

Abstract
Bronchiolar epithelium is postulated to play a critical role in the orchestration of responses to inhaled allergens, and may contribute to the pathogenesis of asthma. Using a murine model of allergic airway inflammation and hyperresponsiveness, we demonstrate in mice sensitized with ovalbumin (OVA) that following a single challenge with nebulized OVA, a rapid and protracted activation of inhibitor of kappa B kinase (IKK) occurred in lung tissue. IKK activation was followed by nuclear localization of nuclear factor (NF)-κB within the bronchiolar epithelium and increased luciferase activity in lungs of mice containing a NF-κB-dependent reporter gene. Challenge of sensitized mice with OVA also induced mRNA expression of the chemokines, macrophage inflammatory protein-2 (MIP-2) and eotaxin in lung tissue, which corresponded temporally with the observed influx of neutrophils and eosinophils, respectively, into the airspaces. Using laser capture microdissection and quantitative polymerase chain reaction, we demonstrated that MIP-2 and eotaxin were predominantly expressed in bronchiolar epithelium, in contrast to distal regions of the lungs, which expressed lower or undetectable levels of these mRNAs. These studies strengthen the potential importance of the bronchiolar epithelial cell as a source of production of NF-κB-dependent mediators that play a role in asthma.
A number of cell types have been implicated in contributing to the pathophysiology of asthma. Important among these are airway smooth muscle, lymphocytes, resident macrophages, inflammatory leukocytes, and epithelium. 1,2 Bronchiolar epithelial cells serve a number of essential functions within the respiratory tract. 3 The airway epithelium forms a physical barrier between the host and the environment and represents an important site of interaction with inhaled material including oxidant gases, microbes, particulate matter, and allergens. The importance of the signaling events induced by allergens within the bronchiolar epithelium and their relevance to asthma remain to be determined. Conceivably, the production of mediators by this cell type contributes to the generation or perpetuation of inflammation that may facilitate the development of asthma. 4,5
Nuclear factor (NF)-κB is a pleiotropic transcription factor that regulates a diversity of responses in mammalian cells. 6,7 NF-κB becomes activated in response to inflammatory cytokines, mitogens, physical and oxidative stress, infection, and microbial products. 8 NF-κB activity is tightly controlled by the inhibitory protein, IκBα, that is normally present in the cytosol complexed to NF-κB dimers, thereby preventing the nuclear localization of NF-κB and ensuring low basal transcriptional activity. On cellular stimulation, IκBα becomes phosphorylated at serines 32 and 36 by the activity of the IκB kinase (IKK) complex, ubiquinated, and degraded through the 26S proteasome pathway. This exposes the nuclear localization sequence of NF-κB, allowing its entry into the nucleus, facilitating DNA binding and the transcriptional up-regulation of genes downstream of the κB motif.
Activation of NF-κB causes enhanced expression of genes encoding inflammatory cytokines, acute phase proteins, immunoreceptors, and chemokines important in the recruitment of neutrophils and eosinophils. For example, eotaxin, 9 interleukin-6 (IL-6), 10 interleukin-8 (IL-8), 11 macrophage inflammatory protein 2 (MIP-2), 12 granulocyte/macrophage colony-stimulating factor (GM-CSF), 13 inducible nitric oxide synthase (iNOS), 14 intercellular adhesion molecule-1 (ICAM-1), 15 cyclooxygenase-2 16 as well as other mediators, all have NF-κB binding sequences in their promoter regions, which are critical to their transcriptional activation. Thus, the induction of NF-κB by pro-inflammatory stimuli may be a critical signal in evoking an inflammatory response in the lung during the pathogenesis of asthma. 3
Numerous investigations have implicated a role for NF-κB in allergic airway disease. For example, mice lacking p50 or c-Rel develop less airway inflammation on antigen challenge, thus demonstrating a causal role of NF-κB in airway inflammation. 17-19 Consequently, avenues to inhibit NF-κB activation are being developed. 4 However, it remains to be determined how rapidly NF-κB becomes activated in response to antigen challenge, whether it involves IKK, at what site within the lung NF-κB activation occurs, and what the consequences of that activation are. In the present study, we used a murine model of ovalbumin (OVA)-driven allergic inflammation to elucidate the timeframe, mechanism, and localization of NF-κB activation, as well as the expression of two chemokines, eotaxin and MIP-2, known to be regulated, at least in part, by NF-κB. Our findings demonstrate that, in response to aerosolized challenge of sensitized mice, the bronchiolar epithelium is an important site of IKK and NF-κB activity, as well as expression of NF-κB-dependent inflammatory mediators.
Materials and Methods
Mice
Six-week-old female BALB/c mice were purchased from the Jackson Laboratories (Bar Harbor, ME). NF-κB-luciferase reporter mice, stably incorporating a transgene containing two copies of the κB motif from the κ light chain enhancer in front of a minimal fos promoter driving luciferase, 20 were a kind gift from Dr. Mercedes Rincon of the Department of Medicine at the University of Vermont. All mice were housed in the University of Vermont Animal Facility and the Institutional Animal Care and Use Committee granted approval for all studies. Mice were administered OVA (20 μg, grade V ovalbumin, Sigma, St. Louis, MO) with Alum (2.25 mg, Imject Alum, Pierce, Rockford, IL) via intraperitoneal injection on days 0 and 14. Mice were challenged using 1 or 3 doses of aerosolized 1% OVA in phosphate-buffered saline (PBS) for 30 minutes on day 21 or on days 21, 22, and 23, as previously described. 21,22 Mice were euthanized by a lethal dose of pentobarbital (Nembutal sodium solution, Abbott Laboratories, North Chicago, IL) via intraperitoneal injection.
