

Abstract
Trophoblastic tumors represent a unique group of human neoplasms because they are derived from fetal tissue. Except for choriocarcinoma, the neoplasms that develop from human trophoblast are poorly characterized. Placental site trophoblastic tumors and epithelioid trophoblastic tumors are thought to arise from intermediate (extravillous) trophoblasts based on histopathological studies, but direct molecular evidence of a trophoblastic origin has not been established. In this study, we performed molecular analysis in an attempt to confirm their presumable trophoblastic origin. We demonstrated that such tumors contain a Y-chromosomal locus and/or new (paternal) alleles not present in adjacent normal uterine tissue in all 31 informative cases. Loss of heterozygosity was found in 60% of tumors and all 42 tumors assessed contained wild-type K-ras. All of the trophoblastic tumors were heterozygous in at least 1 of 10 single-nucleotide polymorphism markers studied in contrast to homozygosity in all 10 single-nucleotide polymorphism markers in most complete hydatidiform moles indicating that these tumors are not related to complete hydatidiform moles. This study provides the first molecular evidence that placental site trophoblastic tumors and epithelioid trophoblastic tumors are of fetal (trophoblastic) origin.
Gestational trophoblastic tumors are a unique group of neoplasms because they are semiallografts that are derived from the conceptus and not from the patient. 1 This is of biological and clinical interest because of the fact that the presence of paternal genetic material distinguishes gestational from nongestational tumors, which may require different therapy. Choriocarcinoma, the most extensively studied trophoblastic tumor, is always derived from a proceeding gestational event, most often a complete hydatidiform mole. 2-4 In contrast, the origins of two other types of trophoblastic tumors, placental site trophoblastic tumor (PSTT) and epithelioid trophoblastic tumor (ETT) have not been established. 2,5 Unlike choriocarcinoma in which a recent gestational event can be clearly documented, the clinical evidence to support a gestational trophoblastic origin of PSTTs and ETTs is usually lacking because the preceding gestational event can be remote. 2,6 The trophoblastic origin of both of these tumors has been proposed based on morphological studies that have demonstrated similarity of the tumor cells in PSTTs and ETTs to the intermediate (extravillous) trophoblastic cells in the normal implantation site and the chorion laeve, respectively. 2,5,7,8 In addition, a recent study has shown that both tumors express a high level of HLA-G, a trophoblast-associated marker. 9
There have been only three molecular studies on PSTTs, and the number of specimens studied was very small. 10-12 There have been no molecular studies of ETTs, a relatively uncommon and only recently described neoplasm. To confirm the trophoblastic origin of PSTTs and ETTs, we analyzed the paternal genomic contribution including the presence of a Y-chromosomal locus (the SRY gene) and the presence of unique (paternal) alleles in a relatively large number of PSTTs and ETTs using a recently developed genotyping technique. 13 Mutational analysis of K-ras oncogenes was also assessed.
Materials and Methods
Tissues and Tumor DNA Samples
After approval by the Joint Committee for Clinical Investigation at Johns Hopkins University, formalin-fixed, paraffin-embedded tissue samples of 23 PSTTs, 19 ETTs, and 20 complete hydatidiform moles were retrieved from the Gestational Trophoblastic Tumor Bank of The Johns Hopkins Hospital, Baltimore, MD. Most of the specimens were consultation cases sent to one of the authors (RJK). Two gynecological pathologists reviewed all of the cases before tissue microdissection. Adjacent normal uterine tissue was present in 12 PSTTs and 13 ETTs. In addition, 10 ovarian serous carcinomas were used as the controls for the genotype analysis. Tumor and the adjacent normal uterine tissues were separately dissected using an inverted microscope with the contamination from nonneoplastic cells estimated at less than 10% of the microdissected tumor component. DNA was purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA) following the manufacturer’s instructions.
