

Abstract
Proximal tubular renal epithelial cells may contribute to the pathogenesis of renal interstitial fibrosis in diabetes by generation of cytokines such as transforming growth factor (TGF)-β1. We have previously demonstrated that proximal tubular renal epithelial cell TGF-β1 synthesis may be modulated by elevated glucose concentration and by cytokines such as platelet-derived growth factor (PDGF). The aim of the current study was to characterize the mechanism by which glucose and PDGF synergistically stimulate the generation of TGF-β1. Addition of either 25 mmol/L of d-glucose or low-dose PDGF increased TGF-β1 mRNA expression without stimulation of TGF-β1 protein synthesis. In contrast sequential stimulation with 25 mmol/L of d-glucose for 48 hours followed by low-dose (25 ng/ml) PDGF led to a significant increase in TGF-β1 synthesis. Elevated glucose concentration stimulated de novo gene transcription as assessed by stimulation of a TGF-β1 promoter-luciferase construct. This led to induction of a poorly translated TGF-β1 transcript determined by polysome analysis. PDGF at low dose did not influence TGF-β1 transcription, but led to alteration in TGF-β1 mRNA stability and translation. Without a previous glucose-induced increase in the amount of TGF-β1 transcript, PDGF did not stimulate significant TGF-β1 protein synthesis. At a high dose (100 ng/ml) PDGF stimulated TGF-β1 synthesis independent of glucose concentration. This was associated with increased TGF-β1 gene transcription and alteration in TGF-β1 mRNA translational efficiency. In conclusion the data suggests that in diabetic nephropathy, the role of glucose is to lower the threshold at which a stimulus such as PDGF stimulates TGF-β1 protein synthesis. The data also suggest that independent regulation of TGF-β1 transcription and translation by glucose and PDGF account for their synergistic effect on TGF-β1 protein synthesis. We hypothesize that the role of glucose in diabetic nephropathy is to prime the kidney for an injurious response to other stimuli.
Diabetic nephropathy is now the commonest cause of end-stage renal disease and accounts for 30 to 40% of all patients requiring renal replacement therapy. Furthermore, the incidence of diabetic nephropathy continues to increase, in part because of the improved survival of type 2 diabetic patients as the cardiovascular mortality in this group declines. 1 Clinically incipient nephropathy is first manifest by the onset of persistent microalbuminuria, after which, overt diabetic nephropathy is heralded by the appearance of persistent proteinuria. Subsequently, there is a progressive decline in glomerular filtration rate resulting, within 5 years, in end-stage renal disease in 50% of those patients with proteinuria. 2 The pathology of the renal lesions are similar in type I and II diabetes, 3 although it has been suggested that there is more heterogeneity in type II diabetes. 4 Studies analyzing structural-functional relationships have demonstrated that the development of proteinuria correlates with the degree of mesangial expansion. 5,6 Although diabetic nephropathy was traditionally considered a primarily glomerular disease, it is now widely accepted that the rate of deterioration of function correlates best with the degree of renal tubulointerstitial fibrosis. 5,7 This suggests that although in the majority of patients the primary event is a condition manifest by glomerular changes resulting in proteinuria, the long-term outcome is determined by events in the renal interstitium. With the increasing awareness of the importance of these pathological interstitial changes, interest has focused on the role of cells, such as the epithelial cells of the proximal tubule (PTC), in the initiation of fibrosis.
Recently epidemiological evidence has indicated that complications of diabetes are related to poor glycemic control. 8,9 Because only 30% of diabetic patients develop nephropathy, however, hyperglycemia alone may be insufficient to initiate pathological changes. A key molecule that has been implicated in the pathogenesis of diabetic nephropathy is the profibrotic cytokine transforming growth factor (TGF)-β1. Elevated glucose concentration as seen in diabetes is known to stimulate TGF-β1 synthesis within the glomerulus. 10 In contrast we have previously demonstrated that exposure of cultured human renal proximal tubular epithelial cells to elevated d-glucose concentrations increased the expression of a poorly translated TGF-β1 transcript without any associated change in TGF-β1 protein synthesis. 11 Although diabetes is primarily a metabolic condition, recent studies have also implicated macrophage infiltration in the pathogenesis of diabetic nephropathy. In vivo studies on streptozotocin-induced diabetic rats have demonstrated prominent macrophage infiltration. 12,13 In addition studies of renal biopsies taken from patients with type II diabetes have suggested that macrophages and their products are involved in the initiation of the pathological changes of human diabetic nephropathy. 14 We have demonstrated that application of macrophage-derived products such as platelet-derived growth factor (PDGF) or interleukin-1β to d-glucose-primed cells, triggered TGF-β1 protein secretion. 15,16 This is consistent with the clinical observation that hyperglycemia alone although leading to adaptive changes within the glomerulus, is not sufficient to initiate interstitial fibrosis and progressive renal disease in all patients. Furthermore, it clearly illustrates the complexity involved in the regulation of TGF-β1 synthesis, in that its generation in a biologically active form may be regulated independently at the level of gene transcription, mRNA processing and translation, secretion in its latent form, and finally its activation, which has been demonstrated in many cell types.
The aim of the current study was to characterize the mechanism by which glucose and PDGF synergistically stimulate the generation of TGF-β1. Initial studies were aimed at examining the regulation of TGF-β1 transcription, mRNA stability, translational efficiency, and de novo protein synthesis in response to either elevated glucose concentrations or PDGF alone. Subsequently we examined how these aspects of the regulation of TGF-β1 synthesis were modified by addition of both stimuli sequentially. The data demonstrate that elevated glucose concentrations stimulated de novo TGF-β1 gene transcription but the increase in the amount of mRNA was not associated with increased protein synthesis because its translational efficiency was poor. At high doses, PDGF increased TGF-β1 gene transcription, translational efficiency, and de novo protein synthesis. PDGF at low doses produced a transient alteration in translational efficiency of TGF-β1 mRNA without any associated increase in TGF-β1 gene transcription, and no increase in TGF-β1 protein synthesis unless cells were pretreated with elevated glucose concentrations. Under these conditions, PDGF altered the stability of the glucose-induced TGF-β1 transcript and caused a sustained increase in its translational efficiency. The data suggest that in diabetes, the role of glucose is to lower the threshold at which a second stimulus such as PDGF may result in the generation of the profibrotic cytokine TGF-β1, and suggests a mechanism by which hyperglycemia may prime the kidney for an injurious response to other stimuli.