Pulmonary Function Assessment
Mice were anesthetized with 90 mg/kg of pentobarbital. Tracheotomized mice from each group were mechanically ventilated for the assessment of pulmonary function, as previously described. 23 Briefly, a tracheotomy tube was inserted and then connected to the inspiratory and expiratory ports of a volume-cycled ventilator (FlexiVent, SCIREQ, Inc., Montreal, PQ, Canada). Mice were ventilated at a rate of 160 to 200 breaths/min, with a tidal volume of 0.2 ml, and frequencies of 2.5 Hz using a computer controlled volume ventilator with 3 cm H2O positive end-expiratory pressure. Pressure, flow, and volume were used to calculate pulmonary resistance after challenge with inhaled doses of aerosolized methacholine (Sigma-Aldrich, St. Louis, MO) ranging from 3.1 to 50 mg/ml in half log increments, as previously described. 22
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) was collected from mice immediately on euthanization by instillation and recovery of 800 μl of 0.9% NaCl. Total cells in BAL were enumerated and 2 × 10 4 cells were centrifuged onto glass slides at 800 rpm. Cytospins were stained using the Hema3 kit (Biochemical Sciences, Inc., Swedesboro, NJ) and differential cell counts were performed on 500 cells.
IKK Enzyme Activity Assay
Lungs from sensitized mice exposed to a single dose of aerosolized OVA were lavaged through a tracheal canula, removed and immediately frozen by immersion in liquid nitrogen and stored at −80°C. Frozen lungs were simultaneously thawed and homogenized in cold Nonidet P-40 (NP-40) immunoprecipitation buffer 24 (0.1% NP-40, 50 mmol/L HEPES (pH 7.4), 1 mmol/L EDTA, 150 mmol/L NaCl, 2 mmol/L MgCl2, 500 μmol/L DTT, 100 μmol/L NaF, 1% aprotinin, 10 μg/ml leupeptin, 10 mmol/L sodium orthovanadate, and 1 mmol/L PMSF using a Tissue Tearor mechanical homogenizer (Dremel, Racine, WI). Lysates were centrifuged at 14,000 rpm at 4°C for 10 minutes and the supernatants removed to clean tubes. Following protein quantitation using the method of Lowrey (DC Protein Assay, BioRad Laboratories, Hercules, CA), the IKK complex was immunoprecipitated from 200 μg of total protein using 0.5 μg of anti-IKKγ (sc-8330, Santa Cruz Biotechnology, Santa Cruz, CA) and 35 μl of twice-washed (with NP-40 immunoprecipitation buffer) protein A agarose beads (GIBCO-BRL, Rockville, MD) for 90 minutes at 4°C on a rotating platform. The washed immunoprecipitates were incubated for 20 minutes at 30°C in 26.5 μl of kinase buffer (20 mmol/L HEPES (pH 7.5), 20 mmol/L β-glycerophosphate, 1 mmol/L MnCl2, 5 mmol/L MgCl2, 2 mmol/L NaF, 1 mmol/L DTT, and 5μCi [γ-32P]ATP), with 1.4 μg GST-IκBα(1–54), the substrate for IKK (kind gift from R.M. Ten, Mayo Clinic, Rochester, MN). Incorporation of 32P into the substrate was visualized by autoradiography after loading of proteins onto 15% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and electrophoresis, and results were quantitated on a BioRad phosphoimage-analyzer.
Immunostaining
Following euthanasia, lungs were instilled with 4% paraformaldehyde in PBS (4% PFA) for 10 minutes at a pressure of 25 cm H2O and placed into 4% PFA at 4°C overnight for fixation of the tissue. Fixed lungs were then mounted in paraffin, and 5-μm sections were prepared, affixed to glass microscope slides, and prepared for immunostaining by deparaffinizing with xylene and rehydrating through a series of ethanols. Alternatively, unfixed lungs inflated with PBS at a pressure of 25 cm H2O were mounted in Tissue-Tek OCT compound (Sakura Finetek, Inc., Torrance, CA) and frozen in liquid nitrogen-chilled isopentane for the preparation of 10-μm frozen sections. Slides were blocked three times for 20 minutes each in PBS containing 1% bovine serum albumin (PBS/1% BSA) and permeabilized with 0.1% Triton X-100 in PBS. Slides were then incubated with an appropriate dilution of primary antibody recognizing the RelA subunit of NF-κB (SC-372, Santa Cruz Biotechnology). Following three washes in PBS, slides were incubated with an Oregon Green-labeled secondary antibody (Molecular Probes, Eugene, OR), washed, RNase treated, and counterstained with propidium iodide (PI) to label DNA, for nuclear localization. Sections were scanned using an Olympus BX50 upright microscope (Olympus America, Lake Success, NY) configured to a Bio-Rad MRX 1000 confocal scanning laser microscope system equipped with a 15 mW mixed-gas krypton-argon laser (Bio-Rad Laboratories) with excitation wavelengths at 588, 568, and 647 nm. Propidium iodide staining of nuclei was detected by exciting fluorescence with the 568 laser whereas Oregon Green was detected following excitation with the 488-laser line.
Ribonuclease Protection Assay
RNA was extracted from frozen lungs using a modified method of Chomczynski, 25 and gene expression was assessed qualitatively and quantitatively using the ribonuclease protection assay (RPA) (Pharmingen, San Jose, CA). Five μg of total lung RNA was hybridized to 32P-labeled probe sets (mCK-5), processed according to the manufacturer’s protocol, and hybridized components were separated on a 5% acrylamide gel. Each specific hybridized product migrates according to its size, thereby allowing identification of individual bands that were assigned to specific mRNA products.
Laser Capture Microdissection and Real-Time Polymerase Chain Reaction
The PixCell II laser capture microdissector from Arcturus Engineering (Mountain View, CA) is a laser-based system designed for the high resolution removal of specific cell types from paraffin-embedded or cryostat tissue sections. Laser capture microdissection (LCM) was used to selectively remove bronchiolar epithelium from frozen tissue on slides. As a control, parenchymal tissue (consisting solely of alveolar wall) was also captured for comparative analysis of gene expression. Frozen lung tissues were sectioned at 10-μm thickness and placed onto uncoated glass slides, stained with hematoxylin, and dehydrated through a series of ethanols and xylenes. Dehydrated sections were placed onto the stage of the microdissector system and captured by multiple pulses of the laser (∼500). The thermoplastic film of the cap containing the captured cells was then inverted onto a microcentrifuge tube and total RNA was extracted using a modified method of Chomczynski. 24 RNA was DNase treated and reverse transcribed into cDNA using SuperscriptII, according to instructions by the manufacturer (GIBCO-BRL). During TaqMan analysis, copy number of cDNA targets was quantified by the point during cycling when the PCR product is first detected. TaqMan probes and primers were designed to bind to the gene of interest. The probes contained a 5′ fluorigenic dye (6-FAM) and a 3′ quencher dye (BHQ-1) in close proximity, which represses fluorescence in the intact probe. As the polymerization of the primers is extended, strand displacement and cleavage of the 5′ reporter from quencher dye increases fluorescence resonance energy transfer from 6-FAM, which was detected and quantified by the ABI Prism 7700 sequence detection system (Perkin Elmer Corp., Foster City, CA). Expression of the housekeeping gene, 18S rRNA, was monitored in parallel, using separate reactions, to normalize differences in starting DNA copy number between samples. Levels of mRNA for MIP-2, eotaxin, or CC10 were normalized to 18S rRNA and presented as relative expression.
