papillary tumor: two tumors related to pancreatic ontogeny. Semin Diagn Pathol 1994, 11:152-164 [PubMed] [Google Scholar]
- 14.Nishihara K, Nagoshi M, Tsuneyoshi M, Yamaguchi K, Hayashi I: Papillary cystic tumors of the pancreas. Assessment of their malignant potential. Cancer 1993, 71:82-92 [DOI] [PubMed] [Google Scholar]
- 15.Cho NH, Go JH, Jung SH, Jung WH, Lee KK: Correlation between proliferating index and prognostic factors in papillary cystic tumors of the pancreas. J Korean Med Sci 1995, 10:342-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson MB, Adams DB, Garen PD, Gansler TS: Aspiration cytologic, ultrastructural, and DNA flow cytometric findings of solid and papillary tumor of the pancreas. Cancer 1992, 69:2235-2243 [DOI] [PubMed] [Google Scholar]
- 17.Greenberg ML, Rennie Y, Grierson JM, Quin JW, Boadle RA: Solid and papillary epithelial tumour of the pancreas: cytological case study with ultrastructural and flow cytometric evaluation. Diagn Cytopathol 1993, 9:541-546 [DOI] [PubMed] [Google Scholar]
- 18.Flejou JF, Boulange B, Bernades P, Belghiti J, Henin D: p53 protein expression and DNA ploidy in cystic tumors of the pancreas. Pancreas 1996, 13:247-252 [DOI] [PubMed] [Google Scholar]
- 19.Lee WY, Tzeng CC, Chen RM, Tsao CJ, Tseng JY, Jin YT: Papillary cystic tumors of the pancreas: assessment of malignant potential by analysis of progesterone receptor, flow cytometry, and ras oncogene mutation. Anticancer Res 1997, 17:2587-2591 [PubMed] [Google Scholar]
- 20.Grant LD, Lauwers GY, Meloni AM, Stone JF, Betz JL, Vogel S, Sandberg AA: Unbalanced chromosomal translocation, der(17)t(13;17)(q14;p11) in a solid and cystic papillary epithelial neoplasm of the pancreas. Am J Surg Pathol 1996, 20:339-345 [DOI] [PubMed] [Google Scholar]
- 21.Maitra A, Weinberg AG, Schneider N, Patterson K: Detection of t(11;22)(q24;q12) translocation and EWS-FLI-1 fusion transcript in a case of solid pseudopapillary tumor of the pancreas. Pediatr Dev Pathol 2000, 3:603-605 [DOI] [PubMed] [Google Scholar]
- 22.Matsubara K, Nigami H, Harigaya H, Baba K: Chromosome abnormality in solid and cystic tumor of the pancreas. Am J Gastroenterol 1997, 92:1219-1221 [PubMed] [Google Scholar]
- 23.Bartsch D, Bastian D, Barth P, Schudy A, Nies C, Kisker O, Wagner HJ, Rothmund M: K-ras oncogene mutations indicate malignancy in cystic tumors of the pancreas. Ann Surg 1998, 228:79-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamaue H, Tanimura H, Shono Y, Onishi H, Tani M, Yamoto H, Kinoshita H, Uchiyama K: Solid and cystic tumor of the pancreas: clinicopathologic and genetic studies of four cases. Int J Pancreatol 2000, 27:69-76 [DOI] [PubMed] [Google Scholar]
- 25.Muller-Hocker J, Zietz CH, Sendelhofert A: Deregulated expression of cell cycle-associated proteins in solid pseudopapillary tumor of the pancreas. Mod Pathol 2001, 14:47-53 [DOI] [PubMed] [Google Scholar]
- 26.Lam KY, Lo CY, Fan ST: Pancreatic solid-cystic-papillary tumor: clinicopathologic features in eight patients from Hong Kong and review of the literature. World J Surg 1999, 23:1045-1050 [DOI] [PubMed] [Google Scholar]
- 27.Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A: Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001, 84:253-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abraham SC, Wu TT, Klimstra DS, Finn L, Lee JH, Yeo CJ, Cameron JL, Hruban RH: Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/β-catenin pathway and chromosome 11p. Am J Pathol 2001, 159:1619-1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, Brennan M, Cameron JL, Klimstra DS: Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/β-catenin pathway. Am J Pathol 2002, 160:953-962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tetsu O, McCormick F: β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398:422-426 [DOI] [PubMed] [Google Scholar]
- 31.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C: Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999, 96:1603-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sellin JH, Umar S, Xiao J, Morris AP: Increased β-catenin expression and nuclear translocation accompany cellular hyperproliferation in vivo. Cancer Res 2001, 61:2899-2906 [PubMed] [Google Scholar]
- 33.Takayasu H, Horie H, Hiyama E, Matsunaga T, Hayashi Y, Watanabe Y, Suita S, Kaneko M, Sasaki F, Hashizume K, Ozaki T, Furuuchi K, Tada M, Ohnuma N, Nakagawara A: Frequent deletions and mutations of the beta-catenin gene are associated with overexpression of cyclin D1 and fibronectin and poorly differentiated histology in childhood hepatoblastoma. Clin Cancer Res 2001, 7:901-908 [PubMed] [Google Scholar]
- 34.Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR: An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994, 172:5-12 [DOI] [PubMed] [Google Scholar]
- 35.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Rhuban RH: Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000, 156:37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 37.Yashima K, Nakamori S, Murakami Y, Yamaguchi A, Hayashi K, Ishikawa O, Konishi Y, Sekiya T: Mutations of the adenomatous polyposis coli gene in the mutation cluster region: comparison of human pancreatic and colorectal cancers. Int J Cancer 1994, 59:43-47 [DOI] [PubMed] [Google Scholar]
