

Abstract
The majority of pediatric anaplastic large cell lymphomas (ALCLs) carry the t(2;5)(p23;q35) chromosomal translocation that juxtaposes the dimerization domain of nucleophosmin with anaplastic lymphoma kinase (ALK). The nucleophosmin-ALK fusion induces constitutive, ligand-independent activation of the ALK tyrosine kinase leading to aberrant activation of cellular signaling pathways. To study the early consequences of ectopic ALK activation, a GyrB-ALK fusion was constructed that allowed regulated dimerization with the addition of coumermycin. Expression of the fusion protein caused a coumermycin-dependent increase in cellular tyrosine phosphorylation and c-Myc immunoreactivity, which was paralleled by a rise in c-myc RNA. To assess the clinical relevance of this observation, c-Myc expression was determined in pediatric ALK-positive and -negative lymphomas. Co-expression of c-Myc and ALK was seen in tumor cells in 15 of 15 (100%) ALK-positive ALCL samples, whereas no expression of either ALK or c-Myc was seen in six of six cases of ALK-negative T-cell lymphoma. C-Myc may be a downstream target of ALK signaling and its expression a defining characteristic of ALK-positive ALCLs.
Lymphoma is the third most common cancer in children and adolescents in the United States, representing 13% of newly diagnosed malignancies in the pediatric age group. 1 Although significant progress has been made in the treatment and outcome of childhood lymphoma, many children still succumb to disease, or suffer acute and long-term toxicity from contemporary multiagent chemotherapy. 2-4 A subtype of pediatric anaplastic large cell lymphoma (ALCL), anaplastic lymphoma kinase (ALK)-positive lymphoma (ALK+ lymphoma), may serve as an ideal model for the study of the molecular mechanisms of malignant transformation. 5-7 ALK+ lymphoma is characterized by a t(2;5)(p23;q35) chromosomal translocation that creates a chimeric fusion protein consisting of the amino-terminal portion of the nucleolar phosphoprotein, nucleophosmin (NPM), and the cytoplasmic domain of the receptor tyrosine kinase, ALK. 8,9 ALK is a recently described receptor tyrosine kinase in the insulin receptor subfamily whose expression is normally restricted to the central nervous system. 10-12 NPM is a ubiquitously expressed homohexameric nucleolar phosphoprotein that shuttles ribosomal proteins between the nucleus and cytoplasm. 13-15 Essential functions of NPM in the chimera are provision of both a promoter that drives ectopic expression of ALK in lymphoid cells, and a dimerization motif that facilitates trans-phosphorylation and hence activation of signaling. 16
The t(2;5) translocation is an essential event in the pathogenesis of ALK+ lymphoma as NPM-ALK potently transforms both rat fibroblast and murine lymphoid cell lines, and induces lymphoid tumors in mice. 17,18 NPM-ALK homodimers autophosphorylate on multiple residues, creating docking sites for several SH2 domain-containing signaling molecules. 16,19-21 Formation of this signaling complex leads to changes in gene expression that underlie the malignant phenotype. One goal of our studies is to understand the changes in cellular transcription that the NPM-ALK fusion protein produces.
Ligand-induced dimerization is thought to directly induce the activation of receptor tyrosine kinases. 22-24 Numerous researchers have generated protein fusions that can be conditionally dimerized in the presence of bivalent compounds such as coumermycin, rapamycin, and FK1012. 25-27 To better define the immediate early effects of ALK activation, we used this technology to generate a conditionally dimerizable kinase construct, designed to mimic the activity of NPM-ALK in vivo. Regulated dimerization produced an increase in tyrosine phosphorylation of the fusion protein, as well as additional cellular proteins. Unexpectedly, we observed an increase in c-Myc protein and RNA expression in response to ALK activation. Supporting the relevance of this finding in vivo, c-Myc was detected in 15 of 15 t(2;5)-positive ALCLs, but in 0 of 6 ALK-negative lymphomas. The data suggest that c-Myc is a specific downstream target of aberrant ALK signaling as well as a novel marker for the detection of ALK+ lymphomas.
Materials and Methods
Construction of Expression Plasmids
6 Myc-GyrB-ALK
For transient expression of myc epitope-tagged GyrB-ALK, the courmermycin-binding domain (codons 2 to 220) of the B subunit of Escherichia coli DNA Gyrase (GyrB) was cloned into pCS2+MT in-frame with the entire cytoplasmic domain of ALK.
Wild-Type and Mutant 6 Myc-NPM-ALK Retroviral Constructs
The 6-myc epitope tag from the pCS2+MT plasmid vector was inserted into the BamHI and EcoRI sites of the pBABE Puro retroviral vector. Full-length NPM-ALK coding sequence was amplified by polymerase chain reaction (PCR) incorporating 5′ and 3′ EcoRI restriction sites (underlined) using forward primer, 5′ CCGAATTCGATGGAAGATTCGATGGAC 3′, and reverse primer 5′ ATGAATTCTCAGGGCCCAGGCTGGTTC 3′. Full-length NPM-ALK was subcloned into the EcoRI site of the 6 Myc-pBABE Puro retroviral vector.
The conserved lysine residue (amino acid 210, nucleotide 628) within the kinase domain of the wild-type NPM-ALK retroviral construct was mutated to alanine (GCC) to abrogate kinase activity using the QuikChange in vitro site-directed mutagenesis system (Stratagene, La Jolla, CA) following the manufacturer’s instructions with the addition of 4% dimethyl sulfoxide.