Real-Time PCR Primers and Probes
Forward (for) and reverse (rev) primers and 6-FAM/BHQ-1 probes (5′−3′) for TaqMan PCR were purchased from Biosearch Technologies (Novato, CA) for the following messages. 18S: for, cggctaccacatccaagga; rev, gagtcctgtattgttatttttcgtcact; probe, cgcgcaaattacccactcccga. Eotaxin: for, agagctccacagcttctatt; rev, cttactggtcatgataaagcagcag; probe, acggtcacttccttcacctcccagg. MIP-2: for, gaacatccagagcttgagtgtga; rev, ttttgaccgcccttgagagt; probe, actgcgcccagacagaagtcatagcc. CC10: for, cctttcaaccctggctcaga; rev, tgggagggtatccaccagtct; probe, caaaatgcgggcacccagctg.
Luciferase Activity Assay
Frozen lung sections were homogenized in 1X Reporter lysis buffer (Promega, Madison, WI) using a TissueTearor homogenizer and cleared by centrifugation. Protein levels were quantitated using the method of Bradford (BioRad, Hercules, CA) and 20-μl aliquots were analyzed in triplicate using a Berthold Lumat LB9501 luminometer (Berthold Australia Pty Ltd, Bundoora, Australia) and 100 μl of luciferin substrate (Promega, Madison, WI). Light emission was measured for 20 seconds and normalized per μg of protein.
Results
Mouse Model of Allergic Airway Inflammation and Hyperresponsiveness
To generate pulmonary inflammation with a number of the functional features of asthma, mice were sensitized with intraperitoneal injections of OVA plus aluminum hydroxide (alum) at days 1 and 14. Control mice were administered intraperitoneal PBS and alum. At days 21, 22, and 23, all mice were challenged with aerosolized OVA. 21,22,26 Only the OVA/OVA protocol resulted in a marked influx of cells into the airways, evidenced by increases in cells recovered by BAL 2 days following the third aerosolized challenge (Figure 1) ▶ . Typical of antigen-induced airway responses, the number of cells recovered from BAL was accounted for in large part by the influx of eosinophils. This murine model of allergic airway inflammation also induced airway hyperresponsiveness, but only in mice that were both sensitized and challenged with allergen, as demonstrated by the increased airway resistance in response to increasing doses of aerosolized methacholine (Figure 2) ▶ .
Figure 1.

Assessment of inflammatory cells in BAL following sensitization and three challenges with antigen (sensitized + challenged). Control mice were unmanipulated, sensitized mice received an intraperitoneal injection of OVA at days 1 and 14. Challenged mice were nebulized with aerosolized OVA at days 21, 22, and 23. BAL was performed 48 hours after the last aerosolized OVA challenge for the assessment of differential cell counts. Data are from four mice per group and are expressed as means ± SEM.
Figure 2.

Responsiveness to methacholine (MC) in sensitized and challenged mice. Sensitized and challenged mice received an intraperitoneal injection of OVA at days 1 and 14 and nebulized OVA at days 21, 22, and 23. Challenged mice received an intraperitoneal injection of vehicle at days 1 and 14 and nebulized OVA at days 21, 22, and 23. Hyperresponsiveness to methacholine was assessed at day 25 (48 hours after the last aerosolized OVA challenge), following administration of increasing doses of aerosolized MC. Data are means ± SEM from eight mice per group.
NF-κB Activation in Bronchiolar Epithelium of Allergen Sensitized and Challenged Mice
The airways, lined by bronchiolar epithelium, represent the first point of contact within the lung to inhaled antigens. Proinflammatory signaling events triggered within these cells in a NF-κB dependent manner may be crucial to the development of inflammation and allergic airway disease. Since the activation of NF-κB is critical to the activation of many chemokine and cytokine genes that control the influx of inflammatory cells, we next determined whether evidence exists for NF-κB activation in the lungs of sensitized and challenged mice. Confocal microscopy was performed on paraffin-embedded lung sections immunostained using an antibody that recognizes RelA, the transcriptionally active subunit of NF-κB. Images in Figure 3 ▶ demonstrate a specific and striking localization of RelA only in bronchiolar epithelium of sensitized and challenged mice. Using propidium iodide as a nuclear marker, nuclear translocation of RelA occurred in OVA/OVA mice, evident from the yellow color formation that occurs when the green (RelA) and red (propidium iodide) color co-localize. Under identical imaging conditions, no marked nuclear RelA was evident in the lungs of control, sensitized only, or challenged only groups. The apparent lack of RelA immunostaining in the cytoplasm of control, sensitized, or challenged lung sections reflects a consequence of tissue preparation previously observed in paraffin-embedded lung tissue, in which active NF-κB was recognized by antibody much better than the inactive NF-κB complex. 27
Figure 3.

Immunolocalization of RelA in bronchiolar epithelial cells of mice after sensitization and challenge with allergen. Control mice were unmanipulated, sensitized mice received an intraperitoneal injection of OVA at days 1 and 14. Challenged mice received an intraperitoneal injection of vehicle at days 1 and 14 and were nebulized with aerosolized OVA at days 21, 22, and 23. Paraffin-embedded lung sections were prepared 48 hours after allergen challenge and stained with an antibody directed against RelA followed by incubation with an Oregon Green-conjugated secondary antibody. Propidium iodide was used as a nuclear counterstain to evaluate nuclear presence of RelA. Sections were scanned by confocal microscopy. Data are representative of results from two separate experiments.
