

Abstract
Types A and B Niemann-Pick disease (NPD) are lysosomal storage disorders resulting from the deficient activity of acid sphingomyelinase (ASM). In this manuscript we report the pathobiology of male gonadal tissue and sperm in a knockout mouse model of NPD and demonstrate the importance of ASM for normal sperm maturation and function. Characteristic lipid-filled vacuoles were evident in light micrographs of testis’ seminiferous tubules and epithelial cells lining the epididymis of −/− mice. Electron micrographs extended these findings and revealed storage vesicles within Sertoli cells of the seminiferous tubules. Mature spermatozoa from −/− mice showed marked ASM deficiency and elevated levels of sphingomyelin and cholesterol. Flow cytometric analysis revealed that affected spermatozoa had disrupted plasma and acrosome membranes, and mitochondrial membrane depolarization. They also did not undergo proper capacitation. Morphological abnormalities such as kinks and bends at the midpiece-principle piece junction were evident in spermatozoa from affected mice, with consequent deficits in motility. Notably, the mutant sperm regained normal morphology on incubation in mild detergent, demonstrating that the bending defects were a direct consequence of membrane lipid accumulation. A mechanism for these abnormalities is proposed that suggests lipid accumulation in the gonads results in regulatory volume decrease defects within the developing sperm, and that regulatory volume decrease defects, in turn, lead to the observed abnormalities in sperm morphology and function. These results provide in vivo evidence that ASM activity plays a critical role in sperm maturation and function, and a basis for similar studies in sexually mature, male NPD patients.
Types A and B Niemann-Pick disease (NPD) are lipid storage disorders caused by mutations in the gene encoding the lysosomal hydrolase, acid sphingomyelinase (ASM). 1 Type A NPD is a severe, neurodegenerative form of the disorder that generally leads to death by ∼3 years of age. In contrast, patients with type B NPD have little or no neurological involvement and may survive into adolescence or adulthood. ASM belongs to a family of sphingomyelinases that catabolize sphingomyelin (SPM) to ceramide and phosphorylcholine. 2,3 It is presumed that the pathophysiology in NPD is primarily because of the accumulation of SPM and other metabolically related lipids (eg, cholesterol) within the cells and tissues of affected patients, although signaling through the ceramide pathway may also be disrupted. 1,2
The full-length human and murine cDNAs and genomic sequences encoding ASM have been isolated, 4-6 and ASM knockout (ASMKO) mice have been constructed. 7,8 The ASMKO mice have no residual ASM activity and present with a clinical and pathological phenotype that is intermediate between types A and B NPD. These models have been extremely valuable for the investigation of the pathophysiological mechanisms underlying types A and B NPD, as well as for the development and evaluation of various treatment approaches. 9-11
Since the construction of the mouse models in 1995, our laboratory has observed reduced fecundity in the affected animals that begins before the onset of behavioral deficits. Reproductive impairment also has been documented in mouse models of Tay-Sachs and Sandhoff diseases, 12-14 related sphingolipid storage disorders that result from deficient β-hexosaminidase activity and the accumulation of GM2 ganglioside. Of particular relevance to the current study, the reproductive pathology and residual enzyme expression in male Tay-Sachs and Sandhoff mice has been characterized, and an increase in the size and number of lysosomes in the epithelia lining the efferent and epididymal ducts was found. Testis weight, morphology, and sperm counts appeared to be unaffected.
The general importance of lysosomal hydrolases in male fertility has been well documented. Evidence exists to suggest that the sperm acrosome is a modified lysosome with active acidic hydrolases, including β-hexosaminidase, β-glucuronidase, α-mannosidase, and β-galactosidase. 15,16 Sperm also contain sphingomyelinases. For example, ram sperm contain a Mg2+-dependent neutral sphingomyelinase in the plasma membrane and an acidic sphingomyelinase in the sperm homogenate. 17,18 Because no acidic activity was found in the membrane fraction of sperm, it was presumed that ASM was a soluble component of the acrosome.
In the present study, the ASMKO mouse model was used to investigate the nature of reproductive pathogenesis in NPD, as well as to decipher the specific role of ASM in normal sperm development and function. Based on the findings presented within, a model is proposed in which SPM and cholesterol accumulation in male gonads of ASMKO mice causes abnormalities in osmolar regulation and subsequent deficits in sperm maturation and function. This likely explains the reduced fecundity of this animal model. These data also suggest that similar abnormalities may occur in sexually mature male NPD patients, and perhaps other disorders in which SPM and/or cholesterol metabolism are perturbed.
Materials and Methods
Maintenance of the Mouse Colony and Sperm Isolation
The ASMKO mouse colony was established from heterozygous (+/−) breeding pairs obtained by gene targeting of 129/SV embryonic stem cells, followed by microinjection into C57BL/6 blastocysts. 7 The animals were maintained on a mixed 129/SV-C57BL/6 genetic background. The biochemical, clinical, and pathological nature of the affected (−/−) animals has been well characterized. 7,8 A polymerase chain reaction genotyping method also has been established to maintain the breeding colony. 11 The colony was maintained in a barrier facility on a 12-hour light/dark cycle and given food and water ad libitum.
Sperm were obtained postmortem from anesthetized mice (Halothane; Halocarbon Laboratories, River Edge, NJ) that were sacrificed by cervical dislocation. Mature sperm were harvested from cauda epididymides (by mincing the tissue) and vas deferentia (by squeezing the duct). All mouse protocols were approved by The Mount Sinai Center for Comparative Medicine and Surgery.
General Pathology
Light Microscopy
Wild-type and −/− mice (2, 4, and 6 months of age) were sacrificed as described above and the gonads removed and placed in 10% formalin (Sigma, St. Louis, MO) for light microscopy. Before fixing, testes were sliced in half and epididymides sectioned into caput and cauda regions. After 48 hours in fixative, all tissues were dehydrated through a graded series of alcohol, cleared in chloroform, and embedded in Para-Plast embedding media (Fisher Scientific, Fair Lawn, NJ). The tissues were microtomed into 5-μm sections and dried at 60°C for 1 hour on a Superfrost/Plus slide (Fisher Scientific). The slides were deparaffinized in xylene, followed by hydration and staining in Harris modified hematoxylin and alcoholic eosin (Fisher Scientific). After an additional dehydration, the sections were mounted with Permount (Fisher Scientific) and a coverslip was added.
Electron Microscopy
For electron microscopy, 6 month old +/+ and −/− mice were anesthetized with Avertin (2.5% per gram body weight) and perfused-fixed through the heart with 0.2 mol/L of cacodylate buffer containing 3% glutaraldehyde (Sigma). After perfusion, the testes and epididymides were excised, cut into 1-mm3 pieces, and placed into fresh fixative and stored at 4°C until further processing. The tissue was subsequently washed, dehydrated in graded ethanol and propylene dioxide, and embedded in Embed (Ems, Fort Washington, PA) 812. One-μm plastic sections were cut, stained with methylene blue, and observed by light microscopy. Selected areas of the block were ultrathin sectioned, mounted on 300-mesh formvar-coated nickel grids, and stained with uranyl acetate and lead citrate (Ems). Sections were then analyzed with a JEM 100C X Transmission Electron Microscope (JEOL, Ltd, Tokyo, Japan).
Gonad Weights and Testicular Sperm Numbers
Testes were harvested from at least six +/+ and −/− mice for each genotype at 2, 4, and 6 months of age. To determine the number of sperm in the testes, each testis was homogenized using a hand-held homogenizer on maximum setting (Tissue Tearor; Biospec Products, Inc., Bartlesville, OK) for 10 seconds in 0.25 mol/L of Thimerosal (Sigma). The homogenates were stained with 0.1% Trypan Blue (1:10) and spermatozoon heads were counted using a Cell-Vue counting chamber (Fertility Technology Resources, Natick, MA). The absolute sum of sperm nuclei from each testis was obtained.
Sperm Morphology
Three +/+ and −/− mice each were sacrificed at 6 months of age. Sperm were harvested into Medium 199 (Specialty Media; CMT, Inc.) conditioned with 25 mmol/L NaHCO3, 4 mg/ml bovine serum albumin (BSA), 30 μg/ml sodium pyruvate, and 4 mmol/L EGTA, for 10 minutes at 37°C and 5% CO2. Cell-Vue counting chambers were used according to the manufacturer’s directions to determine morphology counts according to the following criteria: normal, bent (a kink at the annulus, usually 90°); hairpin (180° kink at the annulus); other (eg, microcephaly, loop bending within the midpiece, coiling of the principal piece, duplication of the tail, or bending at head-to-midpiece junction). At least two fields of 100 sperm each were counted for each mouse; decapitated sperm were not counted.
Sperm suspensions were also used to obtain Nomarski images by diluting the samples to 1 ml and rendering the sperm immobile by submersion in a 65°C water bath for 2 minutes. A small aliquot (3 to 5 μl) was spotted onto a Superfrost/Plus precleaned microscope slide (Fisher Scientific), a coverslip added, and images acquired at room temperature. A ×40 objective was used on an Axiophot 2 Nikon microscope for differential-interference-contrast microscopy.