In Vitro Translation
In vitro translation of 6 Myc-GyrB-ALK was performed with the TNT Quick Coupled Transcription/Translation System with SP6 RNA polymerase (Promega, Madison, WI). Reactions were performed at 30°C for 2 hours. In vitro-translated 6 Myc-GyrB-ALK was then treated with or without varying concentrations of coumermycin A1 (Sigma, St. Louis, MO) prepared in dimethyl sulfoxide for an additional 30 minutes at 30°C.
Cell Culture, DNA Transfection, and Retroviral Infections
Rat1A fibroblasts (kindly provided by Andrew Thorburn, University of Utah), and the retroviral packaging Phoenix cell line (a gift from Nori Matsunami, University of Utah) were grown in Dulbecco’s modified Eagle’s medium (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal calf serum (Hyclone, Logan, UT) and 100 U/ml of penicillin and 100 μg/ml of streptomycin (Life Technologies Inc., Gaithersburg, MD). Cells were maintained at 37°C and 5% CO2 in a humidified incubator. Rat1A fibroblasts were seeded in 6-well (35-mm) plates and transiently transfected with 1 μg of 6 Myc-GyrB-ALK expression construct, 8 μl of lipofectamine, and 6 μl of Plus reagent per well following the manufacturer’s instructions (Life Technologies Inc.). Forty-eight hours after transfection, cell cultures were treated with varying concentrations of coumermycin for 30 minutes at 37°C. Cells were then briefly washed with 2 ml of phosphate-buffered saline and lysed in 200 μl of RIPA with protease inhibitors [150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mmol/L Tris-HCl, pH 7.5, 1.4 mmol/L phenylmethyl sulfonyl fluoride, 1 mmol/L ethylenediaminetetraacetic acid, 1 μg/ml each of leupeptin and pepstatin, 1 mmol/L benzamidine, and 1 mmol/L sodium orthovanadate, pH 10] for 30 minutes on ice. Lysates were clarified by microcentrifugation at 13,000 rpm for 10 minutes at 4°C.
The Phoenix packaging cell line was similarly transfected with the wild-type and mutant 6 Myc-NPM-ALK retroviral constructs. Medium was replaced 24 hours after transfection. Forty-eight hours after transfection the medium was removed, filtered through a 0.45-μm filter, and then polybrene was added to a final concentration of 4 μg/ml. Rat1A fibroblasts were then infected with the viral supernatant. After 48 hours, the medium containing retrovirus was removed and replaced by selection medium containing 5 μg/ml of puromycin.
Immunoblotting
Protein expression was analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting using standard methods. RC20 anti-phosphotyrosine antibodies conjugated to horseradish peroxidase were purchased from Transduction Laboratories (Lexington, KY). Polyclonal anti-phosphotyrosine antibody was a gift from Dr. Steven Wiley, University of Utah. The 9E10 monoclonal antibody recognizing the myc epitope was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Goat anti-rabbit and anti-mouse secondary antibodies coupled to horseradish peroxidase were purchased from Pierce (Rockford, IL).
Immunohistochemical Staining
Immunohistochemical staining for ALK-1 and c-Myc co-expression was performed on paraffin-embedded tissues received from the Children’s Hospital Tissue Network (Columbus, OH) obtained from patients enrolled on the Children’s Cancer Group treatment protocol 5941, which is used currently for the treatment of CD30-positive anaplastic large cell (T cell and null cell) lymphomas in children. Fifteen cases of ALK-positive ALCLs, three cases of ALK-negative T large cell lymphoma, and three cases of T-cell lymphoblastic lymphoma were examined. Paraffin sections were mounted on Fisherbrand/Plus Superfrost slides, heated at 60°C for 30 minutes, deparaffinized, and rehydrated. ALK-1 (DAKO, Carpinteria, CA) staining was performed first after antigen epitope retrieval (3 minutes in an electric pressure cooker followed by 30 minutes of cooling in Tris buffer, pH 9.5). The slides were stained with ALK-1 (1:25 dilution) using a Ventana automated immunostainer (Ventana, Tucson, AZ) using a diaminobenzidine detection. The slides were then double-stained with c-Myc (N-262) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:70 dilution using the Ventana automated immunostainer with the Ventana Blue (alkaline phosphate) detection kit. Appropriate positive and negative controls were performed with each staining procedure.
Staining results were interpreted by one author (SLP). Neoplastic cells were scored on the basis of morphology and ALK-1 staining (brown chromagen staining) in the nucleus and cytoplasm. Co-expression of c-Myc was identified by co-staining of nuclear structures with the blue chromagen.
Double-label immunohistochemistry was also performed on formalin-fixed cell blocks from two ALK-positive ALCL cell lines, Karpas 299 and SUDHL-1, as well as a non-ALK-expressing cell line, MAC2A. The cell lines were kindly provided by Dr. Megan Lim (University of Utah) and were originally obtained from the American Tissue Culture Collection (Rockville, MD). All cell lines were maintained in RMPI 1640 (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/L l-glutamine, and 100 U/ml of penicillin/streptomycin (Life Technologies, Inc., Gaithersburg, MD) at 37°C and 5% CO2. Cells were pelleted and fixed in 10% buffered formalin and embedded in paraffin for cell-block preparations. Staining was performed on sections from the cell blocks as described above using a alkaline-phosphatase blue detection for the c-Myc antibody and diaminobenzidine detection for the ALK-1 antibody.