Time Course of NF-κB Activation
Since NF-κB is considered to play an important role in the regulation of inflammatory cytokine and chemokine production, we reasoned that the activities of this transcription factor might be an early event in bronchiolar epithelium following aerosolized allergen challenge of sensitized mice. Therefore, we studied sensitized mice in response to a single challenge with aerosolized OVA. As is shown in Figure 4 ▶ , analysis of BAL revealed a temporally distinct pattern of cellular recruitment to the airways. At early timepoints following aerosolized OVA challenge (15 minutes to 6 hours), neutrophils were recovered in the BAL, which increased in number over time. At the 24 hour timepoint following challenge, eosinophils were first detected and neutrophil numbers were decreasing (Figure 4) ▶ . These results demonstrate temporally distinct patterns of inflammatory cell recruitment to the airways of sensitized mice following a single aerosolized allergenchallenge.
Figure 4.

Assessment of inflammatory cells in bronchoalveolar lavage following sensitization and single challenge with allergen. Mice received an intraperitoneal injection of OVA at days 1 and 14 and were nebulized with aerosolized OVA at day 21. Control mice were sensitized but not exposed to aerosolized OVA. BAL was performed at the indicated times for the assessment of differential cell counts. Data are from four mice per group and are expressed as means ± SEM.
We next addressed whether the rapid induction of airway inflammation in sensitized mice following a single challenge with OVA was preceded by the activation of NF-κB by examining the activity of the IKK complex. IKK functions to inducibly phosphorylate IκBα at serines 32 and 36, leading to the proteasome-mediated degradation of IκBα and activation of NF-κB. Compared to non-sensitized mice exposed to aerosolized OVA, sensitized mice demonstrated rapid and pronounced induction of lung IKK activity (Figure 5) ▶ . Fifteen minutes following cessation of the 30 minute allergen exposure regimen, IKK activity was fivefold higher in sensitized mice compared to the non-sensitized controls. Importantly, IKK activity remained elevated, relative to control levels, for at least 24 hours.
Figure 5.
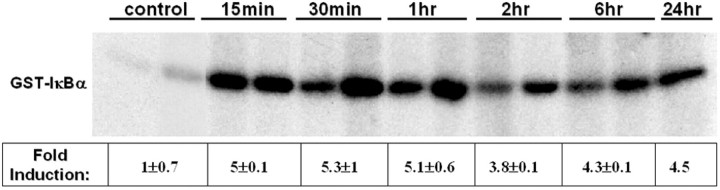
IKK activity assay from whole lungs of mice following sensitization and single challenge with allergen. Mice received an intraperitoneal injection of OVA at days 1 and 14 and were nebulized with aerosolized OVA at day 21. Control mice were sensitized but not exposed to aerosolized OVA. Mice were euthanized at various times following a single allergen challenge, the IKK complex was immunoprecipitated from lung homogenates and subjected to an in vitro kinase assay to measure the incorporation of 32P into GST-IκBα. Presented data are representative of results from two separate experiments, from which band intensities were quantitated by phosphoimage analysis and are presented as fold-induction ± SEM.
We next determined whether the persistent IKK activity culminated in translocation of NF-κB into the nucleus, and exactly which pulmonary cell types were involved. Frozen lung sections obtained 15 or 30 minutes or 1, 2, 6, or 24 hours after OVA challenge were immunostained for RelA and imaged using confocal microscopy. As is seen in Figure 6 ▶ , nuclear localization of RelA was observed preferentially in bronchiolar epithelial cells only of sensitized and challenged mice. Similar to the rapid and prolonged activation of IKK, nuclear translocation of RelA within airway epithelium occurred within 15 minutes following cessation of aerosolized allergen exposure and remained in the nucleus for as long as 24 hours following the single challenge.
Figure 6.
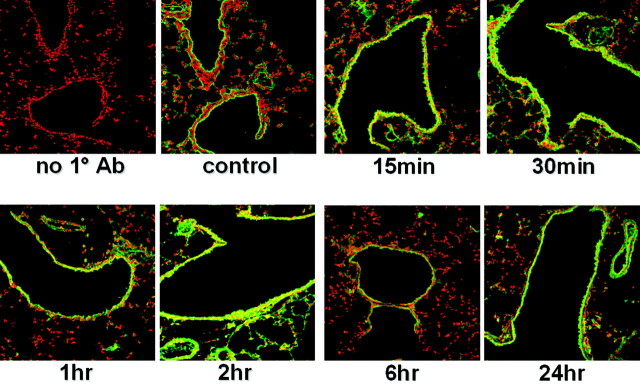
Immunolocalization of RelA in bronchiolar epithelial cells of mice after sensitization and a single challenge with allergen. Mice received an intraperitoneal injection of OVA at days 1 and 14 and nebulized OVA at day 21. Control mice were sensitized but not exposed to aerosolized OVA. Frozen lung sections were prepared at various times following a single antigen challenge and stained with an antibody directed against RelA followed by incubation with an Oregon Green-conjugated secondary antibody. Propidium iodide was used as a nuclear counterstain to evaluate nuclear presence of RelA. All sections were scanned by confocal microscopy using identical settings. Data are representative of results from two separate experiments.
Reporter Gene Expression in Sensitized NF-κB Luciferase Mice following Aerosolized Allergen Challenge
To confirm the induction of NF-κB-dependent gene transcription in lung tissue, NF-κB-luciferase reporter mice were sensitized and exposed to aerosolized OVA. Lungs were harvested 6 hours and 24 hours after a single challenge or 48 hours after three daily challenges and homogenized for the assessment of NF-κB-dependent luciferase enzyme activity. As is demonstrated in Figure 7 ▶ , the luciferase activity in lung homogenates was elevated threefold 6 hours following a single challenge with aerosolized allergen and remained elevated at 24 hours after a single challenge. Similarly, 48 hours following three daily challenges, luciferase activity was elevated as well, thus demonstrating the protracted increases in NF-κB-dependent gene transcription. Taken together, these data demonstrate that NF-κB-dependent gene transcription occurs in the airway epithelial cells of allergic mice in response to aerosolized allergen challenge.