Sperm Demembranation
At least two +/+ and −/− mice at 5 months of age were sacrificed and sperm harvested into CO2-equilibrated Medium 199 conditioned with 25 mmol/L NaHCO3, 4 mg/ml BSA, and 30 μg/ml sodium pyruvate for 10 minutes at 37°C and 5% CO2 before tissue removal. For each genotype, 1-ml aliquots were transferred to individual wells of a 12-well plate. The spermatozoa were then incubated for 40 minutes at 37°C and 5% CO2 in media containing 0.1% Triton X-100 (Sigma). A set of aliquots was also incubated without Triton as a control. The experiment was terminated when the wells were suspended in a 65°C water bath for 2 minutes to immobilize the sperm. Cell-Vue counting chambers were used according to the manufacturer’s directions to determine the morphology counts according to the following criteria: normal, bent (a kink at the annulus, usually 90°); hairpin (180° kink at the annulus). At least two fields of 100 sperm each were counted for each mouse; decapitated sperm were not counted.
Biochemical Assays
Sample Preparations
Mature sperm were collected into phosphate-buffered saline (PBS) (Mg2+- and Ca2+-free) and filtered through a 70-μm nylon mesh filter (Falcon; Becton-Dickinson, Franklin Lakes, NJ) to remove excess tissue and aggregated clumps. The filtrates were then spun at 850 × g for 3 minutes, the supernatants discarded, and the sperm pellets resuspended in 100 μl of 0.2% Triton X-100 and placed on ice for 15 minutes, followed by three freeze-thaw cycles (5 minutes each) using dry ice and a 37°C water bath. These homogenates were spun at 15,000 × g for 5 minutes to obtain a supernatant (ie, detergent-soluble fraction) and pellet (detergent-insoluble fraction). Protein concentrations in each fraction were determined using a commercial kit (BioRad Protein Assay; BioRad, Hercules, CA).
In Vitro SPM Assay
The SPM content of the detergent-solubilized sperm supernatants were determined using a recently developed, fluorescence-based method. 19 Briefly, 5 μl of the supernatants was added to individual wells of a 96-well microplate that contained an enzyme cocktail consisting of 112.5 mU of bacterial sphingomyelinase (Sigma), 400 mU of alkaline phosphatase (Sigma), 120 mU of choline oxidase (Sigma), 200 mU of horseradish peroxidase (Sigma), and 20 nmol of Amplex Red (Molecular Probes Inc., Eugene, OR) in 100 μl of reaction buffer (50 mmol/L Tris-HCl, 5 mmol/L MgCl2, pH 7.4). For each sample, a control well contained the same components without the bacterial sphingomyelinase. After a 20-minute incubation at 37°C, the fluorescence emission of the wells were read by a fluorescent microplate reader (Molecular Devices Corp., Sunnyvale, CA). The excitation and emission wavelengths were set at 560 and 587 nm, respectively. The fluorescence difference between the control and test samples was compared to an SPM standard curve for quantitation.
In Vitro Sphingomyelinase Assays
To determine ASM activity, 3 μl of an acidic enzyme assay buffer was mixed with 3 μl of the sperm supernatants and incubated for 1 hour at 37°C. The buffer contained 200 μmol/L Bodipy-C12-SPM (Molecular Probes), 0.1 mmol/L ZnCl2, 0.6% Triton X-100, 0.1 mol/L sodium acetate buffer, pH 5.0. To terminate the reaction, 114 μl of 100% EtOH was added, mixed, and the total mixture spun for 5 minutes at 15,000 × g. The supernatant was then transferred to a Waters glass sampling vial, and 5% of the original reaction mixture was autosampled by a WIPS 712 autosampler (Waters Corp., Milford, MA) onto a high performance liquid chromatography (HPLC). The HPLC was equipped with a reverse-phase column (Aquasil C-18; Keystone Scientific Inc., Bellefonte, PA) and eluted isocratically with methanol:BTB [99:1, BTB (basic Tris buffer), 0.5 mol/L Tris-HCl, pH 9.0], at a flow rate of 1.0 ml/min. Fluorescence was quantified using a Waters 474 fluorescence detector set to excitation and emission wavelengths of 505 and 540 nm, respectively. The peak corresponding to the product (ie, Bodipy-C12-ceramide) was identified by comparing its retention time with a standard, and the amount of product was calculated using a regression equation that was established from a standard curve using various amounts of Bodipy-C12-ceramide.
In Situ Sphingomyelinase Assays
Thin Layer Chromatography Assay
Mature sperm were collected into Medium 199 conditioned with 25 mmol/L NaHCO3, 4 mg/ml BSA, 30 μg/ml sodium pyruvate, and 4 mmol/L EGTA without phenol red. EGTA was used to prevent or reduce the acrosome reaction. After a 10-minute collection period, the sperm samples were filtered and 200 μl of each was pipetted into individual wells of a 12-well plate. The total volume of the wells was brought to 1 ml with a solution of lissamine-rhodamine-conjugated sphingomyelin (LR-SPM) (final concentration, 10 nmol/ml) in Medium 199. After a labeling (pulse) period of 45 minutes, the sperm were spun at 850 × g for 3 minutes, and the pellets were resuspended in LR-SPM-free media and allowed to incubate at 37°C. This chase period lasted for 2 hours during which the sphingomyelinases present in the living sperm could metabolize the LR-SPM into LR-ceramide.
To quantify the sphingomyelinase activity in living sperm after the chase period, equal numbers of sperm were transferred to microcentrifuge tubes and spun at 13,000 × g for 1 minute. The supernatants were removed and dried by vacuum centrifugation. Lipid extracts were prepared from both the dried supernatants and the pelleted sperm by organic extraction. Four hundred μl of chloroform:methanol (1:1) was added, the mixtures vortexed, and the lipids extracted by incubating at 55°C for 5 minutes. After centrifugation at 10,000 × g for 1 minute, the upper phase was transferred to a new Eppendorf tube. An equal volume of water was added and the samples vortexed again. After centrifugation (as above), the lower chloroform phase was transferred to a glass test tube and the chloroform was evaporated at 55°C under a stream of nitrogen. The remaining residue was then dissolved in a small amount of chloroform:methanol (1:1) and spotted in equal amounts onto a thin layer chromatography plate. The plate was run for 5 minutes in a solvent containing chloroform:methanol:0.2 N HCl (93:7:1). A standard amount of LR-ceramide (0.1 nmol/μl) also was spotted for comparative quantitation.
The plate was then run a second time to visualize the unmetabolized LR-SPM using a solvent of chloroform:methanol:0.2 N HCl (75:25:4). LR-SPM standard (0.1 nmol/μl) was used for quantification. An exposed image of the plate was made on a UV Illuminator and analyzed with Scion Image Software (Scion Corp., Frederick, MD). The intensity of the fluorescent spots in the sample lanes was compared to the standards to determine the amounts of LR-SPM and LR-ceramide produced during the pulse and chase periods.
Flow Cytometry Assay
This assay was similar to that of thin layer chromatography, except that Bodipy-conjugated SPM (Molecular Probes), final concentration 1 nmol/ml, was used. At the conclusion of the chase period, the cells were spun, filtered, and resuspended to 1 ml in warmed PBS. Bodipy (490 nm excitation and 525 nm emission; FL-1)-derived fluorescence was measured with a Becton Dickinson FacsCalibur Cytometer (Becton Dickinson, San Jose, CA). The fluorochrome was excited at 488 nm with an argon laser. Cells (n = 100,000) were acquired after setting a threshold to exclude debris and compensating for spectral overlap. The percentage of these cells and the mean fluorescence channel (N) on a 1024-channel scale were derived. The latter parameter was converted into a mean fluorescence intensity value on a 4-decade (100 to 103) log scale. Analysis of the acquired data were performed with WinMDI 2.8 software (Scripps Institute).
In Situ Cholesterol Assay
Sperm were isolated from at least seven +/+ and −/− mice at 6 months of age into M199 media conditioned with NaHCO3 (25 mmol/L), BSA (4 mg/ml), sodium pyruvate (30 μg/ml), and EGTA (4 mmol/L). After a 10-minute swim-out period at 37°C and 5% CO2, the sperm suspensions were filtered through a 70-μm nylon mesh filter to remove excess tissue and aggregated clumps. The filtrates (1 ml) were incubated for 30 minutes in individual wells of a 12-well plate at 37°C and 5% CO2. The sperm and media were then collected from each well and spun at 15,000 × g for 5 minutes. To quantify the amount of cholesterol, 50 μl of each supernatant was added to an individual well of a 96-well microplate that contained 50 μl of an enzyme cocktail (300 μmol/L Amplex Red reagent, 0.2 U/ml cholesterol esterase, 2 U/ml cholesterol oxidase, 2 U/ml of horseradish peroxidase) in a reaction buffer (125 mmol/L KPO4, 5 mmol/L cholic acid, 50 mmol/L NaCl, and 0.1% Triton X-100, pH 7.4). These reagents are available as an Amplex Red Cholesterol Assay Kit (Molecular Probes). A negative control contained the same enzyme cocktail without sample. The suspensions were mixed and incubated for 30 minutes at 37°C in the dark. The microtiter plate was read by a fluorescent microplate reader (Molecular Devices Corp.). The excitation and emission wavelengths were 560 and 587 nm, respectively. The fluorescence difference between the control and test samples was compared to a cholesterol standard curve for quantitation. Sperm numbers were determined using Cell-Vue counting chambers (Fertility Technology) according to the manufacturer’s directions.