Real-Time PCR
C-myc transcript levels were determined in the wild-type and mutant NPM-ALK-expressing cell lines. Total RNA was isolated from Rat1A cell lines transduced with wild-type and mutant NPM-ALK retroviruses. First strand cDNA was generated in a 40-μl reverse transcription reaction and was purified using a QIAquick PCR purification kit (Qiagen, Chatsworth, CA). Real-time PCR was performed in a fluorescence temperature cycler (LightCycler LC24; Idaho Technology, Idaho Falls, ID). Individual 10-μl reactions contained 2 mmol/L MgCl2, 0.25 mg/ml bovine serum albumin, 50 mmol/L Tris-HCl, pH 8.3, 200 μmol/L dNTPs, 5 μmol/L each primer, 1:30,000 dilution in TE, pH 8, of SYBR Green I (Molecular Probes, Eugene, OR), and 1 μl of an 1:1:8 mix of TaqDNA Polymerase, TaqStart Antibody (Clontech Laboratories, Palo Alto, CA), and Taq Enzyme Diluent (Idaho Technology, Idaho Falls, ID) and 50 ng of purified cDNA template. Rat c-myc primer sequences were as follows. Forward primer: 5′ AGC TCG CCC AAA TCC TGT AC 3′ (exon 2), and reverse primer: 5′ TGC TGG TGA GTA GAG ACA TGG 3′ (exon 3). After an initial 95°C denaturation, the amplification program consisted of 40 four-step cycles of heating at 20°C/second to 95°C with a 0-second hold, cooling at 20°C/second to 55°C with a 0-second hold, heating at 10°C/second to 72°C with a 15-second hold and heating at 20°C/second to 85°C with a 0-second hold.
Standards for determining PCR product copy number were prepared from PCR products that were purified using a QIAquick PCR purification kit (Qiagen, Chatsworth, CA), and diluted. Transcript copy numbers in cell lines were calculated by comparing the fluorescence of PCR products of unknown concentration with the fluorescence of serial dilutions of the external standard. Only fluorescence values measured in the log-linear phase of amplification were used for sample quantification. A best-fit log-linear fluorescence curve was calculated for each sample, and these curves in turn were used to estimate the number of transcripts in individual samples. All samples were run in triplicate, and c-myc expression was normalized to the expression of the housekeeping gene, PER2.
Results
Conditional Dimerization of GyrB-ALK Induces Phosphorylation of the Fusion in Vitro
The GyrB-ALK fusion construct (Figure 1) ▶ was generated using a standard PCR cloning strategy, and consisted of the myc epitope-tagged E. coli DNA GyrB coumermycin-binding domain fused to the cytoplasmic domain of ALK. The portion of ALK contained in this fusion is identical to that present in NPM-ALK chimera. To test the fusion construct, we expressed chimeric protein in an in vitro transcription/translation reaction. Lysates were treated for 30 minutes with varying concentrations of coumermycin. The proteins were then resolved by SDS-PAGE and were visualized by Western blotting with anti-phosphotyrosine and anti-myc antibodies (Figure 2) ▶ . Figure 2 ▶ shows that coumermycin induced tyrosine phosphorylation of a protein the expected size of the Myc-GyrB-ALK fusion. Low levels of kinase tyrosine phosphorylation were observed in the absence of drug (lanes 1 and 2). A marked increase in phosphorylation was seen at 100 nmol/L of coumermycin. This result suggests that a small degree of background kinase tyrosine phosphorylation occurs in the absence of drug. As has been seen for other conditional dimerizing systems, higher levels of drug decrease the degree of ALK activation (lanes 4 and 5). This may result from the altered stoichiometry of GyrB to drug.
Figure 1.

Structure of 6 Myc-GyrB-ALK.
Figure 2.

ALK is activated by the addition of coumermycin in vitro. Top: Myc-GyrB-ALK was transcribed and translated in vitro using reticulocyte lysates. Newly synthesized protein was then either mock-treated or treated with increasing concentrations of coumermycin for 30 minutes at 30°C. After treatment, the level of tyrosine phosphorylation of ALK was determined by immunoblotting with RC-20 anti-phosphotyrosine (α pTyr) antibody. Changes in the level of ALK tyrosine phosphorylation were seen suggesting coumermycin-induced autophosphorylation of GyrB-ALK. Bottom: A 5-μl aliquot of each transcription-translation reaction was analyzed by immunoblotting with 9E10 anti-Myc mAb (α Myc) as a control for the amount of Myc-GyrB-ALK present in each reaction.