Figure 7.

Luciferase activity in lungs from sensitized NF-κB/luciferase mice following challenge with aerosolized allergen. NF-κB/luciferase mice received an intraperitoneal injection of OVA at days 1 and 14 and were nebulized with aerosolized OVA at day 21 or at days 21, 22, and 23 (3× Ova). Control mice were sensitized but not exposed to aerosolized OVA. Lungs were harvested at 6 or 24 hours following a single challenge (Ova 6 hours and Ova 24 hours) or 48 hours following three aerosolized challenges (3× Ova 48 hours), and homogenates were assessed for luciferase reporter activity, which was normalized to total protein. Data are from three mice per group and are expressed as means ± SEM.
Chemokine Expression in Bronchiolar Epithelium of Allergen Sensitized and Challenged Mice
To demonstrate that NF-κB activation in airway epithelium of sensitized and challenged mice initiates a cascade of events that culminates in recruitment of inflammatory cells, we evaluated mRNA expression of several NF-κB-regulated genes in lung tissue using ribonuclease protection assays. Results in Figure 8 ▶ demonstrate marked increases in the mRNA levels of chemokine/cytokine genes that function to specifically recruit granulocytes in the lungs of sensitized mice administered a single challenge with aerosolized OVA, compared to mice that were only sensitized (C). Acute increases in the mRNA expression of MIP-2, MIP-1β, TCA-3, and monocyte chemotactic protein (MCP-1), were observed in sensitized and challenged mice. In contrast, a more delayed increase in expression of eotaxin was apparent, which is consistent with the delayed influx of eosinophils. The pattern of MIP-1α expression also paralleled that of eotaxin. As additional controls, mice that were neither sensitized nor challenged, or challenged alone, did not result in increased expression of NF-κB-regulated genes in the lungs (data not shown), consistent with the lack of nuclear RelA shown in Figure 3 ▶ . It is of interest to note that MIP-2 mRNA was induced very rapidly while the increases in mRNA expression of eotaxin occurred in a more protracted fashion, despite both being transcriptionally regulated by NF-κB. 9,12 Eotaxin was up-regulated by 6 hours and remained elevated in the lungs of OVA/OVA mice. These results demonstrate a rapid and prolonged up-regulation of NF-κB dependent genes that facilitate the differential recruitment of neutrophils and eosinophils in lungs of sensitized and antigen challenged mice.
Figure 8.

Ribonuclease protection analysis of chemokine mRNA expression in lungs of sensitized and challenged mice. Mice received an intraperitoneal injection of OVA at days 1 and 14 and were nebulized with aerosolized OVA at day 21. Control mice (C) were sensitized but not exposed to aerosolized OVA. Lungs were harvested at the indicated times following a single aerosolized OVA challenge, and RNA was prepared for ribonuclease protection assays using a commercial template set (Pharmingen). Data are representative of results from three separate experiments.
To assess whether the increased mRNA expression of NF-κB regulated chemokines occurred selectively in bronchiolar epithelial cells, frozen lung sections were subjected to LCM, to enrich desired cell populations. From hematoxylin-stained frozen sections, bronchiolar epithelium (Figure 9,A–D) ▶ and parenchymal regions (not shown) were captured separately and RNA was isolated. Enrichment of bronchiolar epithelium by LCM was confirmed by analysis of expression of the bronchiolar epithelial specific gene, CC10 (Figure 9E) ▶ by TaqMan analysis. From samples of bronchiolar epithelium and parenchyma enriched by LCM, expression of MIP-2, eotaxin, and 18S rRNA was analyzed in control animals, which were only exposed to aerosolized allergen, and in sensitized animals 1 hour, 6 hours, and 24 hours after aerosolized allergen challenge. Expression of MIP-2 and eotaxin was normalized relative to the housekeeping gene 18S rRNA. Results in Figures 9F and 9G ▶ demonstrate that the early expression of MIP-2 occurred preferentially in bronchiolar epithelium, whereas expression of MIP-2 in the distal pulmonary regions was much lower (Figure 9F) ▶ . In addition, the delayed expression of eotaxin was observed only in bronchiolar epithelium (Figure 9G) ▶ , as eotaxin expression was undetectable in parenchymal tissue. These results demonstrate that genes regulated by NF-κB are selectively expressed by bronchiolar epithelium of sensitized mice exposed to aerosolized allergen and that expression of these products correlates with patterns of inflammatory cell influx into the airways. In summary, the results presented herein support the hypothesis that airway epithelial NF-κB activity plays a role in the development of allergic airway inflammation.
Figure 9.
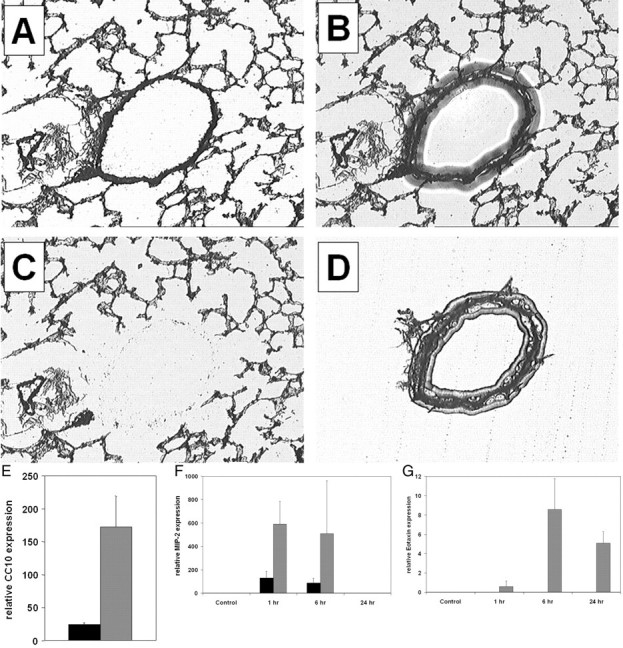
LCM and TaqMan PCR analysis of bronchiolar epithelium and alveolar epithelium following a single nebulized allergen challenge. Mice received an intraperitoneal injection of OVA at days 1 and 14 and were nebulized with aerosolized OVA at day 21. Control mice were sensitized but not exposed to aerosolized OVA. Mice were euthanized at various times following a single allergen challenge, and frozen lung sections were prepared. Sections were visualized by light microscopy (A), a transfer cap was placed atop the section and a laser was pulsed, causing the film to melt and adhere to the section (B). The cap was lifted away, revealing the remaining tissue (C) and the enriched fraction on the cap (D). Bronchiolar epithelial (gray bars) and parenchymal tissues (black bars) were laser captured from mouse lung sections and the RNA expression of the bronchiolar epithelial specific gene product, CC10, was quantitated relative to the expression of the housekeeping gene 18S ribosomal RNA using TaqMan PCR (E). Quantitation of MIP-2 mRNA (F) and eotaxin mRNA (G) expression in bronchiolar epithelium and parenchymal tissue of sensitized mice following a single aerosolized allergen challenge. mRNA levels are normalized to 18s rRNA. Data are from four mice per group and are expressed as means ± SEM.