Polymerase Chain Reaction (PCR) for Genes on Y and X Chromosomes
Identification of the SRY (human sex-determining region Y) gene on the Y-chromosome was used to confirm the Y genetic component 14 and an X-linked protein gene on the X-chromosome was used to confirm the X-chromosomal element. Genomic DNA was added to the PCR cocktail containing pairs of primers that specifically amplified the SRY gene or the X-linked protein gene. For the SRY gene, the sequence for the forward primer was 5′-aagatgctgccgaagaattg-3′ and the reverse primer it was 5′-tcttgagtgtgtggctttcg-3′. For the X-linked protein gene, the sequence for the forward primer was 5′-agaatcctttgcacacgg-3′ and for the reverse primer it was 5′-cacaaaaggaggccacgt-3′. The PCR was performed using the following protocol: 95°C (2 minutes); 50 cycles of 95°C (30 seconds), 53°C (30 seconds), and 72°C (15 seconds); and 72°C (10 minutes). The amplified PCR products with ∼150 bp were visualized by electroseparation on 10% TBE (Tris base, boric acid, ethylenediaminetetraacetic acid) gels (Invitrogen, Carlsbad, CA).
Genotyping Using Single Nucleotide Polymorphism (SNP) PCR Assay
The principles and applications of molecular beacons in allelic determination have been previously reported in detail. 15,16 The genotyping method used in this study was detailed in a previous report. 13 In brief, the SNP markers were randomly selected with a heterozygosity rate greater than 0.38 based on the SNP database (http://lpg.nic.nih.gov). The sequences of the primers and molecular beacons for 10 SNPs including 8118 (at chromosome 1p), 9067 (1p), 1756 (5q), 1745 (8p), 28254 (8p), 1085 (8p), 3833 (8p), 852 (12p), p53 (17p), and 1468 (18q) have been previously reported. 16 Both forward and reverse primers were designed for each SNP, allowing the amplification of ∼100-bp PCR products. The paraffin DNA sample (0.5 to ∼1.5 ng) was distributed into six wells in a 384-well plate allowing at least 50 samples to be included in one plate and analyzed at the same time. In addition to all essential PCR reagents, the PCR cocktail contained a pair of molecular beacons labeled with either fluorescein (green fluorescence) or HEX (red fluorescence) that hybridized with the allele harboring the specific SNP (Gene Link, Thornwood, NY, and Operon Technologies, Inc., CA). 17,18 An excess of the reverse primer allowed generation of single-stranded DNA complementary to the molecular beacon. PCR was performed in a single step with the following protocol: 94°C (1 minute); four cycles of 94°C (15 seconds), 64°C (15 seconds), 70°C (15 seconds); four cycles of 94°C (15 seconds), 61°C (15 seconds), 70°C (15 seconds); four cycles of 94°C (15 seconds), 58°C (15 seconds), 70°C (15 seconds); 60 cycles of 94°C for (15 seconds), 55°C (15 seconds), 70°C (15 seconds); 94°C (1 minute); and 60°C (5 minutes). The fluorescence intensity in each well was then measured using a Galaxy FLUOstar fluorometer (BMG Lab Technologies, Durham, NC) and the ratio of fluorescein/HEX fluorescence intensity was determined from each well and the average from six repeats on each sample was determined. The data were converted into genotypes by a computer program. A novel allele in a tumor was defined as the presence of a new allele in the tumor that was absent in the corresponding normal uterine tissue for a given SNP marker. Accordingly, a novel allele (C for example) could be found in a tumor containing a heterozygous allele (GC for example) as compared to the homozygous alleles (GG) in adjacent normal tissues or in a tumor with homozygous alleles (CC) that were different from the ones (TT) in normal controls.
To determine the confidence level of bipaternal contribution in PSTTs, ETTs and complete moles, the homozygosity rate (fhomo) for each SNP was determined by genotyping normal tissues from 50 individuals. The confidence level was estimated from the cumulative homozygosity frequency (1 − fhomo) from the 10 SNP markers used in this study.
Mutational Analysis of K-ras
K-ras mutations at codon 12 and 13 were analyzed using DNA sequencing of the PCR products amplified from tumor DNA. The DNA was isolated from paraffin sections using the QIAquick PCR purification kit. The sequences of PCR primers and PCR conditions have been previously described. 19 Both forward and reverse primers were used for sequencing and they were forward: 5′-cattgtttttattataaggcctgc-3′ and reverse: 5′-tctgaattagctgtatcgtcaagg-3′. Sequencing was performed using fluorescently labeled Applied Biosystems Big Dye terminators and an Applied Biosystems 377 automated sequencer (Applied Biosystems, Foster City, CA). As a positive control, a low-grade ovarian serous carcinoma that has been known to contain K-ras mutation (GGT to GCT) at codon 12 was included in the assays.