Filipin-Sterol Complex Analysis by Freeze-Fracture Electron Microscopy
Sperm were isolated from two +/+ and −/− mice at 6 months of age as described above. An aliquot of sperm from each mouse was fixed for 1 hour in the dark at 4°C in 0.1% cacodylate buffer (pH 7.4) containing 3% glutaraldehyde and 0.02% filipin. (Filipin is a polyene antibiotic that binds to 3β-hydroxysterols; filipin complex was from Streptomyces filipinensis, Sigma.) A second aliquot from each mouse was incubated under identical conditions but without the addition of filipin. All sperm were then spun at 10,000 × g for 1 minute and the pellets bathed in 25% glycerol (in PBS) for 2 hours at room temperature. The pellets were mounted on gold supports, submerged in freon supercooled by liquid N2, and fractured using a double-replica device in a Balzers BAF-301 apparatus with a stage temperature of −110°C. The specimens were coated with platinum (3 to 5 nm, 45° angle) and carbon (25 nm, 90° angle). The surface replicas were cleaned with Clorox and washed three times in double-distilled water. Specimens were then observed using a JEM 100CXII (JEOL, Ltd., Tokyo, Japan) at 80 kV.
For quantification, Matlab software (Mathworks Inc., Natick, MA) was used to analyze three images from +/+ and −/− mice labeled with filipin, and a single image of a filipin-unlabeled control. For ease of computation, small images (128 × 128 pixels) were used. A two-dimensional Fourier transformation of the surface spatial properties was performed on each image to obtain the corresponding spectrum. To further simplify the computation without losing the spectrum characteristics, the two-dimensional spectrum was converted into a one-dimensional representation by averaging the components on the same frequency line in the 45° direction. In general, a spectrum in a high-frequency area is significantly affected by the image noise, and those in the low-frequency area are dominated by large waveforms that may not fully represent the image texture. Hence, the converted spectrum was truncated at high and low frequencies (range between 10 and 32 on graph) for more reliable results. Also, an image whose surface appears rough may have a more uniform or flat spectrum, whereas a smooth image may have a steeper spectrum (high in the low-frequency area and low in the high-frequency area). The steepness measured by the resulting slope of the spectrum can be used to represent the coarseness of the image’s appearance.
Physiological Measurements
Sperm Motility
Sperm motility and kinametric analysis was performed by Pathology Associates (Frederick, MD). Two mice of each genotype (6 months of age) were sacrificed and an epididymis removed and placed into PBS containing 0.1% BSA. After a 3-minute swim-out, a 100-μm deep cannula was dipped into the suspension and a sample obtained. A total of five fields were analyzed at 200 sperm/field at a cell depth of 10 μm. The analysis was performed using a Hamilton-Thorne IVOS system for 30 frames at 60 Hz. Based on these parameters, sperm motility was categorized as rapid (average velocity of the smoothed cell path >50.0 μm/s), medium (velocity between 7.4 μm/s and 50.0 μm/s), slow (velocity <7.4 μm/s), and static. Epididymal sperm counts were also calculated.
Plasma Membrane Integrity
Sperm from cauda epididymides and vas deferentia of +/+ and −/− mice at 6 months of age were isolated into warmed, CO2-equilibrated, M199 conditioned with NaHCO3 (25 mmol/L), BSA (4 mg/ml), sodium pyruvate (30 μg/ml), and EGTA (4 mmol/L). Sperm were allowed to swim out for 10 minutes at 37°C and 5% CO2. The M199 lacked phenol red. The suspensions were filtered with a 70-μm nylon mesh filter, diluted, and transferred to individual wells of a 12-well plate. The sperm were then incubated for 20 minutes in media containing SYBR-14 (20 nmol/L). After 10 minutes, propidium iodide (PI, 12 μmol/L) was added directly to the wells and the plate placed in a 37°C warm room on a shaker in the dark. SYBR-14 and PI are available commercially in a Live/Dead Sperm Viability Kit (Molecular Probes). The suspensions were then transferred to micro Eppendorf tubes and spun for 3 minutes at 800 × g. The media was aspirated and 1 ml of warmed PBS was used to resuspend the pellet. SYBR-14 (FL-1, 490 nm excitation and 525 nm emission)- and PI (FL-3, 520 nm excitation and 610 nm emission)-derived fluorescence was acquired on a Becton Dickinson FacsCalibur cytometer as described above.
Acrosome Status
Sperm were isolated and filtered as described above. After incubation for 1 hour to allow capacitation, the suspension was transferred to micro Eppendorf tubes and spun for 3 minutes at 800 × g. The media was aspirated and fresh media containing peanut agglutinin-fluorescein isothiocyanate (1 μg/ml, Sigma), but lacking BSA, was added to the sperm, and aliquots were transferred to individual wells of a 12-well plate for 30 minutes at 37°C and 5% CO2. Peanut agglutinin lectin binds specifically to β-galactose moieties on the outer acrosomal membrane. 20 After 20 minutes, PI (12 μmol/L) was added directly to the wells and the plate placed in a 37°C warm room on a shaker in the dark. As a control, duplicate samples were suspended in media containing the Ca2+ ionophore A23187 (10 μmol/L, Sigma) to induce the acrosome reaction. The suspensions was then transferred to micro Eppendorf tubes and spun for 3 minutes at 800 × g. The media was aspirated and 1 ml of warmed PBS was used to resuspend the pellet. Peanut agglutinin-fluorescein isothiocyanate (FL-1, 490 nm excitation and 525 nm emission)- and PI (FL-3, 520 nm excitation and 610 nm emission)-derived fluorescence was acquired on a Becton Dickinson FacsCalibur cytometer as described above.
Mitochondrial Membrane Potential
Sperm were isolated and suspended as indicated above, and incubated for 10 minutes in media containing JC-1 (10 μg/ml) at 37°C and 5% CO2. JC-1 is available commercially (Molecular Probes). The suspension was then transferred to micro Eppendorf tubes and spun for 3 minutes at 800 × g. The media was aspirated and 1 ml of warmed PBS was used to resuspend the pellet. JC-1 monomer (FL-1, 490 nm excitation and 525 nm emission)- and aggregate PI (FL-2, 535 nm excitation and 580 nm emission)-derived fluorescence was acquired on a Becton Dickinson FacsCalibur cytometer as described above.
Nitric Oxide (NO) Production
Sperm were isolated as indicated above and incubated in media M199 conditioned with NaHCO3 (25 mmol/L), BSA (4 mg/ml), sodium pyruvate (30 μg/ml), and Hepes (50 mmol/L), but lacking phenol red. The spermatozoa were then suspended for 30 minutes at 37°C in a 12-well plate on a three-dimensional rotator (low setting) in the dark. Suspension media contained NO-fluorogenic dye (5 μmol/L), which is commercially available in an ApoAlert Nitric Oxide/Annexin-V Dual Sensor Kit (Clontech, Palo Alto, CA). This dye binds to NO and increases nearly 100-fold in quantum yield for fluorescence detection. NO detects oxidative stress and DNA damage. At 20 minutes into the incubation, PI (12 μmol/L) was added. Spermatozoa were transferred to micro Eppendorf tubes and spun for 3 minutes at 800 × g. The media was aspirated and the spermatozoa suspended in 500 μl of Annexin V binding buffer (supplied in ApoAlert Kit). The suspension was filtered with a 40-μm nylon mesh filter and analyzed. NO-fluorgenic dye (FL-1) and PI (FL-3) fluorescence was acquired on a Becton Dickinson FacsCalibur Cytometer as described above.
Phosphatidylserine (PS) Translocation
Sperm were isolated as indicated above and incubated in media M199 conditioned with NaHCO3 (25 mmol/L), BSA (4 mg/ml), sodium pyruvate (30 μg/ml), and Hepes (50 mmol/L). The media was aspirated and the sperm resuspended at 100 μl for 15 minutes at room temperature on 360 degree rotator (low setting) in the dark. Suspension media contained Annexin V-phycoerythrin (5%) and YO-PRO (2 μmol/L). Annexin V-phycoerythrin detects PS translocation from the inner to outer leaflet of the plasma membrane and is commercially available in an ApoAlert Nitric Oxide/Annexin-V Dual Sensor Kit (Clontech); YO-PRO is a monomeric cyanine nucleic acid dye that cannot enter cells with intact plasma membranes and is therefore used in a similar manner to PI (Molecular Probes). After 15 minutes, 400 μl of Annexin V binding buffer (supplied in ApoAlert Kit) was added to the sperm suspension and the total volume was filtered with a 40-μm nylon mesh filter and analyzed. Annexin V-phycoerythrin (FL-2) and YO-PRO (FL-1) fluorescence was acquired on a Becton Dickinson FacsCalibur cytometer as described above.