Discussion
Numerous studies have implicated NF-κB as an important transcription factor that regulates airway inflammation in asthmatics and that may serve as a therapeutic target for the treatment of asthma. 28 Murine models of asthma similar to the one used in these studies have incorporated gene knockout animals to elucidate the role of NF-κB in some aspects of the disease. An essential role for NF-κB activation in the induction of eosinophilia in allergic airway disease was demonstrated in p50 (a heterodimeric partner for the RelA subunit of NF-κB) knockout mice, which do not develop eosinophilia or airway hyperresponsiveness. The p50 knockout mice fail to produce the chemokines eotaxin, MIP-1α and MIP-1β, 19 as well as the Th2 cytokines IL-4, IL-5, and IL-13. 17 Similarly, mice deficient in c-Rel, another member of the NF-κB family that is normally expressed in T lymphocytes, also exhibit reduced circulating IgE levels, airway inflammation, and hyperresponsiveness. 18 These studies, however, do not elucidate the critical cell type responsible for the activation of NF-κB during the elaboration of airway inflammation. Therefore, in the present study we sought to characterize the kinetics and location of NF-κB activation in the lungs of allergen sensitized and challenged mice. Our data demonstrate that the NF-κB pathway is activated rapidly in the lungs of sensitized mice following a single aerosolized antigen challenge and that this is preceded by the activation of IKK, the major regulator of NF-κB activation. Furthermore, using a number of approaches, we have demonstrated that the activation of NF-κB is rapid, persistent, and occurs predominantly in the bronchiolar epithelium. Importantly, the increased expression of the NF-κB regulated neutrophil and eosinophil chemokines, MIP-2 and eotaxin, respectively, by bronchiolar epithelium, provide the necessary gradient for the recruitment of these inflammatory cells into the air spaces.
The bronchiolar epithelium serves an important first line of defense within the airways, and perturbation of these cells by inhaled materials, including allergens, evokes responses that may contribute to the development and maintenance of the asthma phenotype through the activation of NF-κB. 29 In this regard, heaves-affected horses, which are spontaneously sensitized and challenged, display marked airway inflammation and acute airway obstruction with features of asthma. Epithelial cells recovered by bronchial brushing exhibit high levels of NF-κB activity compared to those from healthy horses. 30 Interestingly, the amount of NF-κB activity remaining after recovery from the airway obstruction phase correlated with persistent lung dysfunction, suggesting a prolonged maintenance of NF-κB activity in the bronchial epithelium of affected horses. 31 Similarly, our data demonstrate persistent IKK activity in the lungs and NF-κB activity in the airway epithelium of sensitized mice following a single aerosolized antigen challenge. Importantly, activation of NF-κB has also been observed in airway epithelial cells obtained from asthmatic individuals by bronchial biopsy, 32 evidence of a common mechanism between the murine model and clinical disease.
In our studies, in addition to the nuclear localization of RelA in bronchiolar epithelium of sensitized and challenged mice, enhanced RelA immunostaining was observed in the cytoplasm of bronchiolar epithelium from sensitized mice challenged three times, suggesting elevated expression of RelA in these cells. Although the mechanism of RelA up-regulation observed in these studies is unclear, our findings are complimentary to those demonstrating that lung biopsies from asthmatic patients show enhanced levels of RelA in the bronchiolar epithelium relative to the levels seen in non-asthmatic control subjects. 32 Increases in RelA levels have also been demonstrated in endothelial cells at sites predisposed to development of atherosclerotic lesions, which contribute to enhanced NF-κB activation and expression of NF-κB-inducible genes in these regions. 33 Furthermore, rats exposed to inhaled asbestos also exhibit increased RelA immunoreactivity in bronchiolar epithelium. 27
The NF-κB-regulated chemokines eotaxin and MIP-2 are thought to be important in the development and maintenance of the asthma phenotype. 34 Eotaxin is critical to the recruitment and activation of eosinophils into the airways and potentially also in the development of pulmonary fibrosis as a result of sensitization and challenge with allergen. 34 Human eotaxin is a NF-κB-regulated gene 35 that is inducibly expressed by airway epithelial cells in response to inflammatory cytokines and allergen challenge. 36 The murine eotaxin gene also has a NF-κB binding site (GGAAGCTTCC) −64 to −55 upstream of the transcriptional start site (GenBank accession number U77462). 37 Like most genes, eotaxin in not transcriptionally regulated solely by NF-κB. Both the murine and human eotaxin promoters also contain binding sites for the transcription factor STAT6, which is activated in response to signaling by the Th2 cytokines IL-4, IL-5, and IL-13. 38,39 These cytokines are elevated in asthma and synergistically induce eotaxin mRNA expression. The regulation of eotaxin by STAT6, in cooperation with NF-κB, may explain the delay in eotaxin mRNA expression in sensitized and challenged mice in contrast to the rapid up-regulation of genes that are predominantly regulated by NF-κB. Certainly, the kinetics of STAT6 activation in airway epithelium of sensitized and challenged mice merits further investigation. Unlike eotaxin, which requires both STAT6 and NF-κB for full transcriptional activity, the MIP-2 gene is transcriptionally regulated predominantly by NF-κB and is induced by inflammatory cytokines. 40 MIP-2 (a rodent analog of human IL-8) is a key chemokine necessary for the recruitment of neutrophils into the lung, which can be produced by epithelial cells and normally plays a role in respiratory tract defenses against a variety of insults. 12,41 The temporal dissociation of MIP-2 expression/neutrophil recruitment and eotaxin expression/eosinophil recruitment suggests that the immediate activation of NF-κB by bronchiolar epithelium in response to antigen induces expression of the chemokine MIP-2. Only at later times, in cooperation with the antigen-specific response of local Th2 lymphocytes to generate the cytokines IL-4, IL-5, and IL-13, STAT-6 may become activated, which then would cooperate with NF-κB to induce the expression of eotaxin and subsequently recruit eosinophils to the airway.