Conditional Dimerization of a GyrB-ALK Fusion Induces Phosphorylation in Vivo
To determine whether conditional dimerization occurred in vivo, we transiently transfected Rat1A cells with the Myc-GyrB-ALK expression construct described above. Cells were then treated for 30 minutes with coumermycin concentrations ranging from 0 to 100 μmol/L. Cellular proteins were then separated by SDS-PAGE and the level of phosphorylation was determined by Western blotting with anti-phosphotyrosine antibodies (Figure 3) ▶ . As was seen in vitro (Figure 2) ▶ , basal tyrosine phosphorylation of the ALK fusion was seen in the absence of drug (lane 1). Peak tyrosine phosphorylation occurred at coumermycin concentrations of between 1 to 10 μmol/L. At concentrations more than 10 μmol/L, decreased phosphorylation of GyrB-ALK was seen, most likely because of inhibition of dimer formation with excess drug. The difference in the coumermycin concentration required for maximal tyrosine phosphorylation of GyrB-ALK in vivo and in vitro is likely because of differences in cell permeability, protein expression levels, and/or competing binding reactions. Notably, an increase in tyrosine phosphorylation of additional cellular proteins in the presence of drug (indicated with arrows), paralleled that seen with the ALK fusion.
Figure 3.

ALK dimerization leads to an increase in tyrosine phosphorylation of proteins in vivo. Top: Rat1A fibroblasts were transiently transfected with the Myc-GyrB-ALK expression construct. Forty-eight hours after transfection cells were either mock-treated or treated with increasing concentrations of coumermycin for 30 minutes at 37°C. Cells were then lysed, and tyrosine-phosphorylated proteins were analyzed by immunoblotting with polyclonal anti-phosphotyrosine (α pTyr) antibody. With increasing concentrations of coumermycin, increases in tyrosine phosphorylation of Myc-GyrB-ALK and additional cellular proteins (arrows) were seen. Bottom: An equivalent amount of total cellular protein was analyzed separately for expression of Myc-GyrB-ALK with 9E10 anti-Myc mAb (α Myc) as a control for protein loading.
c-Myc Protein Expression Is Induced by GyrB-ALK Activation
We hypothesized that constitutive ALK tyrosine phosphorylation seen in the absence of drug treatment when GyrB-ALK was highly expressed potentially masked more significant increases in the degree of ALK phosphorylation after exposure to coumermycin. To further explore the mechanisms underlying the constitutive activation of GyrB-ALK, gene dosage was modified during the transient transfections of Myc-GyrB-ALK. Rat1A cells were transiently transfected with between 0 to 12 μg of the Myc-GyrB-ALK expression construct in the absence of coumermycin. Cells were lysed and an equivalent amount of protein from each transfection was resolved by SDS-PAGE and visualized by immunoblotting with anti-phosphotyrosine and anti-myc antibodies. Figure 4 ▶ shows that the degree of ALK autophosphorylation increased with increasing fusion protein expression except at the highest level of expression. This result suggests that the degree of constitutive phosphorylation of the construct will be a percentage of maximal induction after coumermycin treatment. Additionally, while performing the gene dosage studies, we observed an increase in the expression level of another protein in our anti-myc antibody immunoblot (Figure 4 ▶ , open circle). The level of this protein increased with increasing Myc-GyrB-ALK dosage up to 4 μg of DNA, and then its level remained constant. If this protein had been a degradation product of Myc-GyrB-ALK, we would have expected its expression level to remain a constant fraction of the total Myc-GyrB-ALK protein concentration. Because it increased only up to a point and then stabilized after GyrB-ALK tyrosine phosphorylation, we investigated if it was cellular c-Myc detected by the 9E10 antibody.
Figure 4.

c-Myc protein expression is induced by GyrB-ALK activation. To determine basal level of kinase activation in the absence of coumermycin, Rat1A fibroblasts were transiently transfected with 0 to 12 μg of the Myc-GyrB-ALK expression construct. Cells were then lysed and equivalent amounts of lysate were analyzed by immunoblotting with 9E10 anti-Myc mAb (α Myc) and RC-20 anti-phosphotyrosine (α pTyr) antibodies. With increasing expression, increasing autophosphorylation of Myc-GyrB-ALK was seen. While performing gene dosage studies, we observed an increase in a protein the approximate size of c-Myc on 9E10 immunoblots (open circle).
c-myc RNA Expression Is Induced by NPM-ALK Activation
To validate the observations seen in cell line analyses with the Myc-GyrB-ALK fusion (Figure 4) ▶ , we examined the expression of c-myc RNA in cells stably expressing both the wild-type and mutant NPM-ALK chimeras by retroviral transduction. Rat1A cells expressing each of these constructs are shown in Figure 5 ▶ . Cells expressing the wild-type kinase showed a transformed phenotype, whereas those expressing the mutant appeared morphologically identical to the parent Rat1A cell line. Total RNA was isolated from each cell line, and quantitative real-time PCR was performed to determine c-myc transcript number. As shown in Table 1 ▶ , c-myc transcript number was induced 3.4-fold in cells expressing wild-type NPM-ALK compared to cells expressing mutant kinase. This finding confirmed the results observed in cell lines with the GyrB-ALK fusion, and suggests that c-Myc expression is induced by NPM-ALK activation.
Figure 5.

Morphology of cells expressing NPM-ALK. Rat1A fibroblasts were stably transduced with wild-type 6 Myc-NPM-ALK, or mutant 6 Myc-NPM-ALK. A: Rat1A cells transduced with wild-type 6 Myc-NPM-ALK showed morphological features of transformation. B: Rat1A cells transduced with mutant 6 Myc-NPM-ALK were not transformed.