Results
The SRY gene was used to confirm the presence of a Y-chromosomal component. 14 Based on PCR analysis, the SRY gene amplicon was found in 12 of 23 (52%) PSTTs (Figure 1) ▶ and in 11 of 19 (58%) ETTs (Figure 2) ▶ . The SRY gene was confirmed by nucleotide sequencing in representative PCR products (data not shown). As negative controls, all normal uterine tissues adjacent to the tumors were analyzed in parallel and none yielded the SRY amplicons. The absence of the SRY gene in PCR-negative PSTT and ETT specimens was supported further by repeating the PCR assays using higher template concentrations (3× of originals). The results in PCR-negative PSTTs and ETTs were not because of the technical problems or too low copy number of templates in PCR reactions because PCR products were detectable using a pair of X-chromosome-specific primers.
Figure 1.

Genotype analysis and PCR for Y- and X-chromosomes in PSTTs. White- and light-gray-shaded boxes: novel alleles present in the tumor; dark gray boxes: loss of maternal alleles in the tumor. P, PSTT; N, normal uterine tissue; NI, noninformative; NP, not present; X, presence of X-chromosome-specific PCR product; Y, presence of Y-chromosome-specific PCR product; Ch, chromosome.
Figure 2.
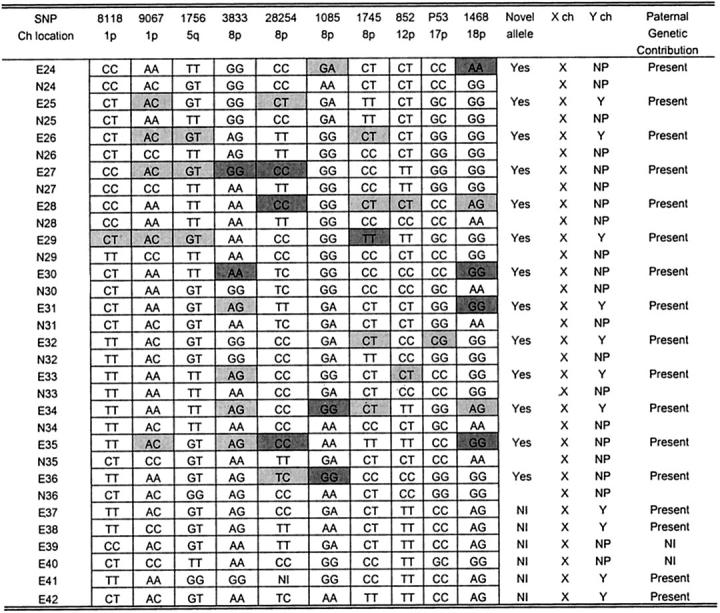
Genotype analysis and PCR for Y- and X-chromosomes in ETTs. White- and light-gray-shaded boxes: novel alleles present in the tumor; dark gray boxes: loss of maternal alleles in the tumor. E, ETT; N, normal uterine tissue; NI, noninformative; NP, not present; X, presence of X-chromosome-specific PCR product; Y, presence of Y-chromosome-specific PCR product; Ch, chromosome.
To further determine the presence of a paternal genetic contribution in PSTTs and ETTs, we performed genotyping using a newly developed technique that overcomes the technical difficulties associated with traditional genotyping methods using microsatellite markers. 13 The genotype results for PSTTs and ETTs are summarized in Figures 1 and 2 ▶ ▶ , respectively. There were 25 specimens (12 PSTTs and 13 ETTs) that contained adjacent normal uterine tissues. This allowed comparison of allele profiles between tumor and normal tissue. Among these informative cases, all PSTTs and ETTs contained at least one novel allele present only in tumor DNA and not in the normal uterine (maternal) tissue controls. Among 12 PSTTs and 13 ETTs with adjacent normal tissues, six PSTTs and nine ETTs showed loss of maternal alleles in at least one SNP marker as evidenced by different homozygous alleles in tumor and the corresponding normal maternal tissue. For example, the SNP 852 genotype in PSTT specimen P1 (Figure 1) ▶ was homozygous T in contrast to homozygous C in the adjacent normal (maternal) tissue N1; therefore the maternal C allele must have been lost during the development of the trophoblastic tumor. There were five PSTTs and two ETTS in which the normal tissues were not available for comparison, and these tumors were negative for the SRY-PCR assay. Therefore, these tumors were not informative to assess paternal genomic contribution. For the genotyping analysis, ovarian carcinomas were included as controls using the same panel of SNP markers. Unlike PSTTs and ETTs, ovarian carcinomas did not contain novel alleles; instead there was frequent loss of one of the alleles, ie, loss of heterozygosity in several SNP markers (data not shown).
