

Abstract
Microsatellite instability (MSI) secondary to loss of DNA mismatch repair (MMR) is present in adenomas and colorectal carcinomas from individuals with hereditary nonpolyposis colorectal cancer (HNPCC). To better characterize when MMR loss occurs during HNPCC progression, the extent of deletions in noncoding polyA sequences were compared between 6 adenomas (all ≤1.0 cm in size) and 10 cancers. Numbers of deleted bases reflect time since loss of MMR because polyA deletions are stepwise. Adenoma deletions were nearly the same (85%) as the cancers with sum total deletions at four different polyA loci of −32.7 bases in adenomas and −38.4 bases in cancers. Intervals between negative clinical examinations and tumor removal (average of 2.1 years) were known for six tumors. There were no significant differences in the extent of deletions in tumors removed under clinical surveillance (−34.8 bases) versus tumors removed without prior negative examinations (−36.5 bases). These findings illustrate that MSI is extensive in both small adenomas, and tumors which appear after negative clinical examinations, consistent with an early loss of MMR in HNPCC, even before a gatekeeper mutation.
Germline mutations in mismatch repair (MMR) loci are present in individuals with HNPCC. 1 Inactivation of the normal allele, typically from loss of heterozygosity, 2 results in MMR deficiency and elevated mutation rates. 1 Mutation rates in simple repeat sequences or microsatellite (MS) loci are increased approximately 100-fold in MMR-deficient cell lines 3,4 and widespread microsatellite instability (MSI) is present in hereditary nonpolyposis colorectal cancer (HNPCC) tumors. 5
Most MS loci are noncoding and selection for these mutations is unlikely. Proportions of altered MS loci have been useful for classification of tumors as either with low or high MSI. 6 High MSI (MSI-H) is strongly associated with loss of MMR. 6 Comparisons between tumor and normal alleles of long polyA repeats are sensitive indicators of MSI because most alterations are deletions. 7 Deletions occur sequentially (stepwise) with loss of a single or a few bases at a time. 3,7-9 Therefore, polyA deletions not only indicate loss of MMR but their extent also provides information on the time since loss of MMR. 10
The MSI of HNPCC adenomas suggests MMR loss occurs early in tumor progression. 11-13 In theory, MMR loss could even precede a gatekeeper mutation because phenotypically normal cells may be MMR-deficient. 14 It is possible to infer the past from the extent of MS mutations or drift from germline for individual MS loci. Patterns of MS mutations in CA repeat dinucleotide MS loci estimate loss of MMR occurs on average approximately 6.3 years before tumor removal (5.5 years for adenomas and 6.6 years for cancers) and can precede a gatekeeper mutation. 15 To further characterize the timing of MMR loss during progression, the extent of deletions at four polyA loci were compared between HNPCC adenomas and cancers. If loss of MMR occurs very early and precedes a gatekeeper mutation, then the extent of polyA deletions should be similar between adenomas and cancers, regardless of prior clinical surveillance.
Materials and Methods
DNA was extracted 10 from formalin-fixed, paraffin-embedded tumor sections of 6 adenomas and 10 carcinomas from 11 Finnish HNPCC patients (Table 1) ▶ . Clinical information was obtained from patient charts. Surveillance intervals are defined as the time between a negative clinical examination (colonoscopy or prior surgery) and removal of a new tumor. Normal DNA was free of tumor and tumor regions were microdissected to obtain greater than 60% of tumor cells. Germline MLH1 or MSH2 mutations were confirmed by sequencing. The t-test (two-tail) was used to determine the significance between means.
Table 1.