Materials and Methods
Cell Culture
All experiments were performed using HK-2 cells (no. CRL-2190; American Type Culture Collection, Rockville, MD) that are human proximal tubular epithelial cells immortalized by transduction with human papilloma virus 16 E6/E7 genes. 17 Cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F12 (Life Technologies, Paisley, UK) supplemented with 10% bovine calf serum (Biological Industries Ltd., Cumbernauld, UK), Hepes, l-glutamine, insulin, transferrin, sodium selenite, and hydrocortisone (Sigma, Poole, UK). Fresh growth medium was added to cells every 3 to 4 days until confluent. All experiments were performed using cells at passage 30 or below and, with the exception of the cells used for transfection and reporter gene analysis, cells were growth-arrested in serum- and insulin-free medium for 72 hours before use in experiments. All experiments were performed in serum- and insulin-free conditions.
Analysis of TGF-β1 mRNA and Protein Quantification
TGF-β1 mRNA was quantified by Northern blotting. Total RNA was extracted from the cells with Trireagent (Sigma). Twenty μg of total RNA were separated by electrophoresis in a denaturing 1% agarose/formaldehyde gel before overnight capillary transfer to a positively charged nylon membrane (Hybond N+; Amersham Pharmacia, Little Chalfont, UK) and crosslinking by ultraviolet irradiation. RNA standards (Promega Ltd., Southampton, UK) were used to determine transcript length. Membranes were hybridized with a 32P internally labeled TGF-β1 cDNA probe synthesized by DNA random hexanucleotide priming. Intensity of signal was detected with high-performance autoradiography film (Hyperfilm, Amersham) quantified using a digital image analyzer (Chemi Doc; Bio-Rad Laboratories, Richmond, CA), before stripping the probe from the blot in boiling 0.1% (w/v) sodium dodecyl sulfate and rehybridizing a GAPDH probe, with signal detection as above.
Total TGF-β1 in the cell culture supernatant was measured by specific enzyme-linked immunosorbent assay (R&D Systems, Abingdon, UK) of cell culture supernatant samples collected from growth-arrested HK-2 cells, stimulated under serum-free conditions. This assay has <1% cross-reactivity for TGF-β2 and TGF-β3. TGF-β1 concentration was normalized to number of cells, determined by cell counting with a hemocytometer. Data are expressed as pg TGF-β1/ml per 105 cells.
Analysis of Transcriptional Activity
The TGF-β1 promoter-luciferase construct pGL3-TGF-β1 + 11/−1362 was generated as previously described. 18 The pSV-β-galactosidase control vector was purchased from Promega.
For transfection of the reporter construct, 1.75 × 106 HK-2 cells were seeded per 35-mm dish (this density of cells produced a 90% confluence monolayer the following day). The next day cells were transfected with 1 μg of plasmid pGL3-TGF-β1 + 11/−1362, and 1 μg of pSV-β-galactosidase plasmid to act as an internal control for transfection efficiency, using the mixed lipofection reagent FuGene 6 (Roche, Lewes, East Sussex, UK) at a ratio of 3 μl of Fugene to 1 μg of DNA in serum-free and insulin-free medium. Twenty-four hours after transfection, cells were stimulated with 25 mmol/L of d-glucose or PDGF for 12 hours. After lysis of the cells in Reporter Lysis Buffer (Promega) β-galactosidase activity was determined by colorimetric assay (β-Galactosidase Enzyme Assay System, Promega), and luciferase content was quantified by glow-type luminance assay (Bright-Glo, Promega) with a standard curve of recombinant luciferase (Promega). Data are expressed as pg luciferase/10 mU β-galactosidase.
Analysis of mRNA Stability
For quantitation of the rate of decay of TGF-β1 mRNA, growth-arrested cells were stimulated with 25 mmol/L of d-glucose for 48 hours. Transcription was then inhibited by the addition of actinomycin D and cells were stimulated with PDGF-AA (25 ng/ml). No PDGF was added in control experiments. Toxicity of actinomycin D was assessed using cellular ATP measurements, and the maximal nontoxic dose was used (0.5 μg/ml). The rate of mRNA degradation was subsequently determined using the PCR MIMIC system (Clontech Laboratories Inc. Cambridge BioScience, Cambridge, UK), using TGF-β1-specific primers and TGF-β1 composite primers were designed that amplified a 342-bp fragment of the BamHI/EcoRI fragment of v-erbB DNA (Table 1) ▶ . The primary polymerase chain reaction (PCR) amplification, using the composite primers and the secondary PCR amplification with the TGF-β1 gene-specific primers were performed for 30 cycles according to the manufacturer’s protocol. The TGF-β1 PCR MIMIC cDNA was purified with the Wizard PCR Preps DNA Purification System (Promega Ltd.) and the concentration of the purified product was measured at an OD260nm. A 10-fold TGF-β1-PCR MIMIC dilution series was set up and standard curves were constructed for each sample of target cDNA to be measured. For each sample, 2 μl of target cDNA was included in each PCR reaction mix together with 2 μl of one of the dilutions from the PCR MIMIC dilution series. The PCR reaction was performed as previously described. 15 From the standard curve generated in this manner the dilution at which the experimental cDNA and the PCR MIMIC cDNA reached equivalence was ascertained for each sample. 19 The minimum concentration of mRNA in the original, extracted sample was then calculated.