Because ∼50% of choriocarcinomas are related to complete hydatidiform moles, 3,4 most commonly the homozygous ones, we addressed whether PSTTs and ETTs were genetically related to a complete hydatidiform mole. Here we assessed the allelic representation in PSTTs and ETTs and compared it to a complete hydatidiform mole. Homozygosity is a common feature in most complete hydatidiform moles because of duplication of one sperm in an empty ovum. 20 As shown in Figures 1 and 2 ▶ ▶ , none of the 22 PSTTs and 19 ETTs was homozygous in all 10 SNP markers. In contrast, 17 of 20 (85%) complete hydatidiform moles were homozygous in all markers (Figure 3) ▶ . Based on the SNP panel used in this study, the confidence level of bipaternal contribution in the homozygous moles was 99.9% (ie, the probability that homozygosity in all 10 SNPs occurs by chance is 0.1%) (Figure 3) ▶ .
Figure 3.
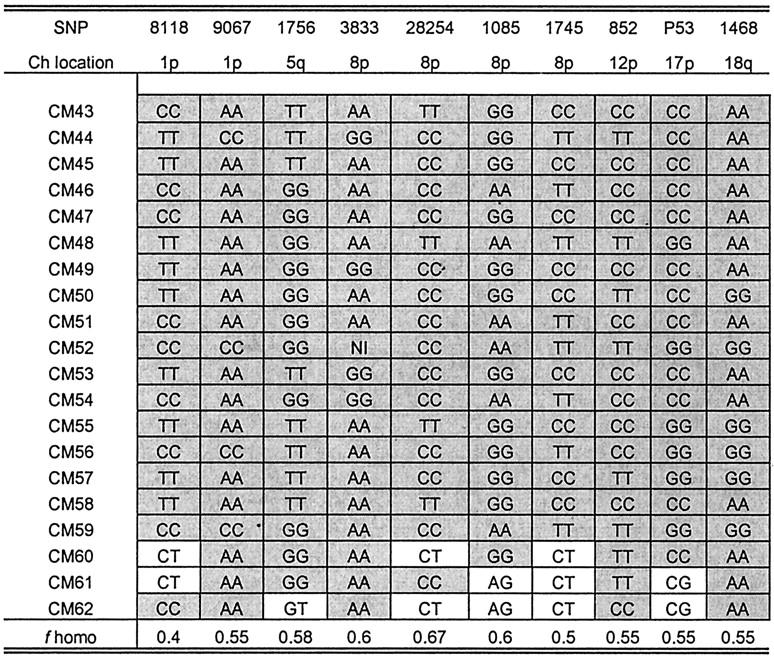
Genotype analysis in complete moles. Shaded boxes: homozygous to a specific SNP marker. f homo, homozygosity rate for a specific SNP marker; CM, complete mole; Ch, chromosome.
Because mutations in K-ras oncogene are commonly associated with the development of a variety of human cancers, 21,22 we attempted to assess the mutation status of K-ras in PSTTs and ETTs. Mutational analysis of K-ras oncogene was assessed in all 42 tumors and none of them showed mutation in either codon 12 or 13. In contrast, a GGT to GCT mutation at codon 12 was found in a low-grade ovarian serous carcinoma, which was included as the positive control in this study.
Discussion
The results of this study confirm the presence of paternal genetic contribution including the presence of Y-chromosomal material and novel (paternal) alleles in PSTTs and ETTs. These findings, together with our previous observation demonstrating HLA-G immunoreactivity in both of these tumors, 9 provide the first molecular evidence of their trophoblastic origin.