Patient Data
| Patient | Age (yr) | Tumor | Size or stage | Germline mutation | Total deletions | Clinical interval |
|---|---|---|---|---|---|---|
| 1 | 46 | Adenoma | 1.0 cm | MLH1 | −32 | NA |
| 46 | Carcinoma | B | MLH1 | −38 | NA | |
| 2 | 45 | Adenoma | 0.3 cm | MLH1 | −34 | 2.8 yr |
| 3 | 43 | Adenoma | 1.0 cm | MLH1 | −39 | NA |
| 43 | Carcinoma | C | MLH1 | −42 | NA | |
| 44 | Carcinoma | B | MLH1 | −49 | 0.5 yr | |
| 4 | 64 | Carcinoma | B | MLH1 | −34 | NA |
| 5 | 57 | Carcinoma | B | MLH1 | −40 | NA |
| 6 | 56 | Adenoma | 0.5 cm | MSH2 | −29 | 2.0 yr |
| 7 | 53 | Adenoma | 0.5 cm | MSH2 | −28 | 2.3 yr |
| 8 | 58 | Adenoma | 1.0 cm | MLH1 | −34 | 3.8 yr |
| 58 | Carcinoma | A | MLH1 | −40 | 3.8 yr | |
| 9 | 43 | Carcinoma | B | MSH2 | −41 | NA |
| 10 | 38 | Carcinoma | D | MLH1 | −26 | NA |
| 11 | 57 | Carcinoma | B | MLH1 | −35 | 1.0 yr |
NA, not available.
PolyA deletions were measured by comparing germline and tumor sizes. 10 Briefly, four polyA repeat loci were examined (BAT20, BAT25, BAT26, and BAT40). Lengths of mononucleotide repeats were estimated by comparing the most intense polymerase chain reaction (PCR) product band between tumor and normal DNA after electrophoresis on 6% sequencing gels, incorporating [33P]-dCTP (NEN Research Products, Boston, MA) with 35 to 38 PCR cycles. In some cases, to better distinguish between tumor and contaminating normal alleles, tumor DNA was diluted before PCR to avoid overlapping stutter bands.
Results
Somatic deletions were present in all four polyA loci (BAT20, BAT25, BAT26, and BAT40) in the HNPCC adenomas and cancers (Figure 1 ▶ and Table 1 ▶ ). The sum of deletions over the four polyA loci ranged from −26 to −49 bases with overlap between adenomas and cancers (Figure 2) ▶ . Adenomas had 85% of the deletions observed in the cancers (average of −32.7 versus −38.4 bases). There was a slight trend for larger deletions in larger adenomas, but among cancers, fewer deletions were present in higher stage cancers (Figure 3) ▶ .
Figure 1.
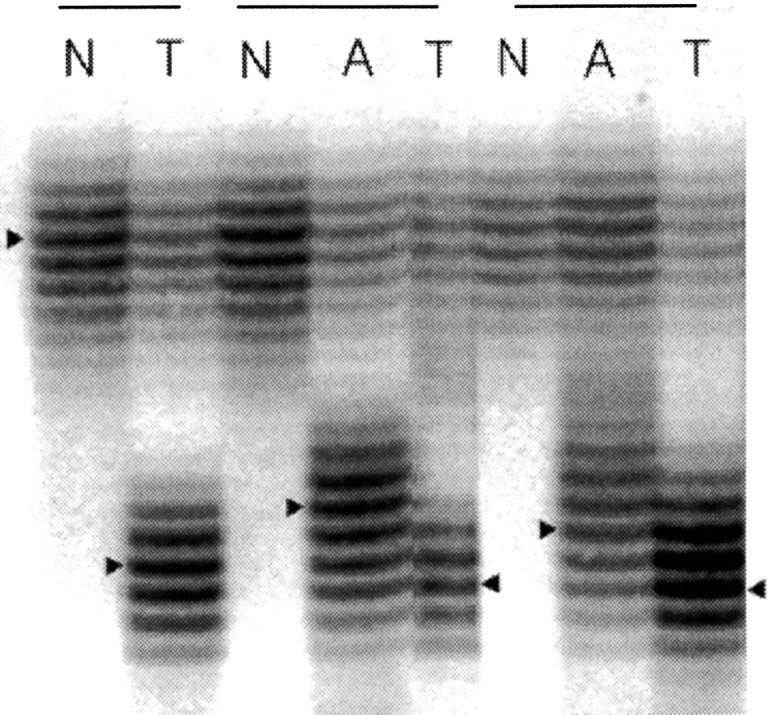
Examples of BAT26 deletions in adenomas (A) and carcinomas (T) from three HNPCC individuals. PolyA repeat deletions are estimated by counting the differences between the densest band (indicated by triangles) from normal (N) and tumor DNA.
Figure 2.

Summary of polyA deletions in adenomas and carcinomas. Average deletions for all tumors were −8.7 bases for BAT20, −6.3 bases for BAT25, −10.8 bases for BAT26, and −10.4 bases for BAT40. Average total deletions were −32.7 bases for the adenomas versus −38.4 bases for the cancers.