Results
Gonad Morphology in ASMKO Male Mice
Evidence of lipid-laden foam cells within the reticuloendothelial system of NPD mice has been observed as part of the etiology of ASM deficiency. 1,10,11 Within male gonads, 6-month-old ASMKO mice exhibited large numbers of lipid vacuoles and multilamellar vesicles both internal to the seminiferous tubules in Sertoli cells and external in interstitia (Se and IS; Figure 1, A and B ▶ ). The accumulation seemed to disrupt the general morphology of the seminiferous tubule, but did not interfere with the production of spermatozoa because sperm were evident in lumena of the caudae epididymidis (Lu; Figure 1A ▶ ), and was of comparative number to that found in normal mice (Table 1) ▶ . There was no evidence of lipid accumulation within germ cells (G; Figure 1, A and B ▶ ), although accumulation was evident in principle, basal, and smooth muscle cells of epididymal epithelia (P, B, and Sm; Figure 1B ▶ ). In addition, many small endocytic vesicles were found proximal to the lumena (Lu, Figure 1B ▶ ). Accumulation also was evident in the intertubular space and stroma of the epididymis (IT and St, Figure 1A ▶ ). Although no lipid accumulation was found in the gonads of wild-type mice, mitochondria were observed in germ cells of the testis (G, Figure 1B ▶ ), and large granular endocytic vesicles were found in the epithelia lining of the lumena (Figure 1B) ▶ . Taken together, these results showed that significant lipid accumulation occurred within the gonads of ASMKO male mice (beginning as early as 8 weeks; data not shown), but did not impair the quantitative process of sperm formation, as confirmed by testis and epididymis sperm counts (Table 1) ▶ .
Figure 1.
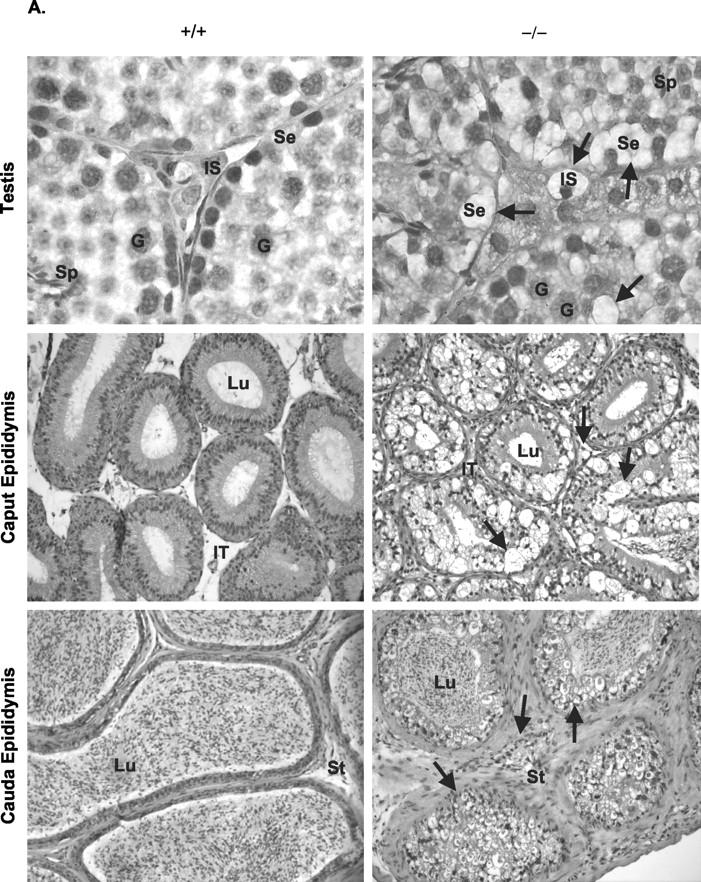
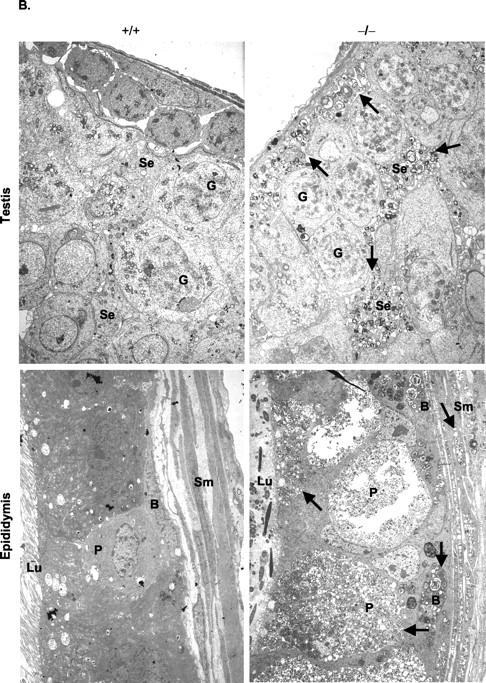
Lipid accumulation in gonads at 6 months of age. A: H&E-stained sections of testis (original magnification, ×1000) and epididymis (original magnification, × 200). Note the areas of lipid accumulation (arrows) in Sertoli cells (Se), interstitial (IS), epididymal lumen (Lu), epididymal intertubular space (IT), and stroma (St). G, germ cells; Sp, spermatozoa. B: Electron micrographs of testis and epididymides. Note multilamellar vesicles (arrows) in all cell types. Sm, smooth muscle cells; B, basal cells; P, principle cells; Lu, lumen. Original magnifications: × 2000.
Table 1.
Effect of ASM Deficiency on Spermatogenesis, Spermatozoon Morphology, and Motility
| +/+* | −/−* | |
|---|---|---|
| Testis sperm number (× 106) | 39.6 ± 8.7 | 39.1 ± 7.6 |
| Epididymal sperm number (× 106) | 20.1 ± 4.4 | 17.6 ± 10.8 |
| Morphology (%) | ||
| Straight† | 87.1 ± 2.8 | 35.7 ± 6.8 |
| Bent† | 1.7 ± 0.9 | 20.2 ± 7.6 |
| Hairpin† | 0.9 ± 0.7 | 30.4 ± 9.9 |
| Other | 10.3 ± 2.8 | 13.7 ± 4.6 |
| Motility (%) | ||
| Rapid | 59.0 ± 5.9 | 26.7 ± 4.5 |
| Medium | 31.2 ± 5.6 | 40.3 ± 4.1 |
| Slow | 3.1 ± 2.0 | 4.8 ± 2.7 |
| Static‡ | 2.2 ± 1.0 | 28.2 ± 3.7 |
*Values represent means or percentage means ± standard deviation.
†P < 0.001.
‡P < 0.01.
To determine whether the gonadal abnormalities observed in male ASMKO mice had an effect on reproductive capacity, a 4-month mating study was performed. As can be seen in Table 2 ▶ , wild-type mating pairs sired more litters, and of a larger size, than mating pairs of wild-type females and ASMKO males. Importantly, this trend was evident even when the mating pairs were 2 months of age, before the onset of a behavioral phenotype (data not shown). Mating outcomes between two ASMKO mice produced the lowest average number and size of litter, suggesting that abnormalities may also develop in the female gonads that affect reproduction (Table 2) ▶ .
Table 2.
Fecundity of Wild-Type and ASMKO Mice
| Mating pairs | Number of litters* | Size of litter* |
|---|---|---|
| +/+× +/+ | 3.4 | 6.4 |
| −/− ♂× +/+ ♀ | 1.6 | 4.7 |
| −/−× −/− | 1.3 | 3.0 |
*Data represent the averages from five mating pairs recorded over a 4-month period beginning at 2 months of age.
Sphingomyelinase Activities and SPM Accumulation in ASMKO Sperm
Figure 2A ▶ shows that mature sperm from 6-month-old ASMKO mice had a 10-fold increase in SPM content relative to +/+ mice, and Figure 2B ▶ shows that the residual ASM activity in these sperm was <5% of normal. The ASM activity was mostly found in the soluble (ie, supernatant) fraction of the sperm homogenate (Figure 2C) ▶ , indicating that ASM is likely to be a component of the acrosome secretory granule and not the spermatozoon plasma membrane. Surprisingly, when sphingomyelinase activity was studied in situ by flow cytometry and thin layer chromatography, spermatozoa from −/− mice exhibited a similar degree of SPM degradation as +/+ mice (Figure 2, D and E) ▶ . For these studies, spermatozoa were incubated in culture media at pH 7.4 conditioned to prevent the acrosome reaction. Thus, these assays revealed the presence of a sphingomyelinase activity in both wild-type and ASMKO sperm that was active at physiological pH and was distinct from the ASM activity.