Table 1.
c-myc Transcript Number in Cell Lines Expressing Wild-Type and Mutant NPM-ALK
| Mean c-myc copy number (normalized) | |
|---|---|
| Rat1A cell line expressing wild-type NPM-ALK | 49.2 |
| Rat1A cell line expressing mutant NPM-ALK | 14.4 |
| Expression ratio: wild-type versus mutant NPM-ALK | 3.4 |
c-Myc and ALK Are Co-Expressed in 100% of Patient ALK+ Lymphomas
At the inception of this study we aimed to elucidate downstream targets of NPM-ALK activation in childhood lymphoma. Therefore, we sought to extend our observations in cell line studies to pediatric patient samples. Formalin-fixed, paraffin-embedded lymphoma biopsies from pediatric patients enrolled on Children’s Cancer Group therapeutic trials for ALCL were examined by immunohistochemistry for expression of c-Myc and ALK (Figure 6) ▶ . Fifteen of 15 (100%) of ALCLs staining positive for ALK also co-expressed c-Myc. In contrast, no non-ALCL specimens (0 of 6) that were ALK-negative had detectable c-Myc by immunohistochemistry.
Figure 6.
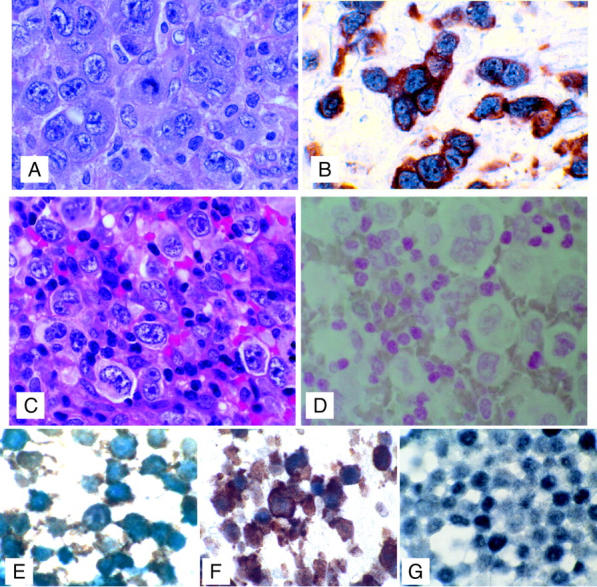
Co-expression of ALK and c-Myc in tumor cells. Top: A: H&E-stained section of CD30-positive, ALK-positive, T-cell ALCL. B: Dual immunohistochemical staining demonstrating co-expression of c-Myc (blue, alkaline phosphatase detection, nuclear stain) with ALK (t2;5) translocation protein product (detected with brown diaminobenzidine, cytoplasmic stain) in tumor cells. Representative of 15 ALK-positive cases. Middle: C: H&E-stained section of CD30-positive, ALK-negative, T-cell ALCL. D: Dual immunohistochemical staining (with nuclear red counterstain) demonstrating lack of c-Myc (blue alkaline phosphatase detection, nuclear stain) and ALK (t2;5 translocation protein product detected with brown diaminobenzidine, cytoplasmic stain) in tumor cells. Representative of six ALK-negative cases (three T-large cell lymphomas and three T-cell lymphoblastic lymphomas). Bottom: Double-label immunohistochemical staining of formalin-fixed cell lines with ALK-1 (diaminobenzidine detection, brown) and c-Myc (alkaline phosphatase blue, blue-black color) showing co-expression of ALK-1 and c-Myc in the ALK-positive ALCL cell lines SUDHL-1 (E) and Karpas 299 (F) but expression of c-Myc only in the non-ALK-expressing T-cell lymphoma cell line MAC2A (G). Original magnifications: ×100 (A–D); ×1000 (E–G).
Immunohistochemical analysis of defined ALK-positive lymphoma cell lines (Karpas 299 and SUDHL-1) and a T-cell lymphoma cell line that is ALK-negative (MAC2A) showed similar results to that seen in the clinical specimens with co-expression of c-Myc and ALK-1 in the Karpas 299 and SUDHL-1 cells and expression of c-Myc only in the MAC2A cells (Figure 6) ▶ .
Discussion
To investigate downstream signaling pathways, we created a regulatable form of ALK, designed to mimic the dimerization and aberrant kinase activation seen in NPM-ALK-expressing lymphomas. Specifically, we developed a conditionally dimerizable GyrB-ALK construct that showed an increase in tyrosine phosphorylation in response to the drug coumermycin both in vitro and in vivo. Coumermycin has been previously used to activate a number of signaling molecules including Raf, ASK1, JAK2, members of the STAT family of transcription factors, and L-selectin. 25 The pattern of ALK autophosphorylation followed a characteristic bell-shaped dose response to coumermycin suggesting that the phosphory-lation was the direct or indirect result of dimerization, and reflected ALK activation. As seen in other systems, maximal kinase activation occurred at coumermycin concentrations ranging from 100 nmol/L to 1 μmol/L. Other cellular proteins were also tyrosine phosphorylated after GyrB-ALK activation with coumermycin, supporting a model in which aberrant phosphorylation of cellular proteins by ALK plays an essential role in lymphomagenesis.