Table 1.
The Sequences of the Amplification Primers for Quantitative and Reverse Transcriptase-PCR
| Gene | Primer sequence | Product size |
|---|---|---|
| TGF-β1 | 5′ ACCGGCCTTTCCTGCTTCTCA 3′ | 288 bp |
| 5′ CGCCCGGGTTATGCTGGTTGT 3′ | ||
| TGF-β1 Mimic | 5′ ACCGGCCTTTCCTGCTTCTCACAGCCGTCCCAAGTTTCGTG 3′ | 342 bp |
| 5′ CGCCCGGGTTATGCTGGTTGTGTGCCCCTGCCCATTTCTGT 3′ | ||
| α-actin | 5′ CCTTCCTGGGCATGGAGTCCT 3′ | 204 bp |
| 5′ GGAGCAATGATCTTGATCTT 3′ |
The influence of de novo protein synthesis on PDGF-mediated alterations in TGF-β1 mRNA stability was examined in parallel experiments with PDGF being added to glucose-primed, actinomycin D-treated cells, either in the presence or absence of cycloheximide. The rate of mRNA degradation was subsequently determined by isolating total cellular RNA, and performing reverse transcriptase-PCR as previously described. 15
Analysis of Efficiency of Translation
Polysome analysis was performed as previously described. 20 Approximately 1.3 × 107 growth-arrested cells per experiment were trypsinized, pelleted, and extracted in 1 ml of ice-cold lysis buffer (10 mmol/L Tris-Cl, pH 8.0, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.5% Nonidet P-40, 500 U/ml−1 recombinant RNAsin (Promega Corp, Madison, WI). Nuclei were removed by centrifugation at 3000 × g for 2 minutes and the supernatant transferred to a new tube supplemented with 100 μg/ml of cycloheximide, 1 mmol/L of phenylmethyl sulfonyl fluoride, 10 mmol/L of dithiothreitol, and 0.5 mg/ml of heparin and then centrifuged at 13,000 × g for 5 minutes to remove mitochondria and membrane debris. The supernatant was layered onto a 10-ml 15 to 40% linear sucrose gradient containing 10 mmol/L Tris-Cl, pH 7.5, 140 mmol/L NaCl, 1.5 mmol/L MgCl2, 10 mmol/L dithiothreitol, 10 mmol/L cycloheximide, and 0.5 mg/ml heparin in a Polyallomer centrifuge tube (Beckman, High Wycome, Bucks, UK) and centrifuged using an SW41Ti rotor at 36,000 rpm for 2 hours at 4°C. The gradient was fractionated into 22 0.5-ml fractions each supplemented with 1% sodium dodecyl sulfate, 10 mmol/L ethylenediaminetetraacetic acid, and 200 μg/ml proteinase K, and incubated at 37°C for 30 minutes to degrade endogenous nucleases. Subsequently the fractions were mixed with phenol:chloroform:isoamyl alcohol (24:24:1) and the aqueous layer containing the RNA removed. A 5% aliquot of each fraction was analyzed by electrophoresis in a 3% agarose gel to ensure that the RNA was not degraded, and that the tRNA and rRNA species were appropriately distributed through the gradient. RNA was precipitated overnight from the remainder of each fraction with 1 ml of 100% ethanol, 50 μl of 3 mol/L sodium acetate, and 1 μl of glycogen (Sigma) and washed once with 70% ethanol before air-drying. Fifty percent of samples of each fraction were run as a single large Northern blot, as above. A single 2.4-kb band was detectable on autoradiography, this was quantified by densitometry (Chemi Doc, Bio-Rad, Hemel Hempstead Herts, UK). Data are expressed as percentage of the total TGF-β1 mRNA for that experiment in each fraction. The actual blot is also shown for comparison.
To further examine the rate of translation of TGF-β1 we measured incorporation of radioactive amino acids into newly synthesized protein by metabolic labeling with detection by TGF-β1 immunoprecipitation and autoradiography. Forty μCi of 3H-radiolabeled amino acid mixture (1000 μCi/ml, Amersham) were added to growth-arrested cells in 25-cm2 flasks. Supernatant samples were subsequently collected for TGF-β1 immunoprecipitation. Before immunoprecipitation supernatant samples were precleared with 25 μl of protein A-Sepharose beads (Sigma) and 0.25 μg of normal rabbit immunoglobulin at 4°C for 1 hour with constant mixing. The supernatant was removed from the beads, and 2 μg of polyclonal anti-TGF-β1 antibody (Autogen Bioclear Calne, Wilts, UK) was added to each 1 ml of cleared supernatant and incubated with constant mixing at 4°C for 2 hours. Subsequently 50 μl of protein A-Sepharose beads were added, and mixing continued for 12 hours. Samples were centrifuged, the supernatant removed, and the beads washed twice with phosphate-buffered saline (PBS). After the final centrifugation and removal of the second PBS wash, 25 μl of sodium dodecyl sulfate/β-mercaptoethanol loading buffer was added, and the samples heated to 95°C for 10 minutes before sodium dodecyl sulfate-polyacrylamide gel electrophoresis in a 10% gel. Gels were fixed by incubating in 10% acetic acid/40% methanol overnight and then soaked in scintillant (Amplify, Amersham Life Science) for 30 minutes and dried before visualization of immunoprecipitated TGF-β1 by autoradiography.