Figure 2.

Biochemical abnormalities of caudal spermatozoa at 6 months of age. A: SPM accumulation in whole cell lysates. B: HPLC quantitation of ASM activity in whole cell lysates. C: Thin layer chromatogram showing ASM activity in homogenates, detergent soluble supernatants, and detergent insoluble pellets. D: Sphingomyelinase activity determined in situ and analyzed by flow cytometry. “Chase” indicates the extent of exogenous SPM degradation after initial labeling of cells (“Pulse”). E, Sphingomyelinase activity determined in situ and assessed by thin layer chromatography. Chase indicates the extent of exogenous SPM degradation after initial labeling of cells (Pulse). Ceramide is a metabolic product of SPM.
Cholesterol Accumulation in ASMKO Sperm
Cholesterol accumulation in mature ASMKO sperm was assessed with the sterol-specific-binding molecule, filipin. Freeze-fracture images of spermatozoon plasma membranes showed discriminating patterns of filipin-sterol complexes for wild-type and ASMKO mice (Figure 3A) ▶ . The homogenous pattern of these complexes exhibited in the plasma membrane of +/+ spermatozoa became heterogeneous in −/− mice. Qualitative analysis of the surface spectral properties (Figure 3B) ▶ showed the steepest decline for −/− spermatozoa (mean slope value, −0.8381 ± 0.006, compared to −0.7687 ± 0.007 for +/+), indicating increased image surface coarseness relative to +/+ spermatozoa. These values were highly significant (P < 0.001). To determine whether cholesterol accumulation in the plasma membrane of spermatozoa affected efflux of cholesterol during capacitation, the cells were incubated in media that promoted capacitation, but prevented the acrosome reaction. The cholesterol released into the media was quantified and the data are shown in Figure 3C ▶ . Note that sperm from −/− mice released twice as much cholesterol than +/+ mice (8.6 ± 0.8 μg versus 3.9 ± 0.3 μg), consistent with the elevated levels of cholesterol in the −/− sperm membranes.
Figure 3.
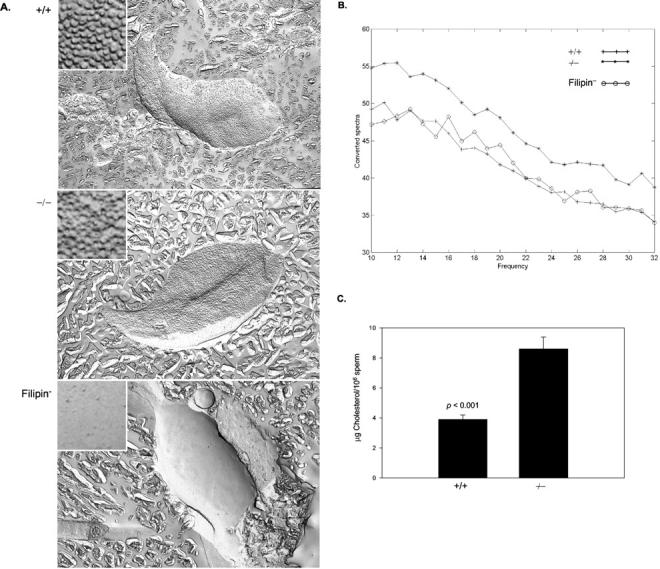
Cholesterol accumulation in caudal spermatozoa at 6 months of age. A: Representative freeze-fracture PF-face images of filipin-sterol complexes, FSC (original magnifications, ×10,000; insets: ×60,000). B: Surface analysis of FSC. The steepness of the spectral slope is indicative of image surface coarseness. C: In situ plasma membrane cholesterol efflux.
Morphological Defects in ASMKO Sperm
Qualitative and quantitative analysis of mature sperm morphology at 6 months of age revealed that −/− mice exhibited less than half the amount of straight spermatozoa than +/+ mice (Figure 4 ▶ and Table 1 ▶ ). Although there were numerous abnormalities, the most prominent was retroflexion of the tail to form kinks or complete bends (hairpins). The kinks appeared to be initiated at the midpiece-principle piece junction of the tail, where a cytoplasmic droplet was often noticeable (Figure 4) ▶ . Other spermatozoon abnormalities observed in −/− mice included microcephaly, bending at the head-midpiece junction, coiling of the tail, decapitation, and small aggregates (Table 1 ▶ , Morphology; and data not shown). Bent or hairpin sperm accounted for 50.6% of spermatozoa from −/− mice relative to 2.6% of spermatozoa from +/+ mice.
Figure 4.

Morphological abnormalities of caudal spermatozoa at 6 months of age. Spermatozoon tail retroflexion: <90° (arrow 1), ∼90° (arrow 2), and 180° (arrow 3). Original magnifications: × 400.
The observed retroflexion of the −/− sperm had a significant impact on their motility, with the percentage of rapidly moving spermatozoa from −/− mice less than half that of +/+ mice (Table 1 ▶ , Motility). Although these data were striking, it is important to again note that there were no significant differences in sperm numbers between mice of each genotype.
To determine whether lipid release from spermatozoon plasma membranes could correct the teratozoospermia observed in ASMKO mice, sperm were incubated in media containing a mild detergent. Bent and hairpin sperm completely reverted to the straight position observed in wild-type mice (Table 3) ▶ . Alsoof note, the residual bodies initially observed at the site of bending of −/− sperm were absent (not shown). Taken together, the data presented in Tables 1 and 3 ▶ ▶ suggest that excess lipid accumulation in the plasma membranes of spermatozoon from ASMKO mice results in morphological abnormalities that retard sperm motility.
Table 3.
Morphologic Assessment of Spermatozoa after Lipid Demembranation
| +/+ | −/− | |||||
|---|---|---|---|---|---|---|
| Straight* | Bent | Hairpin | Straight† | Bent | Hairpin | |
| Untreated | 87.0 ± 4.2 | 4.3 ± 1.4 | 8.7 ± 2.8 | 28.9 ± 13.6 | 25.4 ± 12.4 | 45.8 ± 24.2 |
| 0.1% Triton X-100 | 99.3 ± 1.1 | 0.7 ± 1.1 | 0 | 100.0 ± 0.0 | 0 | 0 |
*Values represent mean percentages ± standard deviation.
†P < 0.01 when comparing the number of straight spermatozoa before and after treatment.
Physiological Defects in ASMKO Sperm
Various assays were used to assess whether physiological abnormalities accompanied the morphological defects observed in ASMKO sperm. To assess plasma membrane integrity, sperm from +/+ and −/− mice were incubated with SYBR-14, a DNA intercalating dye that is able to cross the plasma membrane and stain cells green (FL-1, Figure 5A ▶ ). Membrane integrity is assessed by measuring the ability of a membrane impermeable dye, PI (FL-3), to occlude the green fluorescence of SYBR-14. Therefore, cells with intact plasma membranes will have high FL-1 fluorescence; those with compromised membranes will have high FL-3 fluorescence. 21 The lower right quadrants (FL-1high, FL-3low) of Figure 5A ▶ show that only 13.4% of spermatozoa from −/− mice had intact plasma membranes compared to 59.2% for +/+ mice.
Figure 5.

Physiological abnormalities of caudal spermatozoa at 6 months of age by flow cytometric analysis. A: Plasma membrane integrity. The percentage of healthy, plasma membrane intact spermatozoa are shown in the lower right quadrants of the density dot-blot profiles. B: Acrosome status. The percentage of acrosome-intact spermatozoa is shown in the lower right quadrants of the density dot-blot profiles. C: Mitochondrial membrane potential. The percentage of metabolically active sperm is shown in the upper right quadrants of the density dot-blot profiles. D: NO production. The percentage of spermatozoa producing NO as a function of plasma membrane integrity is shown in the upper right quadrants of the density dot-blot profiles. E: Phosphatidylserine bilayer translocation. The percentage of spermatozoa with intact plasma membranes undergoing PS translocation from the inner to the outer leaflet of the plasma membrane is indicated in the lower left quadrants of the density dot-blot profiles.
We next sought to determine the acrosome status in sperm from the +/+ and −/− animals (Figure 5B) ▶ . Spermatozoa from each genotype were incubated for 1 hour to allow capacitation, then 30 minutes in media containing peanut agglutinin, a lectin that binds specifically to the acrosomal membrane. Acrosome intact sperm show high FL-1 fluorescence derived from fluorescein isothiocyanate-conjugated peanut agglutinin, 20 and low FL-3 fluorescence derived from plasma membrane disruption (as above). In Figure 5B ▶ , the lower right quadrants (FL-1high, FL-3low) of the upper panel show that only 1.2% of sperm from −/− mice are acrosome intact, as compared to 33.3% from +/+ mice. When the acrosome reaction was artificially induced, the percentage of +/+ sperm with intact acrosomes falls to <1%, similar to sperm from −/− mice (lower panel). These data indicate that under physiological conditions the vast majority of spermatozoa from −/− mice have lost acrosome integrity, and complements the above data that show plasma membrane perturbations in these same mice.