In this study, the paternal allelic status of the PSTTs and ETTs was not known because they were diagnosed long after the last known pregnancy and thus the paternal genetic material was not readily available for analysis. Despite this, all of the PSTTs and ETTs examined demonstrated the presence of a Y-chromosomal component (the SRY gene) and/or novel (presumably paternal) alleles in tumors that were not present in adjacent uterine (maternal) tissues. These findings provide clear evidence of the fetal origin of both types of tumors. Sex chromosome analysis in a previous report suggested that the development of PSTTs involves the paternal X-chromosome because no evidence of Y-chromosomal component was identified in five PSTTs, 10 but in our larger series of specimens, we were able to detect Y-chromosomal material in ∼50% of PSTTs and ETTs.
The trophoblastic origin of PSTTs and ETTs is further supported by our previous immunohistochemical study demonstrating strong expression of HLA-G in all cases of PSTTs and ETTs examined, using the 4H84 HLA-G-specific monoclonal antibody. 9,23-25 HLA-G is a nonclassical major histocompatibility class I molecule and plays a role in the escape of host immunosurveillance. It is not expressed in normal adult tissue, only in fetal thymus and normal intermediate trophoblast. Thus, HLA-G expression in PSTTs and ETTs strongly suggests that PSTTs and ETTs are related to intermediate trophoblasts.
Although PSTTs and ETTs both exhibit an intermediate trophoblast phenotype, they have distinctive histological features and gene expression profiles that justify their separate designation. The tumor cells in PSTTs resemble the intermediate trophoblastic cells in the implantation site and express markers specific for these trophoblastic cells. 1,26 In contrast, both histological and immunohistochemical features of an ETT are similar to those of chorionic-type intermediate trophoblastic cells found in the chorion laeve. Thus, PSTTs and ETTs seem to be derived from distinct subpopulations of intermediate trophoblast.
Approximately 50% of choriocarcinomas develop from complete hydatidiform moles, but the relationship of PSTTs and ETTs to complete moles is not clear. 3 In this study, both PSTTs and ETTs demonstrated allelic types heterozygous to at least one SNP marker, confirming that these tumors, unlike choriocarcinoma, 3,4 are not likely related to a complete hydatidiform mole, although a relationship to a heterozygous complete mole cannot be excluded. This finding is consistent with previous clinical observations that both PSTTs and ETTs occur most commonly after a normal pregnancy or nonmolar abortion, whereas in only 5 to 8% of patients is there a history of a complete mole. 7,27,28
We attempted to assess the mutation status of K-ras in PSTTs and ETTs, because mutations in the K-ras oncogene are commonly associated with the development of a variety of human cancers. 21,22 As with choriocarcinomas and complete moles, 29 PSTTs and ETTs contained wild-type K-ras at codons 12 and 13 in all of the cases evaluated, suggesting that the aberration of the K-ras signaling pathway does not play a major role in the development of trophoblastic tumors, although K-ras mutations at codon 61, another mutation hot spot of K-ras, were not analyzed in this report. In this study, we did not attempt to comprehensively assess loss of heterozygosity in PSTTs and ETTs because the corresponding normal fetal tissues from which trophoblastic tumors derived were not available for comparison. Based on the genotype analysis between the tumors and adjacent normal (maternal) uterine tissues, we were able to evaluate loss of heterozygosity by determining whether there was loss of the maternal alleles but not paternal alleles. Thus, the loss of heterozygosity rate in the PSTTs and ETTs was underestimated. Nevertheless, the frequent loss of heterozygosity in PSTTs and ETTs indicates that there is a certain level of genetic instability in some of the tumors.
In recent years the routine use of ultrasound in pregnancy has led to a much earlier clinical diagnosis and evacuation of complete moles, often in the first trimester. As a result the classic histopathological features of complete moles, which in the past were based on examination of specimens obtained in the second trimester, are not as apparent, making the pathological diagnosis more difficult. 2 The genotyping method reported here may provide another molecular diagnostic tool for identification of early complete moles. It has at least two advantages as compared to the classic techniques using microsatellite markers and gel-based assays. First, because molecular beacons are used to hybridize the PCR products with identical length (∼100 bp) for both alleles, DNA degradation of the larger microsatellite alleles in paraffin tissues does not pose a problem. 30 Second, our method is based on paraffin sections and does not require fresh tissues or special instruments for analysis.