Results
Alteration in TGF-β1 Protein Generation
The addition of 25 mmol/L of d-glucose for up to 96 hours did not increase TGF-β1 concentration in the cell culture supernatant (Figure 1) ▶ . Similarly, although there was a trend toward increase in TGF-β1 after addition of 25 ng/ml of either PDGF-AA (Figure 1A) ▶ or PDGF-BB (Figure 1B) ▶ , this was not statistically significant (control, 140.1 ± 21.4 pg/ml/105 cells; 25 mmol/L glucose, 164.3 ± 19.7 pg/ml/105 cells; 5 mmol/L glucose + 25 ng/ml PDGF-AA, 190.8 ± 37.2 pg/ml/105 cells; 5 mmol/L glucose + 25 ng/ml PDGF-BB, 177.8 ± 14.7 pg/ml/105 cells; P = NS for all versus control).
Figure 1.
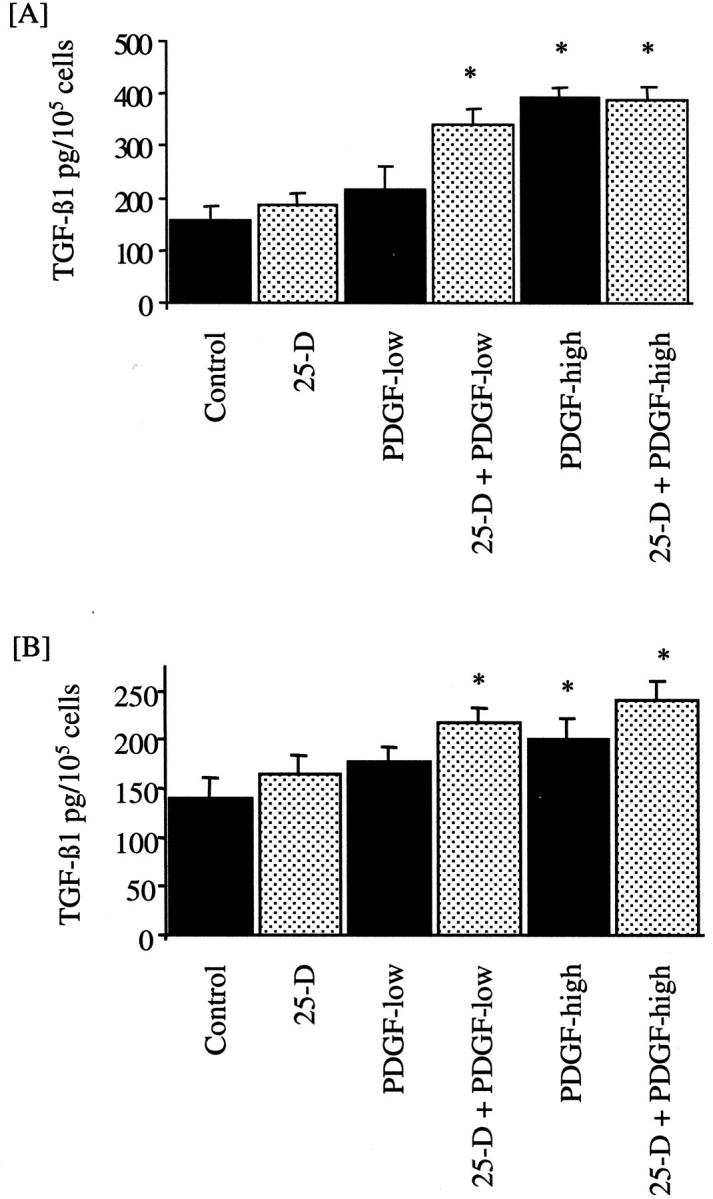
Effect of 25 mmol/L of d-glucose and PDGF on TGF-β1 generation. d-glucose (25 mmol/L) was added to confluent monolayers of growth-arrested HK-2 cells under serum-free conditions (dotted bars). After 48 hours of exposure to 25 mmol/L of d-glucose, the medium was changed and cells exposed to 25 mmol/L of d-glucose for a further 48 hours or 25 mmol/L of d-glucose together with PDGF [PDGF-AA (A) or PDGF-BB (B)] at a dose of 25 ng/ml (25D-PDGF-low) or 100 ng/ml (25D-PDGF-high). Subsequently TGF-β1 was quantified by enzyme-linked immunosorbent assay. In parallel experiments 5 mmol/L of d-glucose was added to confluent monolayers of growth-arrested HK-2 cells under serum-free conditions (solid bars). After 48 hours the medium was changed and cells exposed to 5 mmol/L of d-glucose for a further 48 hours or 5 mmol/L of d-glucose together with PDGF [PDGF-AA (A) or PDGF-BB (B)] at a dose of either 25 ng/ml (PDGF-low) or 100 ng/ml (PDGF-high). Cell counts of the monolayer were performed and the results expressed as TGF-β1 per 105 cells. Results represent mean ± SE of six individual experiments.
Addition of either isoform of PDGF at a dose of 100 ng/ml led to significant stimulation of TGF-β1 in the cell culture supernatant (Figure 1) ▶ , which was independent of glucose concentration (100 ng/ml PDGF-AA and 5 mmol/L glucose, 343.7 ± 15.9 pg/ml TGF-β/105 cells; 100 ng/ml PDGF-AA and 25 mmol/L glucose, 342.0 ± 19.2 pg/ml TGF-β/105 cells; 100 ng/ml PDGF-BB and 5 mmol/L glucose, 201.9 ± 20.5 pg/ml TGF-β/105 cells; 100 ng/ml PDGF-BB and 25 mmol/L glucose, 240.7 ± 20.5 pg/ml TGF-β/105 cells; P < 0.05 versus control for each, n = 6 for each). TGF-β1 was only detected after acidification of the samples indicating that TGF-β1 was produced in its latent form.