To determine whether the plasma membrane abnormalities we observed in ASMKO sperm affected mitochondrial membrane potential, JC-1, a lipophilic dye, was used. In healthy cells, where mitochondrial membrane potential is high, JC-1 aggregates form in the mitochondria with a consequent increase in FL-2 fluorescence; if mitochondrial membrane potential is low, FL-2 fluorescence is low; 21 FL-1 indicates the fluorescence of JC-1 monomers. The upper right quadrants of Figure 5C ▶ show that only approximately half of spermatozoa from −/− mice have intact mitochondrial membrane potential relative to +/+ mice (45.5% versus 87.5%, respectively). In relation to the data presented on motility (Table 1) ▶ , it is not surprising that ASMKO spermatozoa exhibit reduced motility because metabolically active mitochondria are necessary for flagellar motion. In addition, the accumulation of lipids within the plasma membrane (Figure 2A ▶ and Figure 3 ▶ ) may affect mitochondrial membrane proton pumps.
To assess the ability of ASMKO sperm to undergo capacitation, NO production and PS translocation were studied. Production of NO and consequent PS translocation from the inner to the outer layer of the plasma membrane is required for sperm capacitation. NO production can be detected with a fluorescein isothiocyanate-conjugated dye that increases in FL-1 fluorescence as it binds NO; 22 FL-3 fluorescence increases as cells lose plasma membrane integrity. Figure 5D ▶ (upper right quadrants) shows spermatozoa from −/− mice experience a sixfold decrease in NO production relative to spermatozoa from +/+ mice (6.7% versus 41.2%, respectively).
Phosphatidylserine translocation was assessed using Annexin V, a PS-specific molecule that increases in FL-2 fluorescence on binding; 23 FL-1 fluorescence increases as cells lose plasma membrane integrity. Figure 5E ▶ shows approximately half as many membrane-intact spermatozoa from −/− mice undergo PS translocation relative to spermatozoa from +/+ mice (31.4% versus 61.5%; lower left quadrants). Taken together, Figure 5 ▶ describes physiological deficiencies that may accompany the morphological defects observed above (Table 1 ▶ , Figure 4 ▶ ). In addition, the premature loss of the acrosome granule observed in sperm from ASM-deficient mice, accompanied by mitochondrial membrane defects, may influence the fertilizing ability of these mice (Table 2) ▶ .
Discussion
Effect of SPM Accumulation on Cholesterol Efflux, Acrosome Reaction, and Capacitation
SPM and cholesterol are abundant in somatic cell and sperm plasma membranes, 24,25 and are hypothesized to form lipid rafts that alter the lateral distribution of proteins. 26 The removal of SPM from membranes (eg, by hydrolysis with exogenous sphingomyelinase) results in the efflux of cholesterol to extracellular acceptors. 27,28 Notably, under physiological conditions, the loss of cholesterol from the sperm plasma membrane is a defining step in the process of capacitation, 28 and recently it was shown that the SPM content of sperm membranes influences the rate of capacitation and the release of acrosomal enzymes. 28 In addition, treatment of sperm with exogenous sphingomyelinase accelerated capacitation. 27
In our ASM-deficient mice, lipid accumulation was histologically evident in somatic cells of the gonads (Figure 1) ▶ , as well as in fully formed caudal spermatozoa (Figure 2) ▶ . Cholesterol has been localized to the acrosome region of the sperm head and the tail midpiece, 29,30 and our data showed that spermatozoa from ASMKO mice had an increase in filipin-sterol complexes over the acrosome (Figure 3, A and B) ▶ .
The vast majority of sperm from ASMKO mice also showed loss of plasma, acrosome, and mitochondria membrane integrity (Figure 5; A, B, and C) ▶ . The remaining membrane-intact sperm (∼13.4% in 6-month-old mice, Figure 5A ▶ ) were unable to undergo proper capacitation, as assessed by NO production and PS translocation (Figure 5, D and E) ▶ . The latter finding may be a direct consequence of SPM accumulation, which affects the formation/fluidity of sperm membranes. Because published work demonstrates that NO production induces or facilitates capacitation and acrosome reaction, 31,32 these results also suggest that lack of NO production in ASMKO sperm may result from the loss of NO synthase function, which has been localized to the sperm acrosome. 32
Effect of SPM/Cholesterol Accumulation of Mitochondrial Function
Physiologically, the normal efflux of cholesterol that produces increased fluidity of the plasma membrane and initiates capacitation/acrosome reaction involves signal transduction pathways. 33 These pathways may be regulated by changes in ion channel activity and/or the activity of membrane-associated enzymatic and nonenzymatic proteins and plasma membrane receptors. 33 It has been shown that sperm that fail to capacitate in vitro exhibited weak hyperpolarization and did not relieve voltage-dependent inactivation of protein channels. Moreover, permeability of mitochondrial membranes is inversely related to their cholesterol content. 34 Our data are in agreement with these findings because spermatozoa from ASMKO mice exhibited decreased mitochondrial membrane potential (Figure 5C) ▶ , presumably directly because of the SPM and cholesterol accumulation. Accumulation of other lipids may also contribute to this finding, because it has been found that increased levels of sphingosylphosphorylcholine inhibit mitochondrial ATPase. 35 Notably, it has been shown that this lipid accumulates within tissues of NPD patients. 36
Regulatory Volume Decrease (RVD) Defects and Abnormal Morphology of ASMKO Sperm
Previous work has shown that sperm with motility defects (asthenozoospermia) may result from swelling of mitochondria that leads to the discharge of a proton gradient across mitochondrial membranes. 35 The induction of hypo-osmotic swelling is a common phenomenon caused by a defect in RVD, and often leads to cell necrosis. Wild-type sperm are able to undergo normal RVD, 37,38 and thus exhibit a straight shape and normal mitochondrial membrane potential. In contrast, sperm bending can be induced in vitro by disrupting RVD through blockage of potassium and chloride channels. 39 The physical bending of sperm to produce kinks or bends (teratozoospermia) is known to occur to relieve increased surface area because of RVD abnormalities. The fact that ASMKO mice are teratozoospermic (Figure 4 ▶ , Table 1 ▶ ) suggested that a defect in RVD had occurred and supported the flow cytometric data indicating a disruption in mitochondrial membrane potential (Figure 5C) ▶ .
Teratozoospermia because of RVD defects is thought to occur in the epididymis. 40,41 Evidence for this comes from the c-ros knockout mouse that shows teratozoospermia similar to our ASMKO mice. 42 C-ros is a proto-oncogene that encodes an orphan receptor with an intracellular tyrosine kinase domain that, in adults, is uniquely expressed in the initial segment (caput) of the epididymis. 43 In contrast to wild-type sperm, which have straight flagella, 46 to 86% of c-ros knockout sperm displayed severe tail retroflexion at the midpiece/principal piece junction associated with swollen cytoplasmic droplets, even when motile. 39 In addition, motile knockout sperm showed a reduction in swimming velocities (flagellar strength and forward progression) when compared with straight, wild-type sperm. 39
These results are similar to what we observed for ASMKO mice (Figure 4 ▶ , Table 1 ▶ ). The authors speculate that teratozoospermia in the c-ros knockout mice occurs because of the inability of the sperm to properly mature in the epididymis. Lack of sperm maturation produces asthenozoospermia, which in turn contributes to reduced fecundity. The authors suggest that c-ros teratozoospermia may result when osmotic swelling of the spermatozoa occurs to avoid increases in surface area. They cite previous work that describes the straightening of c-ros knockout sperm on treatment with Triton X-100, 39 indicating that RVD is not an inherent flagellar defect but a cell-swelling phenomenon induced by membrane abnormalities. Hence, an unstated implication is that epididymal sperm maturation defects may result from improper volume regulation because of changes in plasma membrane fluidity and protein function. We assessed this possibility in ASMKO mice by incubating sperm in a dilute detergent solution according to the previously published protocol, and found that dysmorphic spermatozoa reverted to a normal, straight position (Table 2) ▶ . This supports the aforementioned notion that RVD defects may result from the pathological accumulation of lipid in the sperm plasma membrane.