Figure 3.
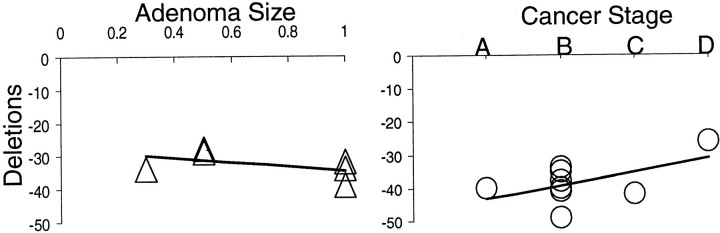
Extensive total polyA deletions regardless of adenoma size or cancer stage.
Some of the HNPCC patients were under surveillance with six known intervals (average of 2.1 years) between tumor removal and a prior negative clinical examination (Table 1) ▶ . There was a slight trend for fewer deletions in tumors with longer surveillance intervals (Figure 4) ▶ . Surveillance did not significantly reduce mutations in the polyA sequences because tumors removed after negative clinical examinations had average deletions of −34.8 bases versus −36.5 bases (P = 0.66) for tumors removed without prior surveillance.
Figure 4.

Few differences in total polyA deletions regardless of interval after a negative examination, or whether under clinical surveillance.
Discussion
Loss or inactivation of the functional MMR allele in HNPCC marks a “second-hit” and starts the accumulation of mutations in MS loci. Frame-shift mutations characteristic of MMR deficiencies are found in a number of tumor suppressor loci such as TGFBRII and BAX in HNPCC tumors, 16,17 suggesting loss of MMR precedes these mutations. However, designating exactly when MMR loss occurs during progression is difficult because “time” is typically based on morphological and frequency criteria. Mutations present in both adenomas and cancers are “early” mutations whereas “late” mutations are found more frequently in cancers. 18 The extensive MSI observed in HNPCC adenomas and carcinomas suggest loss of MMR occurs early in progression. 3,11-13
Determining when a mutation occurs may be confounded by the requirement that only mutations accompanied by a gatekeeper mutation are detectable. A gatekeeper mutation 1 is defined here as the first mutation that allows for visible clonal expansion. Progenitors with mutations other than gatekeeper mutations are “invisible”. Such scenarios are illustrated in Figure 5 ▶ using an “imaginary” observer with the omniscient ability to see a MMR “clock” which may start whether or not it is accompanied by a tumor. Imaginary and real observers will agree on late MMR loss because tumors with and without MMR deficiency can be sampled. In contrast, only the imaginary observer can see the early MMR loss and accumulation of MS mutations that precede a gatekeeper mutation because a real observer will see nothing. However, a real observer may be just as knowledgeable because the clock can be examined when the tumor is removed. Assuming an ability to recognize and interpret this clock, a real observer can infer when it started.
Figure 5.
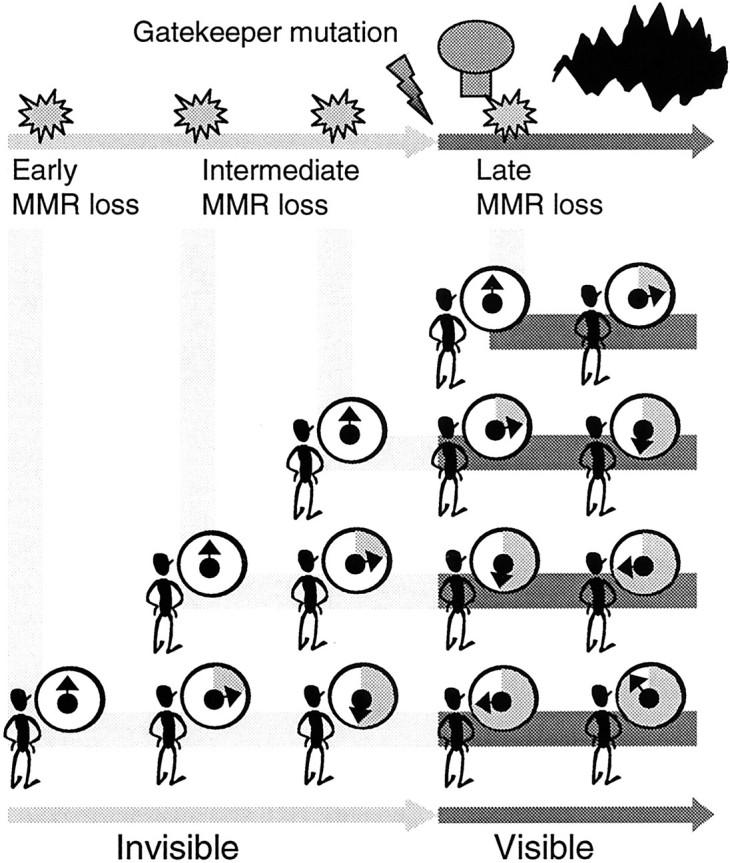
Possible MMR loss scenarios. Loss of MMR can only be physically detected after a gatekeeper mutation because otherwise no tumor is present. However, an “imaginary” omniscient observer can watch for the accumulation of MS mutations (depicted here as the shaded portion of a MMR clock) whether or not a visible tumor is present. When MMR loss precedes a gatekeeper mutation, mutations accumulate in clinically occult progenitors. If MMR loss occurs very early relative to a gatekeeper mutation, both adenomas and cancers will have extensive MSI. Tumors that appear shortly after negative clinical examinations will also have extensive MSI because most mutations accumulate in occult progenitors. A real observer who can only examine tumors may infer when loss of MMR occurs by realizing the clock-like nature of polyA deletions.