Although addition of either 25 mmol/L of d-glucose or low-dose PDGF did not affect the generation of TGF-β1, sequential stimulation with 25 mmol/L of d-glucose for 48 hours followed by a low dose (25 ng/ml) of either PDGF-AA or PDGF-BB led to a significant increase in TGF-β1 concentration in the cell culture supernatant (25 mmol/L of d-glucose followed by 25 ng/ml of PDGF-BB, 217.9 ± 15.7 pg/ml/105 cells; 25 mmol/L of d-glucose followed by 25 ng/ml of PDGF-AA, 300.9 ± 25.6 pg/ml/105 cells; P < 0.05 for both PDGF isoforms versus control). As was the case with high-dose PDGF, TGF-β1 was only detected after acidification of the samples indicating that TGF-β1 was produced in its latent form. Sequential addition of an osmotic control (5 mmol/L d-glucose + 20 mmol/L l-glucose) and PDGF did not result in stimulation of TGF-β1 generation (data not shown). In contrast to the effect seen after sequential stimulation by glucose and low-dose PDGF, sequential stimulation with 25 mmol/L of d-glucose followed by the higher dose of PDGF did not augment the PDGF response (Figure 1) ▶ .
Influence of 25 mmol/L of d-Glucose
Although the addition of 25 mmol/L of d-glucose did not alter total TGF-β1 concentration in the cell culture supernatant, there was an increase in the amount of TGF-β1 mRNA (Figure 2) ▶ . Northern blotting showed increased expression of a 2.4-kb TGF-β1 transcript apparent 96 hours after addition of 25 mmol/L of d-glucose. Addition of 25 mmol/L of d-glucose to cells transiently transfected with the TGF-β1 promoter-luciferase construct led to a 2.9-fold increase in relative luciferase activity (Figure 3) ▶ suggesting that the increased amount of TGF-β1 transcript was the result of de novo gene transcription (n = 4, P < 0.002). The efficiency of translation of glucose-induced TGF-β1 mRNA was determined by polysome analysis to assess the number of ribosomes associated with the mRNA. The results suggest that the induced 2.4-kb TGF-β1 mRNA is poorly translated with 38% of the total TGF-β1 mRNA localizing to a region of the gradient corresponding to polysomes (Figure 4) ▶ . This was not significantly different to translational efficiency of TGF-β1 mRNA in control unstimulated cells in which 25% of the total TGF-β1 mRNA localized to a region of the gradient corresponding to polysomes.
Figure 2.

Effect of glucose and PDGF on amount of TGF-β1 mRNA. Confluent monolayers of HK-2 cells were growth-arrested and exposed to 25 mmol/L of d-glucose (96 hours) or PDGF-AA alone (48 hours), or exposed sequentially to 25 mmol/L of d-glucose for 48 hours followed by PDGF-AA for a further 48 hours. After RNA isolation TGF-β1 mRNA was detected by Northern analysis. Equal mRNA loading was confirmed by stripping the blot and reprobing it for GAPDH.
Figure 3.

Alteration in TGF-β1 promoter activity. Ninety percent confluent monolayers of HK-2 cells were transfected with the TGF-β1 promoter-luciferase plasmid construct pGL3-TGF-β1 + 11/−1362 and the control plasmid pSV-β-galactosidase before exposure to 5 mmol/L of d-glucose (control), 25 mmol/L of d-glucose (dotted bar), 25 ng/ml of PDGF-AA (hatched bar), or 100 ng/ml of PDGF-AA (solid bar). After 12 hours total cell protein was extracted and analyzed for luciferase and β-galactosidase activity. Data are expressed as luciferase activity divided by β-galactosidase activity normalized to the values obtained for control cells. Results represent mean ± SE for four individual experiments.
Figure 4.

Effect of 25 mmol/L d-glucose on translational efficiency of TGF-β1 mRNA. Confluent monolayers of HK-2 cells were growth-arrested and exposed to 5 mmol/L of d-glucose (B) or 25 mmol/L of d-glucose (C) for 12 hours before extraction of the cytosol and separation of mRNA on a sucrose gradient as described in the Materials and Methods section. A: Five percent sample of each fraction of a gradient examined by 3% agarose gel electrophoresis and detection of RNA by UV fluorescence in the presence of ethidium bromide. Fractions 1 to 11 represent tRNAs, free ribosomal subunits, monosomes, and untranslated mRNAs in the form of messenger ribonucleoprotein particles. Fractions 12 to 22 represent polysomes of increasing size. B and C: Northern blot of the fractionated mRNA, probed for TGF-β1. D: Graphical representation of the polysome distribution of TGF-β1 mRNA, expressed as percentage of the total TGF-β1 mRNA detected on a given blot in each fraction with control. Five mmol/L of d-glucose shown as shaded area, and 25 mmol/L of d-glucose enclosed by solid line.
Effect of High-Dose PDGF
Addition of PDGF-AA for 48 hours at a dose of 100 ng/ml led to an increase in the amount of the 2.4-kb TGF-β1 mRNA transcript (Figure 2) ▶ . Similarly at this dose PDGF-AA led to a twofold increase in relative luciferase activity of the TGF-β1 promoter-luciferase construct (n = 4, P < 0.02) (Figure 3) ▶ . In contrast to the effect of elevated d-glucose, high-dose PDGF-AA also altered translation efficiency of the TGF-β1 mRNA, because 49% of the total TGF-β1 mRNA 12 hours after addition of PDGF localized to a region of the gradient corresponding to heavy polysomes. Forty-eight hours after addition of PDGF this trend was reversed with 75% of the total TGF-β1 mRNA localizing to the nonpolysome fractions (Figure 5) ▶ .
Figure 5.