The mechanism by which airway epithelial cells from sensitized and challenged mice become stimulated to activate NF-κB in response to allergen remains enigmatic. Our data clearly support the notion that specific antigen recognition is required, as mice sensitized only with adjuvant fail to rapidly activate NF-κB in response to aerosolized challenge with antigen. Interestingly, stimulation of bronchiolar epithelium from allergic asthmatics with allergen in vitro causes the release of cytokines, implicating antigen-specific recognition. 42 The rapid activation of the IKK complex observed in our studies also supports direct recognition of allergen by the airway epithelium. Elucidating the pathways by which bronchiolar epithelium participate in specific responses to allergens is the subject of our ongoing studies. The studies presented herein point to an important role of NF-κB activation in bronchiolar epithelial cells in the development of inflammation and pathophysiological alterations in a murine model of allergic airway disease.
Acknowledgments
We thank Dr. Mercedes Rincon for providing the NF-κB luciferase reporter mice and Dr. Scott Wagers for technical support with assessment of pulmonary mechanics.
Footnotes
Address reprint requests to Yvonne M.W. Janssen-Heininger, Department of Pathology, University of Vermont, Burlington, VT 05405. E-mail: yjanssen@zoo.uvm.edu.
Supported by an NRSA Individual Postdoctoral Fellowship and a Senator Proctor Research Fund grant from the American Lung Association of Vermont and the Vermont Department of Health (to M.E.P.), by National Institutes of Health grant RO1 HL60014 and Public Health Service grants P20 RL15557 and PO1 HL67004 (to Y.J-H.), and by National Institutes of Health grant RO1 HL56638 and Public Health Service grants P20 RR15557 and P01 HL67004 (to C.G.I.).
References
- 1.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM: Asthma: from bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 2000, 161:1720-1745 [DOI] [PubMed] [Google Scholar]
- 2.Boushey HA, Fahy JV: Basic mechanisms of asthma. Environ Health Perspect 1995, 103:229-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes PJ, Chung KF, Page CP: Inflammatory mediators of asthma: an update. Pharmacol Rev 1998, 50:515-596 [PubMed] [Google Scholar]
- 4.Christman JW, Sadikot RT, Blackwell TS: The role of nuclear factor-kappa B in pulmonary diseases. Chest 2000, 117:1482-1487 [DOI] [PubMed] [Google Scholar]
- 5.Polito AJ, Proud D: Epithelial cells as regulators of airway inflammation. J Allergy Clin Immunol 1998, 102:714-718 [DOI] [PubMed] [Google Scholar]
- 6.Janssen-Heininger YM, Poynter ME, Baeuerle PA: Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic Biol Med 2000, 28:1317-1327 [DOI] [PubMed] [Google Scholar]
- 7.Pahl HL: Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18:6853-6866 [DOI] [PubMed] [Google Scholar]
- 8.Ghosh S, May MJ, Kopp EB: NF-κB and rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998, 16:225-260 [DOI] [PubMed] [Google Scholar]
- 9.Hein H, Schluter C, Kulke R, Christophers E, Schroder JM, Bartels J: Genomic organization, sequence, and transcriptional regulation of the human eotaxin gene. Biochem Biophys Res Commun 1997, 237:537-542 [DOI] [PubMed] [Google Scholar]
- 10.Sanceau J, Kaisho T, Hirano T, Wietzerbin J: Triggering of the human interleukin-6 gene by interferon-gamma and tumor necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J Biol Chem 1995, 270:27920-27931 [DOI] [PubMed] [Google Scholar]
- 11.Harant H, de Martin R, Andrew PJ, Foglar E, Dittrich C, Lindley IJ: Synergistic activation of interleukin-8 gene transcription by all-trans-retinoic acid and tumor necrosis factor-alpha involves the transcription factor NF-kappaB. J Biol Chem 1996, 271:26954-26961 [DOI] [PubMed] [Google Scholar]
- 12.Driscoll KE, Hassenbein DG, Howard BW, Isfort RJ, Cody D, Tindal MH, Suchanek M, Carter JM: Cloning, expression, and functional characterization of rat MIP-2: a neutrophil chemoattractant and epithelial cell mitogen. J Leukoc Biol 1995, 58:359-364 [DOI] [PubMed] [Google Scholar]
- 13.Dunn SM, Coles LS, Lang RK, Gerondakis S, Vadas MA, Shannon MF: Requirement for nuclear factor (NF)-kappa B p65 and NF-interleukin-6 binding elements in the tumor necrosis factor response region of the granulocyte colony-stimulating factor promoter. Blood 1994, 83:2469-2479 [PubMed] [Google Scholar]
- 14.Kleinert H, Euchenhofer C, Ihrig-Biedert I, Forstermann U: In murine 3T3 fibroblasts, different second messenger pathways resulting in the induction of NO synthase II (iNOS) converge in the activation of transcription factor NF-kappaB. J Biol Chem 1996, 271:6039-6044 [DOI] [PubMed] [Google Scholar]
- 15.Roebuck KA, Rahman A, Lakshminarayanan V, Janakidevi K, Malik AB: H2O2 and tumor necrosis factor-alpha activate intercellular adhesion molecule 1 (ICAM-1) gene transcription through distinct cis-regulatory elements within the ICAM-1 promoter. J Biol Chem 1995, 270:18966-18974 [DOI] [PubMed] [Google Scholar]
- 16.Schmedtje JF, Jr, Ji YS, Liu WL, DuBois RN, Runge MS: Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem 1997, 272:601-608 [DOI] [PubMed] [Google Scholar]
- 17.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A: A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol 2001, 2:45-50 [DOI] [PubMed] [Google Scholar]