PolyA deletions were similar between HNPCC adenomas and cancers, and between tumors regardless of clinical surveillance. The scenario most consistent with these observations is MMR loss that greatly precedes a gatekeeper mutation. With this scenario (Figure 5) ▶ , adenomas have as nearly as many polyA deletions as cancers because most MS mutations occur before a gatekeeper mutation. Tumors arising with or without prior negative clinical examinations would have similar numbers of deletions because most deletions accumulate in occult progenitors.
With early MMR loss, the final extent of MSI in HNPCC adenomas or cancers would not be surprising for an imaginary omnipresent observer (Figure 5) ▶ , but a real observer might conclude that MMR loss is associated with catastrophic MSI because intermediate MSI states are seldom observed. 19,20 A real observer may also question whether polyA deletions reflect times since loss of MMR because proliferation kinetics vary during progression. However, a key feature of very early MMR loss is that phenotypic differences between adenomas or cancers would have relatively minor effects on the polyA clock because most of their histories are written in normal colon. Most MS mutations accumulate unobserved in phenotypically normal progenitors that are likely to have similar proliferation rates. In addition, polyA repeat sequences exhibit molecular clock properties in many different MMR deficient cells including normal murine intestines and human cancer cell lines. 3,7-10 Total deletions are not precise time measures because mutations are stochastic 10 and confidence intervals (not calculated here) would be expected to be large.
The polyA repeat tumor clock analysis also concurs with a quantitative analysis of CA-dinucleotide repeats, 15 with similar and small differences between adenomas and cancers, with or without surveillance (Table 2) ▶ . Although the current polyA sequence analysis is not formally quantitative, both CA- and polyA-repeat mutation patterns support the conclusion that loss of MMR often precedes a gatekeeper mutation. The mutation spectrum of APC is also consistent with MMR loss preceding APC mutation in most MSI-H cancers. 21 APC mutations likely lag BAT polyA repeat deletions because APC coding regions lack long repeats (≤A8) and mutation rates are markedly lower for shorter polyA repeats. 9
Table 2.
Total PolyA Deletions versus CA-Repeat Clock Data
| Specimen | PolyA deletions | CA-repeat tumor clock: intervals since MMR loss* |
|---|---|---|
| Adenoma | −32.7 bases | 2000 divisions |
| Cancer | −38.4 bases | 2400 divisions |
| Ratio | 0.85 | 0.83 |
| Interval tumors | −34.8 bases | 6.0 years |
| Non-interval tumors | −36.5 bases | 6.2 years |
| Ratio | 0.95 | 0.97 |
*From Tsao et al. 15
The start of progression has been usually defined as a visible change in phenotype. 22 However, the start of genetic progression (the accumulation of mutations) does not need to coincide with visible HNPCC tumor progression because MMR deficient tissues may accumulate MS mutations and remain phenotypically normal. 14,23 Many or most mutations may potentially accumulate before the onset of visible neoplasia. Loss of MMR may not even mark the start of genetic progression because some mutations found in MSI-H tumors lack features characteristic of MMR loss. 21 Further systematic studies of tumor mutations may help better characterize the frequencies and lengths of occult genetic progression periods.