Interestingly, we saw a significant amount of ALK phosphorylation in the absence of coumermycin. This basal tyrosine phosphorylation may be because of intra- or intermolecular autophosphorylation. Because similar phosphorylation was also observed when the fusion protein was expressed in reticulocyte lysates, it seems less likely to be because of phosphorylation by other tyrosine kinases present in the cell. Because the level of fusion protein phosphorylation decreased with high doses of coumermycin, the basal phosphorylation in the absence of drug was likely because of aberrant dimerization and autophosphorylation in trans. This inhibition of autophosphorylation with high doses of coumermycin has been observed with other GyrB fusions, and may be because of altered stoichiometry when the concentration of drug greatly exceeds the number of GyrB binding sites. 28
In addition to changes in the phosphorylation status of ALK, we saw an increase in the level of tyrosine phosphorylation of other cellular proteins after the addition of coumermycin. Furthermore, a cross-reacting protein was detected by the antibody directed against the myc epitope tag of the fusion construct that we hypothesized was c-Myc. To determine whether these results could be validated in cell lines and tumor samples expressing the actual wild-type NPM-ALK chimera, c-myc transcript copy number was quantitated by real-time PCR, and patterns of c-Myc and ALK expression were determined by directly immunostaining tumor samples. A 3.4-fold increase in c-myc transcript number was seen in cell lines expressing wild-type NPM-ALK. Most importantly, ALK and c-Myc were co-expressed in 100% of ALK-positive tumor samples, and in none of the ALK-negative lymphomas.
Several groups have identified candidate signaling molecules that mediate the effects of ALK. A direct physical association between NPM-ALK and the adaptor signaling proteins, GRB2, SHC, PLC-γ, and IRS-1 has been seen. 16,19,20 Further, the interaction between ALK and PLC-γ has been shown to be essential because mutation of the ALK residue responsible for PLC-γ binding eliminates the transforming potential of NPM-ALK. 19 NPM-ALK has also recently been shown to initiate anti-apoptotic signaling through its activation of the phosphatidylinositol 3-kinase/Akt pathway. 21 Increased expression of c-myc, fos, and jun gene products have been seen in cell lines transformed with NPM-ALK. 17 Our analysis suggests a specific role for c-Myc not only in cell lines, but also in ALK-mediated lymphomagenesis.
c-Myc plays an important role in regulating cellular growth and in mediating apoptosis. 29,30 Deregulated c-Myc expression has been implicated in the pathogenesis of many hematopoietic and nonhematopoietic malignancies including Burkitt’s lymphoma, follicular lymphomas, invasive ductal breast carcinoma, prostate and colon carcinoma, melanoma, and medulloblastoma. 31-33 The expression of c-Myc protein and RNA, as well as genomic alterations involving the myc gene locus have been studied in many lymphomas. Although immunohistochemical analyses have demonstrated frequent expression of c-Myc protein in Hodgkin’s disease, 34 reports of c-Myc expression in non-Hodgkin’s lymphomas have been variable. Mitani and colleagues 35 reported intense c-Myc immunostaining in 86% of cases of non-Hodgkin’s lymphoma, however most analyses have shown low levels of c-Myc protein expression by immunohistochemistry in the majority of non-Hodgkin’s lymphomas. 36-39 Even in the case of Burkitt’s lymphoma in which gene overexpression occurs by chromosomal translocation, c-Myc protein staining has been infrequently observed in tumor cells. 36 It has been hypothesized that protein degradation leads to a rapid decline in intracellular levels of protein. 40 Benharroch and colleagues 38 found c-Myc protein expression by immunohistochemistry in 72.4% of cases of Hodgkin’s disease compared to only 36.8% of cases of non-Hodgkin’s lymphoma, which included a spectrum of low- to high-grade tumors.
In a report by Sakalidou and colleagues 39 analyzing the expression of c-Myc protein in childhood lymphomas, expression again was most frequent in Hodgkin’s disease in which 29% of cases showed c-Myc expression in greater than 20% of cells, compared to expression in only 1 of 18 cases of non-Hodgkin’s lymphoma (interestingly, this was the only case of anaplastic T-cell lymphoma in this report). Similarly, variable patterns of c-Myc immunoreactivity in lymphomas were reported by Jack and colleagues 37 in 1986. In that report, the expression pattern of c-Myc was determined in 51 lymphomas, the majority of which were large cell lymphomas and Hodgkin’s disease. C-Myc expression occurred with the greatest frequency in T-cell immunoblastic lymphoma (5 of 5 cases), and Hodgkin’s disease (7 of 12 cases). Although in that study ALK expression was not determined, the overall frequency of ALK expression in ALCLs is 83% for pediatric tumors and 31% for adult tumors. 9 Therefore, it is likely some or all of those c-Myc-expressing T-cell lymphomas were in fact ALK+ ALCLs.
The functional consequence of c-Myc expression in ALK+ lymphoma is unclear. c-Myc is a transcription factor that drives expression of a wide range of genes associated with proliferation, including cyclin D1, telomerase, Cdc25A, and ornithine decarboxylase. 41-43 Our results are consistent with previous studies, which suggest that increased c-Myc protein is not common to all lymphomas, but rather restricted to certain subtypes. Whereas c-Myc expression has been associated with a poor prognosis in adult diffuse large B-cell lymphoma, 44 ALK+ lymphomas have a favorable prognosis. Presumably the favorable prognosis in ALK+ lymphomas arises from the complete pattern of aberrant gene activation, rather than solely from c-Myc expression.