Although bending of spermatozoa because of RVD defects may occur in the epididymis, evidence suggests that the process could begin earlier in Sertoli cells. In ASMKO mice, it was observed that the vast majority of bent spermatozoa contained cytoplasmic droplets at the midpiece-principle junction of the sperm tail (Figure 4) ▶ . Murine epididymal spermatozoa, swollen hypotonically or upon treatment with potassium channel blockers, have been shown to bend at the site of the cytoplasmic droplet located at the end of the midpiece. 44 Indeed, a function of the Sertoli cell is to phagocytose the residual bodies from mature spermatids, 45 and retention of the residual bodies is considered a midpiece abnormality. 46 Hence, improper processing of cytoplasmic remnants by the lysosomes of Sertoli cells in ASMKO mice may prejudice fully formed spermatozoa from undergoing proper RVD once they enter the epididymal lumen. As already noted, Sertoli cells of ASMKO mice show multilamellar vesicles and areas of gross lipid accumulation (Figure 1) ▶ that may prevent the processing of spermatid residual bodies. When these cytoplasmic droplets were dissociated by incubation of sperm in mild detergent, ASMKO teratozoospermia was eliminated (Table 3) ▶ . Consequently, spermatozoa with lipid-filled cytoplasmic droplets may experience a loss of RVD, undergo morphological bending, and experience severe limitations in motility. 47
Mechanism Underlying the RVD Defect
Lipid accumulation in gonads occurs in several other mouse models of sphingolipid storage disorders, most notably Tay-Sachs and Sandhoff diseases. 12,13 In these mice, testis weight and sperm counts were unaffected, as we observed here. The epididymis of the Tay-Sachs mice showed a large increase in the size and number of lysosomes in the initial segment/intermediate zone of the epididymis, whereas Sandhoff mice had more extensive lipid accumulation. Although both mice were initially fertile, Tay-Sachs male mice experienced reduced litter sizes, and Sandhoff male mice became sterile after 10 to 12 weeks of age. Although epididymal sperm were not analyzed in these mice, the authors also concluded that accumulation of lipids in lysosomes of the epididymis lead to epididymal dysfunction and abnormalities in the luminal environment that supports sperm maturation. The authors further noted the fusion of lysosomes in epididymal cells of these mice and suggest that it may be due to increased intracellular calcium levels because lysosomal fusion can be induced by agents that elevate intracellular calcium levels. 12,48 In ASMKO mice, lysosome fusion within the epididymis is also noticeable (Figure 1) ▶ .
Lysosomes and lysosomal hydrolases are prominent in testicular and epididymal epithelial cells of the mammalian male reproductive tract, with postulated roles in degradation of extruded cytoplasmic remnants and abnormal sperm, and other complex endocytotic and exocytotic processes. 16 In addition, multiple protein pumps and channels maintain luminal volume and osmolarity, and help to generate a low luminal pH that is involved in sperm maturation and maintaining sperm in an immobile state during their passage through the epididymis and vas deferens. 49 Voltage-gated potassium channels, aquaporins, and Na+/H+ exchangers regulate epididymal volume, 47,50,51 and volume-sensitive anion channels regulate the flow of organic osmolytes into the lumen. 51 These proteins may be expressed in high levels on the luminal plasma membranes, as well as on intracellular vesicles throughout the epididymis. 49,50 Hence, disturbances in the above-mentioned regulatory proteins might affect the acidity and osmolarity of the epididymal lumen, and such disturbances may occur from membrane lipid accumulation. Because capacitation and acrosome reaction is initiated by an increase in intracellular calcium and pH, 15 it is not surprising that spermatozoa isolated from cauda epididymides of ASMKO mice exhibited compromised plasma membrane integrity (Figure 5A) ▶ and increased acrosome reaction (Figure 5B) ▶ . One may therefore propose that increased lipid accumulation in epididymal epithelial cells of ASMKO mice impairs the regulation of luminal fluid constituents by disrupting the ion and transport protein channels of these cells. As a result, the epididymal lumen may be transformed into a lipid sink with microenvironments that retard the physiological and morphological maturation of spermatozoa. It is of further interest to note that a mechanism proposed for characteristic Purkinje cell loss observed in ASMKO mice is the disruption of α-1a voltage-sensitive calcium channels, 52 where deregulation of chemiosmotic potential may result in RVD defective necrosis.
Conclusions
From the studies reported in this article we conclude that the gross accumulation of lipid within the male gonads of ASMKO mice affects epididymal sperm maturation and fertilizing ability. Lipid accumulation in the epididymal epithelia of ASMKO mice may interfere with regional maintenance of luminal fluid components and directly or indirectly influence several mandatory aspects of sperm capacitation and acrosome reaction. Based on these investigations, we propose that the reproductive impairment observed in ASMKO mice is a direct consequence of lipid accumulation within gonadal tissue, and that similar abnormalities may occur in sexually mature male NPD patients. These data show that ASM is an important hydrolase involved in the process of normal SPM and cholesterol metabolism in sperm development and function, and provide an impetus for similar investigations in sexually mature, male NPD patients.
Acknowledgments
We thank Drs. Jon Gordon and Kevin Kelly of the Mount Sinai School of Medicine for their important comments and assistance throughout the course of this work.
Footnotes
Address reprint requests to Edward H. Schuchman, Ph.D., Department of Human Genetics, Mount Sinai School of Medicine, 1425 Madison Ave., Rm. 14-20A, New York, NY 10029. E-mail: edward.schuchman@mssm.edu.
Supported by the National Institutes of Health (grant R01 HD28607).
References
- 1.Schuchman E, Desnick R: Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. Scriver C Beaudet A Sly W Valle D eds. The Metabolic and Molecular Basis of Inherited Disease, ed 8 2001:p 3589 McGraw-Hill, New York
- 2.Levade T, Jaffrezou JP: Signaling sphingomyelinases: which, where, how and why? Biochim Biophys Acta 1999, 1438:1-17 [DOI] [PubMed] [Google Scholar]
- 3.Tomiuk S, Zumbansen M, Stoffel W: Characterization and subcellular localization of murine and human magnesium-dependent neutral sphingomyelinase. J Biol Chem 2000, 275:5710-5717 [DOI] [PubMed] [Google Scholar]
- 4.Schuchman EH, Suchi M, Takahashi T, Sandoff K, Desnick RJ: Human acid sphingomyelinase. Isolation, nucleotide sequence, and expression of the full-length and alternatively spliced cDNAs. J Biol Chem 1991, 13:8531-8539 [PubMed] [Google Scholar]
- 5.Schuchman EH, Levran O, Pereira LV, Desnick RJ: Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics 1992, 12:197-205 [DOI] [PubMed] [Google Scholar]
- 6.Horinouchi K, Sakiyama T, Pereira L, Lalley PA, Schuchman EH: Mouse models of Niemann-Pick disease: mutation analysis and chromosomal mapping rule out the type A and B forms. Genomics 1993, 18:450-451 [DOI] [PubMed] [Google Scholar]
- 7.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandoff K, Desnick RJ, Stewart CL, Schuchman EH: Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet 1995, 10:288-293 [DOI] [PubMed] [Google Scholar]
- 8.Otterbach B, Stoffel W: Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease). Cell 1995, 81:1053-1061 [DOI] [PubMed] [Google Scholar]
- 9.Miranda SRP, Erlich S, Friedrich VL, Haskins ME, Gatt S, Schuchman EH: Biochemical, pathological, and clinical response to transplantation of normal bone marrow cells into acid sphingomyelinase deficient mice. Transplantation 1998, 65:884-892 [DOI] [PubMed] [Google Scholar]
- 10.Miranda SRP, Erlich S, Visser JWM, Gatt S, Dagan A, Friedrich VL, Schuchman EH: Bone marrow transplantation in acid sphingomyelinase deficient mice: engraftment and cell migration into the brain as a function of radiation, age, and phenotype. Blood 1997, 90:444-452 [PubMed] [Google Scholar]
- 11.Erlich S, Miranda SRP, Visser JWM, Dagan A, Gatt S, Schuchman EH: Fluorescence-based selection of gene-corrected hematopoietic stem and progenitor cells from acid sphingomyelinase-deficient mice: implications for Niemann-Pick disease gene therapy and the development of improved stem cell gene transfer procedures. Blood 1999, 93:80-86 [PubMed] [Google Scholar]