In conclusion, this study has provided the first molecular evidence of the fetal (trophoblastic) origin of PSTTs and ETTs. PSTTs and ETTs are uncommon tumors, but because they represent semiallografts, being derived from the conceptus and not from the patients, they provide a unique tumor system to study the immunological aspects of human cancer.
Acknowledgments
We thank Ms. S. Cho and Dr. Tian-Li Wang for excellent technical support and Dr. B. Ronnett and Dr. G. Singer from the Division of Gynecological Pathology at the Johns Hopkins Medical Institution for critical reviews.
Footnotes
Address reprint requests to Ie-Ming Shih, M.D., Ph.D., Department of Pathology, Johns Hopkins Medical Institution, 418 N. Bond St., B-315, Baltimore, MD 21231. E-mail: ishih@jhmi.edu.
Supported by the Richard TeLinde Research Fund from the Department of Gynecology and Obstetrics, and the 2002 Provost Award, The Johns Hopkins University.
References
- 1.Shih IM, Kurman RJ: Molecular basis of gestational trophoblastic diseases. Curr Mol Med 2002, 2:1-12 [DOI] [PubMed] [Google Scholar]
- 2.Shih IM, Mazur MT, Kurman RJ: Blaustein’s Pathology of the Female Genital Tract. Kurman RJ eds. Chapter 22. Gestational Trophoblastic Disease and Related Lesions. 2002:pp 1193-1247 Springer-Verlag, New York
- 3.Hertig AT: Atlas of Tumor Pathology, section 9, fascicle 33. 1956. Armed Forces Institute of Pathology, Washington DC
- 4.Lurain JR, Brewer JI, Torok EE, Halpern B: Gestational trophoblastic disease: treatment results at the Brewer Trophoblastic Disease Center. Obstet Gynecol 1982, 60:354-360 [PubMed] [Google Scholar]
- 5.Shih IM, Kurman RJ: The pathology of intermediate trophoblastic tumors and tumor-like lesions. Int J Gynecol Pathol 2001, 20:31-47 [DOI] [PubMed] [Google Scholar]
- 6.Coulson LE, Kong CS, Zaloudek C: Epithelioid trophoblastic tumor of the uterus in a postmenopausal woman: a case report and review of the literature. Am J Surg Pathol 2000, 24:1558-1562 [DOI] [PubMed] [Google Scholar]
- 7.Shih IM, Kurman RJ: Epithelioid trophoblastic tumor—a neoplasm distinct from choriocarcinoma and placental site trophoblastic tumor simulating carcinoma. Am J Surg Pathol 1998, 22:1393-1403 [DOI] [PubMed] [Google Scholar]
- 8.Shih IM, Seidman JD, Kurman RJ: Placental site nodule and characterization of distinctive types of intermediate trophoblast. Hum Pathol 1999, 30:687-694 [DOI] [PubMed] [Google Scholar]
- 9.Singer G, Kurman RJ, McMaster M, Shih IM: HLA-G immunoreactivity is specific for intermediate trophoblast in gestational trophoblastic disease and can serve as a useful marker in differential diagnosis. Am J Surg Pathol 2002, 26:914-920 [DOI] [PubMed] [Google Scholar]
- 10.Hui P, Parkash V, Perkins AS, Carcangiu ML: Pathogenesis of placental site trophoblastic tumor may require the presence of a paternally derived X chromosome. Lab Invest 2000, 80:965-972 [DOI] [PubMed] [Google Scholar]
- 11.Fisher RA, Khatoon R, Paradinas FJ, Roberts AP, Newlands ES: Repetitive complete hydatidiform mole can be biparental in origin and either male or female. Hum Reprod 2000, 15:594-598 [DOI] [PubMed] [Google Scholar]
- 12.Arima T, Imamura T, Sakuragi N, Higashi M, Kamura T, Fujimoto S, Nakano H, Wake N: Malignant trophoblastic neoplasms with different modes of origin. Cancer Genet Cytogenet 1995, 85:5-15 [DOI] [PubMed] [Google Scholar]