In summary, we have created a regulatable system to identify downstream pathways dysregulated in ALK+ lymphoma. Our analyses in ALK-expressing cell lines, and tumor samples revealed an increase in c-myc mRNA and protein. C-Myc immunoreactivity was specifically increased in ALK+ lymphomas. These findings suggest a specific role for c-Myc in ALK-mediated lymphomagenesis, and may provide a new immunohistochemical marker for this subset of ALCLs.
Acknowledgments
We thank the University of Utah Research Foundation for Seed Grant support, the members of the Virshup Laboratory for helpful discussions, Stephan Morris for NPM-ALK constructs, Michael Farrar for Gyrase B constructs, Sheryl Tripp (MT-ASCP) and the ARUP Institute for performing immunostaining, Megan Lim for provision of ALK-expressing cell lines, and Erica Vielhaber for her assistance with the retroviral constructs.
Footnotes
Address reprint requests to Elizabeth A. Raetz, M.D., Mount Sinai Hospital, Division Pediatric Hematology-Oncology, 1 Gustave Levy Place, New York, NY 10029. E-mail: elizabeth.raetz@mssm.edu.
Supported by the National Institutes of Health (Mentored Clinical Scientist Development Award K08 HD01215-01 to E. A. R.), the Huntsman Cancer Foundation, and core facility support was provided by the Cancer Center (support grant 2P30CA42014).
References
- 1.Sandlund JT, Downing JR, Crist WM: Non-Hodgkin’s lymphoma in childhood. N Engl J Med 1996, 334:1238-1248 [DOI] [PubMed] [Google Scholar]
- 2.Brugieres L, Deley MC, Pacquement H, Meguerian-Bedoyan Z, Terrier-Lacombe MJ, Robert A, Pondarre C, Leverger G, Devalck C, Rodary C, Delsol G, Hartmann O: CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood 1998, 92:3591-3598 [PubMed] [Google Scholar]
- 3.Reiter A, Schrappe M, Parwaresch R, Henze G, Muller-Weihrich S, Sauter S, Sykora KW, Ludwig WD, Gadner H, Riehm H: Non-Hodgkin’s lymphomas of childhood and adolescence: results of a treatment stratified for biologic subtypes and stage—a report of the Berlin-Frankfurt-Munster Group. J Clin Oncol 1995, 13:359-372 [DOI] [PubMed] [Google Scholar]
- 4.Sandlund JT, Pui CH, Santana VM, Mahmoud H, Roberts WM, Morris S, Raimondi S, Ribeiro R, Crist WM, Lin JS, Mao L, Berard C, Hutchison R: Clinical features and treatment outcome for children with CD30+ large-cell non-Hodgkin’s lymphoma. J Clin Oncol 1994, 12:895-898 [DOI] [PubMed] [Google Scholar]
- 5.Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A: ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999, 93:2697-2706 [PubMed] [Google Scholar]
- 6.Kadin ME, Morris SW: The t(2;5) in human lymphomas. Leuk Lymphoma 1998, 29:249-256 [DOI] [PubMed] [Google Scholar]
- 7.Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, Haralambieva E, Pulford K, Pileri S, Morris SW, Mason DY, Delsol G: ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998, 91:2076-2084 [PubMed] [Google Scholar]
- 8.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT: Fusion of a kinase gene, ALK, to a nucleolar protein gene NPM, in non-Hodgkin’s lymphoma. Science 1994, 263:1281-1284 [DOI] [PubMed] [Google Scholar]
- 9.Drexler HG, Gignac SM, von Wasielewski R, Werner M, Dirks WG: Pathobiology of NPM-ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia 2000, 14:1533-1559 [DOI] [PubMed] [Google Scholar]
- 10.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T: Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14:439-449 [DOI] [PubMed] [Google Scholar]
- 11.Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP: ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14:2175-2188 [DOI] [PubMed] [Google Scholar]
- 12.Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi T, Takeshita M, Mori N, Fujimoto J, Miyauchi J, Mikata A, Nanba K, Takami T, Yamabe H, Takano Y, Izumo T, Nagatani T, Mohri N, Nasu K, Satoh H, Katano H, Fujimoto J, Yamamoto T, Mori S: Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood 1995, 86:1954-1960 [PubMed] [Google Scholar]
- 13.Borer RA, Lehner CF, Eppenberger HM, Nigg EA: Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989, 56:379-390 [DOI] [PubMed] [Google Scholar]
- 14.Chan WY, Liu QR, Borjigin J, Busch H, Rennert OM, Tease LA, Chan PK: Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28:1033-1039 [DOI] [PubMed] [Google Scholar]
- 15.Dumbar TS, Gentry GA, Olson MO: Interaction of nucleolar phosphoprotein B23 with nucleic acids. Biochemistry 1989, 28:9495-9501 [DOI] [PubMed] [Google Scholar]
- 16.Bischof D, Pulford K, Mason DY, Morris SW: Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol 1997, 17:2312-2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wellmann A, Doseeva V, Butscher W, Raffeld M, Fukushima P, Stetler-Stevenson M, Gardner K: The activated anaplastic lymphoma kinase increases cellular proliferation and oncogene up-regulation in rat 1a fibroblasts. EMBO J 1997, 11:965-972 [DOI] [PubMed] [Google Scholar]