- 18.Donovan CE, Mark DA, He HZ, Liou HC, Kobzik L, Wang Y, De Sanctis GT, Perkins DL, Finn PW: NF-kappa B/Rel transcription factors: c-Rel promotes airway hyperresponsiveness and allergic pulmonary inflammation. J Immunol 1999, 163:6827-6833 [PubMed] [Google Scholar]
- 19.Yang BL, Cohn L, Zhang D-H, Homer R, Ray A, Ray P: Essential role of nuclear factor κB in the induction of eosinophilia in allergic airway inflammation. J Exp Med 1998, 188:1739-1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voll RE, Jimi E, Phillips RJ, Barber DF, Rincon M, Hayday AC, Flavell RA, Ghosh S: NF-kappa B activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity 2000, 13:677-689 [DOI] [PubMed] [Google Scholar]
- 21.Cieslewicz G, Tomkinson A, Adler A, Duez C, Schwarze J, Takeda K, Larson KA, Lee JJ, Irvin CG, Gelfand EW: The late, but not early, asthmatic response is dependent on IL-5 and correlates with eosinophil infiltration. J Clin Invest 1999, 104:301-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, Gelfand EW: Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med 1997, 186:449-454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irvin CG, Tu Y-P, Sheller JR, Funk CD: 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Physiol 1997, 272:L1053-L1058 [DOI] [PubMed] [Google Scholar]
- 24.Korn SH, Wouters EFM, Vos N, Janssen-Heininger YMW: Cytokine-induced activation of nuclear factor-κB is inhibited by hydrogen peroxide through oxidative inactivation of IκB kinase. J Biol Chem 2001, 276:35693-35700 [DOI] [PubMed] [Google Scholar]
- 25.Chomczyski P, Sacchi N: Single-step method of RNA isolation by guanidium thiocynate-phenol-chloroform extraction. Anal Biochem 1987, 72:156-159 [DOI] [PubMed] [Google Scholar]
- 26.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW: Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 1997, 156:766-775 [DOI] [PubMed] [Google Scholar]
- 27.Janssen YMW, Driscoll KE, Howard B, Quinlan TR, Treadwell M, Barchowsky A, Mossman BT: Asbestos causes translocation of p65 protein and increases NF-κB DNA binding activity in rat lung epithelial and pleural mesothelial cells. Am J Pathol 1997, 151:389-401 [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JI, Burckart GJ: Nuclear factor kappa B: important transcription factor and therapeutic target. J Clin Pharmacol 1998, 38:981-993 [DOI] [PubMed] [Google Scholar]
- 29.Hamilton LM, Davies DE, Wilson SJ, Kimber I, Dearman RJ, Holgate ST: The bronchial epithelium in asthma: much more than a passive barrier. Monaldi Arch Chest Dis 2001, 56:48-54 [PubMed] [Google Scholar]
- 30.Bureau F, Bonizzi G, Kirschvink N, Delhalle S, Desmecht D, Merville MP, Bours V, Lekeux P: Correlation between nuclear factor-kappaB activity in bronchial brushing samples and lung dysfunction in an animal model of asthma. Am J Respir Crit Care Med 2000, 161:1314-1321 [DOI] [PubMed] [Google Scholar]
- 31.Bureau F, Delhalle S, Bonizzi G, Fievez L, Dogne S, Kirschvink N, Vanderplasschen A, Merville MP, Bours V, Lekeux P: Mechanisms of persistent NF-kappa B activity in the bronchi of an animal model of asthma. J Immunol 2000, 165:5822-5830 [DOI] [PubMed] [Google Scholar]
- 32.Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF: Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med 1998, 158:1585-1592 [DOI] [PubMed] [Google Scholar]
- 33.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI: The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 2000, 97:9052-9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampson AP: The role of eosinophils and neutrophils in inflammation. Clin Exp Allergy 2000, 30(Suppl 1):22-27 [DOI] [PubMed] [Google Scholar]
- 35.Jedrzkiewicz S, Nakamura H, Silverman ES, Luster AD, Mansharamani N, In KH, Tamura G, Lilly CM: IL-1beta induces eotaxin gene transcription in A549 airway epithelial cells through NF-kappaB. Am J Physiol 2000, 279:L1058-L1065 [DOI] [PubMed] [Google Scholar]
- 36.Lilly CM, Nakamura H, Kesselman H, Nagler-Anderson C, Asano K, Garcia-Zepeda EA, Rothenberg ME, Drazen JM, Luster AD: Expression of eotaxin by human lung epithelial cells: induction by cytokines and inhibition by glucocorticoids. J Clin Invest 1997, 99:1767-1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Zepeda EA, Rothenberg ME, Weremowicz S, Sarafi MN, Morton CC, Luster AD: Genomic organization, complete sequence, and chromosomal location of the gene for human eotaxin (SCYA11), an eosinophil-specific CC chemokine. Genomics 1997, 41:471-476 [DOI] [PubMed] [Google Scholar]
- 38.Matsukura S, Stellato C, Georas SN, Casolaro V, Plitt JR, Miura K, Kurosawa S, Schindler U, Schleimer RP: Interleukin-13 up-regulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am J Respir Cell Mol Biol 2001, 24:755-761 [DOI] [PubMed] [Google Scholar]
- 39.Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, Casolaro V, Schleimer RP: Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol 1999, 163:6876-6883 [PubMed] [Google Scholar]
- 40.Widmer U, Manogue KR, Cerami A, Sherry B: Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J Immunol 1993, 150:4996-5012 [PubMed] [Google Scholar]
- 41.Driscoll KE: Macrophage inflammatory proteins: biology and role in pulmonary inflammation. Exp Lung Res 1994, 20:473-490 [DOI] [PubMed] [Google Scholar]
- 42.Stacey MA, Sun G, Vassalli G, Marini M, Bellini A, Mattoli S: The allergen Der p1 induces NF-kappaB activation through interference with IkappaB alpha function in asthmatic bronchial epithelial cells. Biochem Biophys Res Commun 1997, 236:522-526 [DOI] [PubMed] [Google Scholar]