- 13.Chang HW, Yen CY, Liu SY, Singer G, Shih IM: Genotype analysis using human hair shaft. Cancer Epidemiol Biomarkers Prev (in press) [PubMed]
- 14.Su H, Lau YF: Identification of the transcriptional unit, structural organization, and promoter sequence of the human sex-determining region Y (SRY) gene, using a reverse genetic approach. Am J Hum Genet 1993, 52:24-38 [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, Galizia G, Lieto E, Goodman SN, Romans KE, Kinzler KW, Vogelstein B, Choti MA, Montgomery EA: Counting alleles reveals a connection between chromosome 18q loss and vascular invasion. Nat Biotechnol 2001, 19:78-81 [DOI] [PubMed] [Google Scholar]
- 16.Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B: Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res 2001, 61:818-822 [PubMed] [Google Scholar]
- 17.Tyagi S, Bratu DP, Kramer FR: Multicolor molecular beacons for allele discrimination. Nat Biotechnol 1998, 16:49-53 [DOI] [PubMed] [Google Scholar]
- 18.Tyagi S, Kramer FR: Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 1996, 14:303-308 [DOI] [PubMed] [Google Scholar]
- 19.Vogelstein B, Kinzler KW: Digital PCR. Proc Natl Acad Sci USA 1999, 96:9236-9241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassilakos P, Ritton G, Kajii T: Hydatidiform mole: two entities. Am J Obstet Gynecol 1977, 127:167-170 [DOI] [PubMed] [Google Scholar]
- 21.Vogelstein B, Kinzler KW: The Genetic Basis of Human Cancer. 1998. McGraw-Hill Health Professions Division, New York
- 22.Vogelstein B, Kinzler KW: The multistep nature of cancer. Trends Genet 1993, 9:138-141 [DOI] [PubMed] [Google Scholar]
- 23.Blaschitz A, Hutter H, Leitner V, Pilz S, Wintersteiger R, Dohr G, Sedlmayr P: Reaction patterns of monoclonal antibodies to HLA-G in human tissues and on cell lines: a comparative study. Hum Immunol 2000, 61:1074-1085 [DOI] [PubMed] [Google Scholar]
- 24.Goldman-Wohl D, Ariel I, Greenfield C, Hochner-Celnikier D, Lavy Y: A study of human leukocyte antigen G expression in hydatidiform moles. Am J Obstet Gynecol 2001, 185:476-480 [DOI] [PubMed] [Google Scholar]
- 25.Rabreau M, Rouas-Freiss N, Landi M, Le Danff C, Carosella ED: HLA-G expression in trophoblast cells is independent of embryonic development. Hum Immunol 2000, 61:1108-1112 [DOI] [PubMed] [Google Scholar]
- 26.Shih IM, Kurman RJ: The pathology of intermediate trophoblastic tumors and tumor-like lesions. Int J Gynecol Pathol 2001, 20:31-47 [DOI] [PubMed] [Google Scholar]
- 27.Kurman RJ: The morphology, biology, and pathology of intermediate trophoblast: a look back to the present. Hum Pathol 1991, 22:847-855 [DOI] [PubMed] [Google Scholar]
- 28.Rutgers JL, Baergen RN, Young RH, Scully RE: Placental site trophoblastic tumor: clinicopathologic study of 64 cases. Mod Pathol 1995, 8:96A [Google Scholar]
- 29.Fulop V, Mok SC, Genest DR, Szigetvari I, Cseh I, Berkowitz RS: c-myc, c-erbB-2, c-fms and bcl-2 oncoproteins. Expression in normal placenta, partial and complete mole, and choriocarcinoma. J Reprod Med 1998, 43:101-110 [PubMed] [Google Scholar]
- 30.Liu J, Zabarovska VI, Braga E, Alimov A, Klein G, Zabarovsky ER: Loss of heterozygosity in tumor cells requires re-evaluation: the data are biased by the size-dependent differential sensitivity of allele detection. FEBS Lett 1999, 462:121-128 [DOI] [PubMed] [Google Scholar]