- 18.Kuefer MU, Look AT, Pulford K, Behm FG, Pattengale PK, Mason DY, Morris SW: Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood 1997, 90:2901-2910 [PubMed] [Google Scholar]
- 19.Bai RY, Dieter P, Peschel C, Morris SW, Duyster J: Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol 1998, 18:6951-6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimoto J, Shiota M, Iwahara T, Seki N, Satoh H, Mori S, Yamamoto T: Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci USA 1996, 93:4181-4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J: Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 2000, 96:4319-4327 [PubMed] [Google Scholar]
- 22.Weiss A, Schlessinger J: Switching signals on or off by receptor dimerization. Cell 1998, 94:277-280 [DOI] [PubMed] [Google Scholar]
- 23.Schlessinger J: Cell signaling by receptor tyrosine kinases. Cell 2000, 103:211-225 [DOI] [PubMed] [Google Scholar]
- 24.van der Geer P, Hunter T, Lindberg RA: Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 1994, 10:251-337 [DOI] [PubMed] [Google Scholar]
- 25.Farrar MA, Olson SH, Perlmutter RM: Coumermycin-induced dimerization of GyrB-containing fusion proteins. Methods Enzymol 2000, 327:421-429 [DOI] [PubMed] [Google Scholar]
- 26.Amara JF, Clackson T, Rivera VM, Guo T, Keenan T, Natesan S, Pollock R, Yang W, Courage NL, Holt DA, Gilman M: A versatile synthetic dimerizer for the regulation of protein-protein interactions. Proc Natl Acad Sci USA 1997, 94:10618-10623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR: Controlling signal transduction with synthetic ligands. Science 1993, 262:1019-1024 [DOI] [PubMed] [Google Scholar]
- 28.Farrar MA, Alberol I, Perlmutter RM: Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 1996, 383:178-181 [DOI] [PubMed] [Google Scholar]
- 29.Pelengaris S, Rudolph B, Littlewood T: Action of Myc in vivo—proliferation and apoptosis. Curr Opin Genet Dev 2000, 10:100-105 [DOI] [PubMed] [Google Scholar]
- 30.Prendergast GC: Mechanisms of apoptosis by c-Myc. Oncogene 1999, 18:2967-2987 [DOI] [PubMed] [Google Scholar]
- 31.Hecht JL, Aster JC: Molecular biology of Burkitt’s lymphoma. J Clin Oncol 2000, 18:3707-3721 [DOI] [PubMed] [Google Scholar]
- 32.Nesbit CE, Tersak JM, Prochownik EV: MYC oncogenes and human neoplastic disease. Oncogene 1999, 18:3004-3016 [DOI] [PubMed] [Google Scholar]
- 33.Bruggers CS, Tai KF, Murdock T, Sivak L, Le K, Perkins SL, Coffin CM, Carroll WL: Expression of the c-Myc protein in childhood medulloblastoma. J Pediatr Hematol Oncol 1998, 20:18-25 [DOI] [PubMed] [Google Scholar]
- 34.Jiwa NM, Kanavaros P, van der Valk P, Walboomers JM, Horstman A, Vos W, Mullink H, Meijer CJ: Expression of c-myc and bcl-2 oncogene products in Reed-Sternberg cells independent of presence of Epstein-Barr virus. J Clin Pathol 1993, 46:211-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitani S, Sugawara I, Shiku H, Mori S: Expression of c-myc oncogene product and ras family oncogene products in various human malignant lymphomas defined by immunohistochemical techniques. Cancer 1988, 62:2085-2093 [DOI] [PubMed] [Google Scholar]
- 36.Hermann M, Scholman HJ, Marafioti T, Stein H, Schriever F: Differential expression of apoptosis, Bcl-x and c-Myc in normal and malignant lymphoid tissues. Eur J Haematol 1997, 59:20-30 [DOI] [PubMed] [Google Scholar]
- 37.Jack AS, Kerr IB, Evan G, Lee FD: The distribution of the c-myc oncogene product in malignant lymphomas and various normal tissues as demonstrated by immunocytochemistry. Br J Cancer 1986, 53:713-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benharroch D, Yermiahu T, Geffen DB, Prinsloo I, Gopas J, Segal S, Aboud M: Expression of c-myc and c-ras oncogenes in the neoplastic and non-neoplastic cells of Hodgkin’s disease. Eur J Haematol 1995, 55:178-183 [DOI] [PubMed] [Google Scholar]
- 39.Sakalidou A, Kanavaros P, Tzardi M, Kalmanti M: The expression of myc and ras oncogene protein in childhood lymphomas. Anticancer Res 1996, 16:487-491 [PubMed] [Google Scholar]
- 40.Ciechanover A, DiGiuseppe JA, Bercovich B, Orian A, Richter JD, Schwartz AL, Brodeur GM: Degradation of nuclear oncoproteins by the ubiquitin system in vitro. Proc Natl Acad Sci USA 1991, 88:139-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR: Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 2000, 97:3260-3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang CV: c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999, 19:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuhmacher M, Kohlhuber F, Holzel M, Kaiser C, Burtscher H, Jarsch M, Bornkamm GW, Laux G, Polack A, Weidle UH, Eick D: The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res 2001, 29:397-406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang CC, Liu YC, Cleveland RP, Perkins SL: Expression of c-Myc and p53 correlates with clinical outcome in diffuse large B-cell lymphomas. Am J Clin Pathol 2000, 113:512-518 [DOI] [PubMed] [Google Scholar]