Effect of 100 ng/ml of PDGF on TGF-β1 translation. Confluent monolayers of HK-2 cells were growth-arrested and exposed to 5 mmol/L of d-glucose for 12 hours (A) or 100 ng/ml of PDGF-AA for 12 hours (B) or 48 hours (C) before extraction of the cytosol and separation of mRNA on a sucrose gradient as described in the Materials and Methods section. D: Graphical representation of the polysome distribution of TGF-β1 mRNA, expressed as percentage of the total TGF-β1 mRNA detected on a given blot in each fraction. Shaded area represents control, solid line represents PDGF at 12 hours, and dashed line represents PDGF at 48 hours.
These data suggest that PDGF caused a rapid but transient increase in TGF-β1 mRNA translation. The time course of TGF-β1 protein synthesis was therefore further analyzed by metabolic radiolabeling of TGF-β1 with 3H amino acids after PDGF stimulation to characterize kinetics of de novo TGF protein synthesis. These experiments demonstrated stimulation of de novo TGF synthesis during the first 24-hour period after addition of PDGF (Figure 6A) ▶ . Beyond this time point de novo radiolabeled TGF generation returned to baseline, thus confirming the transient nature of induction of translational efficiency of TGF-β1 mRNA demonstrated by polysome analysis.
Figure 6.

Time course of TGF-β1 synthesis after glucose and PDGF stimulation. Confluent monolayers of HK-2 cells were growth-arrested before stimulation with 100 ng/ml of PDGF-AA (A), 25 ng/ml PDGF-AA (B), 25 mmol/L d-glucose (C), or stimulated sequentially with 25 mmol/L of d-glucose for 48 hours before addition of 25 ng/ml of PDGF-AA (D). 3H-labeled amino acids were added to the culture medium at the start of each time point shown. At the end of the time point the medium was removed and the cells discarded. TGF-β1 protein was extracted from the supernatant by immunoprecipitation as described in the Materials and Methods section. After immunoprecipitation radiolabeled TGF-β1, representative of de novo synthesized and secreted TGF-β1, was detected by autoradiography.
Effect of PDGF at Low Dose
Northern analysis demonstrated increased expression of the 2.4-kb TGF-β1 transcript after addition of 25 ng/ml of PDGF (Figure 2) ▶ . In contrast to the higher dose of PDGF, the increase in TGF-β1 mRNA expression was not associated with any change in TGF-β1 transcription as assessed by luciferase activity of the TGF-β1 promoter construct (Figure 3) ▶ . As with the higher dose of PDGF there was a rapid and transient increase in translational efficiency as 51% of total TGF-β1 mRNA localized to the heavy polysome fraction 12 hours after the addition of PDGF (Figure 7) ▶ . Metabolic labeling experiments confirmed lack of stimulation of de novo TGF-β1 synthesis after addition of low-dose PDGF (Figure 6B) ▶ .
Figure 7.

Effect of 25 ng/ml of PDGF on TGF-β1 translation. Confluent monolayers of HK-2 cells were growth-arrested and exposed to 5 mmol/L D-glucose for 12 hours (A) or 25 ng/ml PDGF-AA for 12 hours (B) or 48 hours (C) before extraction of the cytosol and separation of mRNA on a sucrose gradient as described in the methods section. (D) Graphical representation of the polysome distribution of TGF-β1 mRNA, expressed as percentage of the total TGF-β1 mRNA detected on a given blot in each fraction. Shaded area represents control, solid line PDGF at 12hours and dashed line PDGF at 48 hours.
Our previous studies using primary cultures of human renal proximal tubular epithelial cells have demonstrated a synergistic effect of low-dose PDGF and elevated glucose concentration on TGF-β1 protein generation. 11 To confirm the relevance of the above mechanistic observations to primary cell cultures, translational efficiency of TGF-β1 mRNA in response to low-dose PDGF-AA was examined. Primary proximal tubular epithelial cells were isolated, characterized, and cultured as previously described. 11 As with HK-2 cells, addition of 25 ng/ml of PDGF-AA led to a rapid increase in translational efficiency because 40% of total TGF-β1 mRNA localized to the heavy polysome fraction 12 hours after addition of PDGF in comparison to 11% in the control (Figure 8) ▶ .
Figure 8.

Effect of 25 ng/ml of PDGF on TGF-β1 translation in primary cultures. Confluent monolayers of primary cultures of renal proximal tubular epithelial cells were growth-arrested and exposed to 5 mmol/L of d-glucose for 12 hours (A) or 25 ng/ml of PDGF-AA for 12 hours (B) before extraction of the cytosol and separation of mRNA on a sucrose gradient as described in the Materials and Methods section. C: Graphical representation of the polysome distribution of TGF-β1 mRNA, expressed as percentage of the total TGF-β1 mRNA detected on a given blot in each fraction. Shaded area represents control, solid line represents PDGF at 12 hours.
Sequential Stimulation with 25 mmol/L of d-Glucose and Low-Dose PDGF
The predominant effect of glucose was to stimulate TGF-β1 gene transcription, whereas at the low-dose PDGF principally influences translational efficiency. We therefore postulated that induction of TGF-β1 after sequential stimulation by 25 mmol/L of d-glucose and low-dose PDGF was because of glucose-dependent transcription leading to an increase in total TGF-β1 mRNA, followed by PDGF-dependent improved efficiency of its translation. In support of this although 48 hours after addition of 25 ng/ml of PDGF alone there was no alteration in translational efficiency as assessed by polysome analysis, sequential stimulation with 25 mmol/L of d-glucose for 48 hours followed by 25 ng/ml of PDGF for a further 48 hours led to increased TGF-β1 mRNA translational efficiency as 59% of total TGF-β1 mRNA localized to the heavy polysome fraction (Figure 9) ▶ . In comparison stimulation with 25 ng/ml of PDGF resulted in only 20% of total TGF-β1 mRNA localizing to the heavy polysome fraction.
Figure 9.