Acknowledgments
We thank Sylvia I. Lambrechts and Sarka Cernosek for technical assistance.
Footnotes
Address reprint requests to Darryl Shibata, M.D., Department of Pathology, University of Southern California School of Medicine, 1200 N. State St., Unit I, Room 2428, Los Angeles, CA 90033. E-mail: dshibata@hsc.usc.edu.
Supported in part by a postdoctoral fellowship to K.-M. Kim from the Korea Science and Engineering Foundation.
References
- 1.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 2.Hemminki A, Peltomaki P, Mecklin JP, Jarvinen H, Salovaara R, Nystrom-Lahti M, de la Chapelle A, Aaltonen LA: Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet 1994, 8:405-410 [DOI] [PubMed] [Google Scholar]
- 3.Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M: Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet 1994, 6:273-281 [DOI] [PubMed] [Google Scholar]
- 4.Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, Meuth M: Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci USA 1994, 91:6319-6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapelle A: Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260:812-816 [DOI] [PubMed] [Google Scholar]
- 6.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S: A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 7.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363:558-561 [DOI] [PubMed] [Google Scholar]
- 8.Percesepe A, Pedroni M, Sala E, Menigatti M, Borghi F, Losi L, Viel A, Genuardi M, Benatti P, Roncucci L, Peltomaki P, Ponz de Leon M: Genomic instability and target gene mutations in colon cancers with different degrees of allelic shifts. Genes Chromosomes Cancer 2000, 27:424-429 [PubMed] [Google Scholar]
- 9.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA: Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 1997, 17:2859-2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blake C, Tsao J-L, Wu A, Shibata D: Stepwise deletions of polyA sequences in mismatch repair deficient colorectal cancers. Am J Pathol 2001, 158:1867-1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aaltonen LA, Peltomaki P, Mecklin JP, Jarvinen H, Jass JR, Green JS, Lynch HT, Watson P, Tallqvist G, Juhola M, Sistonen P, Hamilton SR, Kinzler KW, Vogelstein B, de la Chapelle A: Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res 1994, 54:1645-1648 [PubMed] [Google Scholar]
- 12.Iino H, Simms L, Young J, Arnold J, Winship IM, Webb SI, Furlong KL, Leggett B, Jass JR: DNA microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer. Gut 2000, 47:37-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeb LA: A mutator phenotype in cancer. Cancer Res 2001, 61:3230-3239 [PubMed] [Google Scholar]
- 14.Parsons R, Li GM, Longley M, Modrich P, Liu B, Berk T, Hamilton SR, Kinzler KW, Vogelstein B: Mismatch repair deficiency in phenotypically normal human cells. Science 1995, 268:738-740 [DOI] [PubMed] [Google Scholar]
- 15.Tsao JL, Yatabe Y, Salovaara R, Jarvinen HJ, Mecklin JP, Aaltonen LA, Tavare S, Shibata D: Genetic reconstruction of individual colorectal tumor histories. Proc Natl Acad Sci USA 2000, 97:1236-1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV: Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268:1336-1338 [DOI] [PubMed] [Google Scholar]
- 17.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M: Somatic frame-shift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275:967-969 [DOI] [PubMed] [Google Scholar]
- 18.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 19.Thibodeau SN, Bren G, Schaid D: Microsatellite instability in cancer of the proximal colon. Science 1993, 260:816-819 [DOI] [PubMed] [Google Scholar]
- 20.Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley CW, Michels VV, Farr GH, Jr, O’Connell MJ: Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998, 58:1713-1718 [PubMed] [Google Scholar]
- 21.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, Kinzler KW, Vogelstein B, Dunlop MG: APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA 1996, 93:9049-9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nowell PC: The clonal evolution of tumor cell populations. Science 1976, 194:23-28 [DOI] [PubMed] [Google Scholar]
- 23.Vilkki S, Tsao JL, Loukola A, Poyhonen M, Vierimaa O, Herva R, Aaltonen LA, Shibata D: Extensive somatic microsatellite mutations in normal human tissue. Cancer Res 2001, 61:4541-4544 [PubMed] [Google Scholar]