- 12.Trasler J, Saberi F, Somani IH, Adamali HI, Huang JQ, Fortunato SR, Ritter G, Gu M, Aebersold R, Gravel RA, Hermo L: Characterization of the testis and epididymis in mouse models of human Tay Sachs and Sandhoff diseases and partial determination of accumulated gangliosides. Endocrinology 1998, 139:3280-3288 [DOI] [PubMed] [Google Scholar]
- 13.Adamali HI, Somani IH, Huang JQ, Mahuran D, Gravel RA, Trasler JM, Hermo LI: Abnormalities in cells of the testis, efferent ducts, and epididymis in juvenile and adult mice with beta-hexosaminidase A and B deficiency. J Androl 1999, 20:779-802 [PubMed] [Google Scholar]
- 14.Kapur DK, Gupta GS: Immunocytochemical localization of β-N-acetyl glucosaminidase in human reproductive organs. Biol Reprod 1988, 39:373-376 [DOI] [PubMed] [Google Scholar]
- 15.Tulsiani DR, Abou-Haila A, Loeser CR, Pereira BM: The biological and functional significance of the sperm acrosome and acrosomal enzymes in mammalian fertilization. Exp Cell Res 1998, 240:151-164 [DOI] [PubMed] [Google Scholar]
- 16.Yanagimachi R, Harper MJK, de Kretser DM, Kerr JB: Mammalian fertilization, gamete and zygote transport, and cytology of the testis. Knobil E Neill JD eds. The Physiology of Reproduction ed 2 1994:pp 189-206 Raven Press, New York 125–127, 1224–1225
- 17.Hinkovska VT, Petkova DH, Koumanov KS: A neutral sphingomyelinase in spermatozoal plasma membranes. Biochem Cell Biol 1987, 65:525-528 [DOI] [PubMed] [Google Scholar]
- 18.Hinkovska-Galcheva VTS, Petkova DH, Nikolova MN: Sphingomyelin and ceramide: phosphoethanolamine synthesis in ram spermatozoa plasma membrane. Int J Biochem 1989, 21:1153-1156 [DOI] [PubMed] [Google Scholar]
- 19.He X, Chen F, McGovern MM, Schuchman EH: A fluorescence-based, high-throughput sphingomyelin assay for the analysis of Niemann-Pick disease and other disorders of sphingomyelin metabolism. Anal Biochem 2002, 306:115-123 [DOI] [PubMed] [Google Scholar]
- 20.Toa J, Critsler ES, Critser JK: Evaluation of mouse sperm acrosomal status and viability by flow cytometry. Mol Reprod Dev 1993, 36:183-194 [DOI] [PubMed] [Google Scholar]
- 21.Gravance CG, Garner DL, Miller MG, Berger T: Fluorescent probes and flow cytometry to assess rat sperm integrity and mitochondrial function. Reprod Toxicol 2001, 15:5-10 [DOI] [PubMed] [Google Scholar]
- 22.Kojima H, Sakurai K, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T: Development of a fluorescence indicator for nitric oxide based on the fluorescein chromophore. Chem Pharm Bull 1998, 46:373-375 [DOI] [PubMed] [Google Scholar]
- 23.Raynal P, Pollard HB: Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium and phospholipid-binding proteins. Biochim Biophys Acta 1994, 1197:63-93 [DOI] [PubMed] [Google Scholar]
- 24.Nikolopoulou M, Soucek DA, Vary JC: Changes in the lipid content of boar sperm membranes during epididymal maturation. Biochim Biophys Acta 1985, 815:486-498 [DOI] [PubMed] [Google Scholar]
- 25.Parks JE, Arion JW, Foote RH: Lipids of plasma membrane and outer acrosomal membranes from bovine spermatozoa. Biol Reprod 1987, 37:1249-1258 [DOI] [PubMed] [Google Scholar]
- 26.Simons K, Ikonen E: Sphingolipid-cholesterol rafts in membrane trafficking and signaling. Nature 1997, 387:569-572 [DOI] [PubMed] [Google Scholar]
- 27.Cross NL: Sphingomyelin modulates capacitation of human sperm in vitro. Biol Reprod 2000, 63:1129-1134 [DOI] [PubMed] [Google Scholar]
- 28.Cross NL: Role of cholesterol in sperm capacitation. Biol Reprod 1998, 59:7-11 [DOI] [PubMed] [Google Scholar]
- 29.Suzuki F: Changes in the distribution of intramembranous particles and filipin-sterol complexes during epididymal maturation of golden hamster spermatozoa. J Ultrastruct Mol Struct Res 1988, 100:39-54 [DOI] [PubMed] [Google Scholar]
- 30.Lin Y, Kan FW: Regionalization and redistribution of membrane phospholipids and cholesterol in mouse spermatozoa during in vitro capacitation. Biol Reprod 1996, 55:1133-1146 [DOI] [PubMed] [Google Scholar]
- 31.Herrero MB, de Lamirande E, Gagnon C: Nitric oxide regulates human sperm capacitation and protein-tyrosine phosphorylation in vitro. Biol Reprod 1999, 61:575-581 [DOI] [PubMed] [Google Scholar]
- 32.Herrero MB, Perez Martinez S, Viggiano JM, Polak JM, de Gimeno MF: Localization by indirect immunofluorescence of nitric oxide synthase in mouse and human spermatozoa. Reprod Fertil Dev 1996, 8:931-934 [DOI] [PubMed] [Google Scholar]
- 33.Visconti PE, Ning X, Fornes MW, Alvarez JG, Stein P, Connors SA, Kopf GS: Cholesterol efflux-mediated signal transduction in mammalian sperm: cholesterol release signals an increase in protein tyrosine phosphorylation during mouse sperm capacitation. Dev Biol 1999, 214:429-443 [DOI] [PubMed] [Google Scholar]
- 34.Baggetto LG, Clottes E, Vial C: Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res 1992, 52:4935-4941 [PubMed] [Google Scholar]
- 35.Strasberg PM, Callahan JW: Lysosphingolipids and mitochondrial function. II. Deleterious effects of sphingosylphosphorylcholine. Biochem Cell Biol 1988, 66:1322-1332 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Lafrasse C, Vanier MT: Sphingosylphosphorylcholine in Niemann-Pick disease brain: accumulation in type A but not in type B. Neurochem Res 1999, 24:199-205 [DOI] [PubMed] [Google Scholar]
- 37.Gschwentner M, Nagl UO, Wöll E, Schmarda A, Ritter M, Paulmichl M: Antisense oligonucleotides suppress cell-volume induced activation of chloride channels. Pfluegers Arch 1995, 430:464-470 [DOI] [PubMed] [Google Scholar]
- 38.Clapham DE: The list of potential volume-sensitive chloride currents continues to swell (and shrink). J Gen Physiol 1998, 111:623-624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeung CH, Sonnenberg-Riethmacher E, Cooper TG: Receptor tyrosine kinase c-ros knockout mice as a model for the study of epididymal regulation of sperm function. J Reprod Fertil Suppl 1998, 53:137-147 [PubMed] [Google Scholar]
- 40.Drevius L: Osmotic behavior, membrane permeability and mobility mechanisms in bull spermatozoa. Acta Univ Ups Abstr Upps Diss Sci 1972, 195:1-20 [Google Scholar]
- 41.Jeyendran RS, Van der Ven HH, Perez-Pelaez M, Crabo BG, Zaneveld LJD: Development of an assay to assess the functional integrity of the human sperm membrane and its relationship to other semen characteristics. J Reprod Fertil 1984, 70:219-228 [DOI] [PubMed] [Google Scholar]
- 42.Yeung CH, Wagenfeld A, Nieschlag E, Cooper TG: The cause of infertility of male c-ros tyrosine kinase receptor knockout mice. Biol Reprod 2000, 63:612-618 [DOI] [PubMed] [Google Scholar]
- 43.Cooper TG: Epididymis. Neill JD Knobil E eds. Encyclopedia of Reproduction, 1998, vol 2.:pp 1-17 Academic Press, New York [Google Scholar]
- 44.Yeung CH, Sonnenberg-Riethmacher E, Cooper TG: Infertile spermatozoa of c-ros tyrosine kinase receptor knockout mice show flagellar angulation and maturational defects in cell volume regulatory mechanisms. Biol Reprod 1999, 61:1062-1069 [DOI] [PubMed] [Google Scholar]
- 45.Morales CR, Clermont Y: Phagocytosis and endocytosis in Sertoli cells of the rat. Bull Assoc Anat 1991, 75:157-162 [PubMed] [Google Scholar]
- 46.Albert M, Roussel C: Changes from puberty to adulthood in the concentration, motility and morphology of mouse epididymal spermatozoa. Int J Androl 1983, 6:446-460 [DOI] [PubMed] [Google Scholar]
- 47.Yeung CH, Cooper TG: Effects of the ion-channel blocker quinine on human sperm volume, kinematics and mucus penetration, and the involvement of potassium channels. Mol Hum Reprod 2001, 7:819-828 [DOI] [PubMed] [Google Scholar]
- 48.Pryor PR, Mullock BM, Bright NA, Gray SR, Luzio JP: The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J Cell Biol 2000, 149:1053-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herak-Kramberger CM, Breton S, Brown D, Kraus O, Sabolic I: Distribution of the vacuolar H+ ATPase along the rat and human male reproductive tract. Biol Reprod 2001, 64:1699-1707 [DOI] [PubMed] [Google Scholar]
- 50.Pastor-Soler N, Bagnis C, Sabolic I, Tyszkowski R, McKee M, Van Hoek A, Breton S, Brown D: Aquaporin 9 expression along the male reproductive tract. Biol Reprod 2001, 65:384-393 [DOI] [PubMed] [Google Scholar]
- 51.Emma F, McManus M, Strange K: Intracellular electrolytes regulate the volume set point of the organic osmolyte/anion channel VSOAC. Am J Physiol 1997, 272:C1766-C1775 [DOI] [PubMed] [Google Scholar]
- 52.Sarna J, Miranda SR, Schuchman EH, Hawkes R: Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur J Neurosci 2001, 13:1873-1880 [DOI] [PubMed] [Google Scholar]