Effect of sequential stimulation with 25 mmol/L of d-glucose and 25 ng/ml of PDGF on TGF-β1 translational efficiency. Confluent monolayers of HK-2 cells were growth-arrested and exposed to 25 ng/ml of PDGF-AA for 48 hours with (A) or without (B) previous exposure to 25 mmol/L of d-glucose for 48 hours. Cytosol extraction and separation of mRNA on a sucrose gradient was done as described in the Materials and Methods section. C: Graphical representation of the polysome distribution of TGF-β1 mRNA, expressed as percentage of the total TGF-β1 mRNA detected on a given blot in each fraction. Shaded area represents exposure to PDGF alone, solid line represents sequential stimulation with 25 mmol/L of d-glucose and PDGF.
To further investigate this hypothesis the kinetics of de novo TGF-β1 protein synthesis was characterized after sequential stimulation. There was no increase in de novo radiolabeled TGF-β1 generation during the initial 72-hour period of glucose stimulation (Figure 6C) ▶ . In contrast to the transient effect of high-dose PDGF, after addition of low-dose PDGF to 25 mmol/L of d-glucose-pretreated cells, there was a sustained increase in de novo radiolabeled TGF generation throughout the next 48-hour period (Figure 6D) ▶ .
To further examine the mechanism by which low-dose PDGF increased TGF-β1 protein synthesis in 25 mmol/L of d-glucose-primed cells, we examined its effects on TGF mRNA decay (Figure 10) ▶ . Quantification of TGF-β1 mRNA by MIMIC PCR demonstrated steady decline in TGF-β1 mRNA levels after stimulation with 25 mmol/L of d-glucose alone 12 hours after addition of actinomycin D (15.56 ± 2.36 pmol/L to 3.21 ± 1.96 pmol/L; mean ± SEM, n = 3, P = 0.007). In contrast, after induction of TGF-β1 mRNA by 25 mmol/L of d-glucose, and inhibition of transcription by actinomycin D for 1 hour, quantitation of TGF-β1 mRNA revealed that the amount of mRNA remained unchanged for up to 12 hours after addition of PDGF (preactinomycin, 16.8 ± 2.6 pmol/L; 12 hours after PDGF, 14.4 ± 1.9 pmol/L; mean ± SEM, n = 3). Increased stability of glucose-induced TGF-β1 mRNA by PDGF was further supported by Northern analysis demonstrating a synergistic effect of combined stimulation on the amount of total TGF-β1 mRNA. The mechanism by which PDGF altered the rate of TGF-β1 mRNA decay was examined by stimulation of glucose-primed, actinomycin D-treated cells with PDGF-AA (25 ng/ml) in the presence of cycloheximide, or addition of cycloheximide alone (Figure 11) ▶ . PDGF in the presence of cycloheximide prevented TGF-β1 mRNA degradation, whereas cycloheximide treatment alone did not stabilize TGF-β1 mRNA. These data suggest that PDGF increased the stability of glucose-induced TGF-β1 mRNA, and that this effect was not related to new protein synthesis.
Figure 10.

Effect of PDGF on stability of glucose-induced TGF-β1 mRNA. TGF-β1 mRNA was induced by addition of 25 mmol/L of d-glucose to confluent monolayer of HK2 cells for 48 hours. Subsequently transcription was inhibited by the addition of actinomycin D (5 μg/ml). Subsequently cells were exposed to 25 ng/ml of PDGF-AA (solid diamonds) or 5 mmol/L of d-glucose alone (open boxes) and total cellular RNA was isolated throughout the following 12 hours. Stability of TGF-β1 mRNA was determined by quantitative mimic PCR (B). Data represents mean ± SEM, n = 3; *, P < 0.05.
Figure 11.

Effect of cycloheximide on PDGF increased stability of TGF-β1. Confluent cells were growth-arrested before exposure to 25 mmol/L of d-glucose for 48 hours. Further gene transcription was inhibited with actinomycin D treatment, after which cells were exposed to 25 ng/ml of PDGF-AA, 25 ng/ml of PDGF-AA and cycloheximide, or cycloheximide alone. At 0, 6, and 12 hours RNA was extracted. After reverse transcriptase-PCR, TGF-β1 and α-actin PCR products were separated by flat bed electrophoresis in 3% agarose gels (B) and the results expressed as the densitometric ratios of TGF-β1 mRNA to α-actin normalized for the unstimulated ratio (C). One representative experiment of three is shown.
Discussion
An important goal in understanding the etiology of diabetic nephropathy is to determine factors that trigger the initiation of progressive renal dysfunction because this may allow us to understand why only 30% of diabetic patients develop nephropathy. TGF-β1 has emerged as a mediator that is implicated in the pathogenesis of fibrosis in both glomerular and interstitial compartments of the kidney and has been particularly associated with diabetic nephropathy. 21 Much of the work in our laboratory has focused on the control of TGF-β1 gene expression and protein synthesis by PTC. We have demonstrated that TGF-β1 production may be controlled at the levels of transcription, translation, secretion of preformed protein, and activation of the latent protein to its active form. 11,15,22,23 Furthermore once generated the consequent alteration in cell function is also influenced by regulation of events beyond receptor-ligand binding, such as expression of the Smad family of signaling intermediates. 24 Previously we have demonstrated that the addition of 25 mmol/L of d-glucose to PTC caused an increase in TGF-β1 mRNA, but no increase in TGF-β1 protein production. 11,16 The mechanism by which glucose increased the amount of TGF-β1 mRNA was however not defined. In the current study we have demonstrated transcriptional activation of the TGF-β1 promoter by an elevated concentration of d-glucose. Numerous studies, including those performed with renal mesangial cells have demonstrated increased transcription of the TGF-β1 gene in response to glucose. 25 Interestingly in these cells, unlike the data derived from human proximal tubular epithelial cells, transcriptional activation was also associated with stimulation of protein synthesis. It is clear however that regulation of TGF-β1 may be cell-type-specific and our data demonstrate that for human proximal tubular epithelial cells transcriptional activation does not necessarily equate with stimulation of translation and de novo protein synthesis. This is consistent with the clinical observation that not all diabetic patients develop progressive renal fibrosis, which would be the likely outcome if hyperglycemia per se stimulated the generation of increased amounts of TGF-β1 protein.