- 38.Barth AI, Nathke IS, Nelson WJ: Cadherins, catenins, and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 1997, 9:683-690 [DOI] [PubMed] [Google Scholar]
- 39.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W: Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature 1996, 382:638-642 [DOI] [PubMed] [Google Scholar]
- 40.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P: Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996, 272:1023-1026 [DOI] [PubMed] [Google Scholar]
- 41.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P: Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA 1995, 92:3046-3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R: β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997, 16:3797-3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW: Serine phosphorylation-regulated ubiquitination and degradation of β-catenin. J Biol Chem 1997, 272:24735-24738 [DOI] [PubMed] [Google Scholar]
- 44.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancers by mutations in β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 45.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler W, Vogelstein B, Clevers H: Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon cancer. Science 1997, 275:1784-1787 [DOI] [PubMed] [Google Scholar]
- 46.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 47.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998, 58:1130-1134 [PubMed] [Google Scholar]
- 48.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-myc as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 49.Hruban RH, Goggins M, Parsons J, Kern SE: Progression model for pancreatic cancer. Clin Cancer Res 2000, 6:2969-2972 [PubMed] [Google Scholar]
- 50.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53:549-554 [DOI] [PubMed] [Google Scholar]
- 51.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero Jr RA, Meltzer PS, Hahn SA, Kern SE: DPC4 gene in various tumor types. Cancer Res 1996, 56:2527–2530 [PubMed]
- 52.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]
- 53.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 54.DiGiuseppi JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allsion DC, Cameron JL, Offerhaus GJA: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 55.Takahashi H, Hashimoto K, Hayakawa H, Kusakawa M, Okamura K, Kosaka A, Mizumoto R, Katsuta K: Solid cystic tumor of the pancreas in elderly men: report of a case. Surg Today 1999, 29:1264-1267 [DOI] [PubMed] [Google Scholar]
- 56.Ohashi K, Nakajima Y, Hisanaga M, Nakano H, Tsutsumi M, Kondoh S, Konishi Y: A solid and papillary (solid-cystic) tumor of the pancreas occurring in a 36-year-old man: report of a case. Surg Today 1993, 23:551-555 [DOI] [PubMed] [Google Scholar]
- 57.Tomioka T, Inoue K, Yamamoto T, Motojima K, Tsunoda T, Kanematsu T: Solid and cystic tumor of the pancreas occurring without cyst formation in an adult male. Int J Pancreatol 1993, 14:195-200 [DOI] [PubMed] [Google Scholar]
- 58.Jung SE, Kim DY, Park KW, Lee SC, Jang JJ, Kim WK: Solid and papillary epithelial neoplasm of the pancreas in children. World J Surg 1999, 23:233-236 [DOI] [PubMed] [Google Scholar]
- 59.Chao HC, Kong MS, Lin SJ, Lou CC, Lin PY: Papillary cystic neoplasm of the pancreas in children: report of three cases. Acta Paediatr Taiwan 2000, 41:101-105 [PubMed] [Google Scholar]
- 60.Abraham SC, Montgomery EA, Giardiello FM, Wu TT: Frequent beta-catenin mutations in juvenile nasopharyngeal angiofibromas. Am J Pathol 2001, 158:1073-1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abraham SC, Reynolds C, Lee JH, Montgomery EA, Baisden BL, Krasinskas AM, Wu TT: Fibromatosis of the breast and mutations involving the APC/β-catenin pathway. Hum Pathol 2002, 33:39-46 [DOI] [PubMed] [Google Scholar]
- 62.Klimstra DS, Wenig BM, Adair CF, Heffess CS: Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol 1995, 19:1371-1389 [DOI] [PubMed] [Google Scholar]
- 63.Klimstra DS, Heffess CS, Oertel JE, Rosai J: Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol 1992, 16:815-837 [DOI] [PubMed] [Google Scholar]
- 64.Terhune PG, Heffess CS, Longnecker DS: Only wild-type c-Ki-ras codons 12, 13, and 61 in human pancreatic acinar cell carcinomas. Mol Carcinog 1994, 10:110-114 [DOI] [PubMed] [Google Scholar]
- 65.Hoorens A, Lemoine NR, McLellan E, Morohoshi T, Kamisawa T, Heitz PU, Stamm B, Ruschoff J, Wiedenmann B, Kloppel G: Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol 1993, 143:685-698 [PMC free article] [PubMed] [Google Scholar]
- 66.Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE, Solcia E, Ranzani GN: K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 1994, 54:1556-1560 [PubMed] [Google Scholar]
- 67.Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, Nagahara N, Sasaki F, Kitagawa N, Nakatani Y, Kobayashi Y: Frequent beta-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res 2001, 61:8401-8404 [PubMed] [Google Scholar]