It is now clear that elements within both the 5′UTR and the 3′UTR of mRNA are involved in control of the initiation of translation. 25 Significantly this is also true for the control of translation of TGF-β1 mRNA. 26-28 The three transcripts that have been reported for TGF-β1 (2.4, 1.9, and 1.4 kb) differ in their translational efficiency and also differ in the length of their GC-rich 5′UTR. 29-31 The predominant TGF-β1 transcript is 2.4 kb, with the other truncated forms being found after injury or in malignantly transformed cells. 29,30,32,33 This suggests that under physiological conditions the regulation of translation of the larger transcript is a key step in control of TGF-β1 synthesis and activity. In other systems 5′UTR with a rich GC content tend to be highly structured and are thought to inhibit translation by impeding the progress of the 40S scanning ribosomal subunit toward the initiation codon. It is thought that translation of the truncated TGF-β1 transcripts may therefore be more efficient owing to the deletion of inhibitory elements in the 5′ leader sequence. In our experiments exposure of PTC to elevated glucose concentrations led to induction of the poorly translated 2.4-kb transcript. Polysome analysis confirmed poor translational efficiency because the transcript localized to a region of the sucrose gradient corresponding to polysomes with few associated ribosomes as has previously described for endogenous TGF-β1 mRNA in mammary carcinoma cells. 34 For an average ribosome spacing of 80 to 100 nucleotides, the 1200 bases of the TGF-β1 ORF would have 12 to 15 ribosomes if it were well translated. This suggests that the renal proximal tubular epithelial cell has a large excess capacity for synthesis of the TGF-β1 protein that is unused. The poor translatability of the predominant TGF-β1 mRNA in the human proximal tubular epithelial cells may explain the discrepancy between mRNA and protein level, as has been described in other cell types. 35,36
In addition to TGF-β1, PDGF has also been implicated in the pathogenesis of glomerulosclerosis, fibrosis/wound repair, 37-39 as well as the microvascular complications associated with diabetes mellitus. 40,41 Autocrine and paracrine interactions between TGF-β1 and PDGF have been demonstrated in numerous cell types. In this respect TGF-β1 is known to regulate PDGF receptor expression in mesangial cells and fibroblasts, 42-44 as well as its secretion in fibroblasts. 45 Likewise induction of TGF-β1 synthesis by PDGF has been reported in cells of diverse origin. 46,47 In the current study we present evidence that at high doses PDGF may influence numerous facets of TGF-β1 synthesis. It induced de novo gene transcription, increased translational efficiency of the transcript generated, and therefore stimulated de novo protein synthesis. In contrast to the effect of PDGF at high doses, at reduced doses we have demonstrated that the predominant effect was to increase TGF-β1 mRNA translational efficiency. The small nonsignificant increase in TGF-β1 protein concentration in cell culture supernatant after stimulation by PDGF at the lower dose suggests that in the absence of a stimulus that concurrently increases the steady state of TGF-β1 mRNA, there is insufficient mRNA to lead to a significant increase in protein synthesis. This is further supported by the observation that when the amount of TGF-β1 mRNA was increased by glucose, modification of its translational efficiency by subsequent addition of PDGF lead to significant and sustained de novo synthesis of TGF-β1 protein. Mechanistically it seems that increased TGF-β1 protein synthesis by PDGF was related to alteration TGF-β1 mRNA stability, as the low dose of PDGF had a positive effect on the stability of glucose-induced TGF-β1 mRNA. This was associated with an increase in TGF-β1 mRNA translational efficiency and was not affected by cycloheximide. Interestingly PDGF has also been shown to mediate increased translational efficiency of nerve growth factor, by a mechanism involving mRNA stabilization. 48 Previous data have also suggested that alteration of TGF-β1 mRNA stability may be involved in facilitation of glucose-induced mRNA translation. Sequential stimulation by 25 mmol/L of d-glucose and interleukin-1β also increased the stability of d-glucose-induced TGF-β1 mRNA, as assessed by actinomycin D chase experiments. 15 In contrast incubation of d-glucose-primed cells with TNF-α did not result in TGF-β1 production, and unlike interleukin-1β and PDGF, TNF-α did not influence TGF-β1 mRNA stability. These observations suggest that posttranscriptional modification of TGF-β1 mRNA may be limited to specific cytokine stimulation, which may therefore play a crucial role in the control of TGF-β1 protein synthesis. The data also suggests that TGF-β1 is produced predominantly in its latent form at least in vitro. At present the mechanisms of activation of latent TGF-β1 in vivo are poorly understood. Activation of TGF-β in vitro occurs by extremes of pH, heat, and deglycosylation, these are however unlikely to be physiologically important. More recent studies have identified activation mechanisms involving the action of either plasmin 49-51 or thrombospondin. 52-54 In addition activation of latent TGF-β1 has been characterized in heterotypic co-culture systems. 51,55,56 Overall it is therefore difficult to make generalizations regarding the generation of biologically active TGF-β1 from in vitro results generated using homotypic cell culture systems.
Two PDGF receptor subunits denoted α and β have been described and the corresponding genes cloned. It is proposed that these receptor subunits either exist separately or form rapidly reversible complexes in the absence of PDGF but associate into stable dimers when PDGF is present. Depending on the isoform of PDGF present, αα, αβ, or ββ receptor dimers are formed. PDGF-BB can bind to all three receptor combinations, whereas PDGF-AA binds only to αα receptors. 57 The data in the current study demonstrate a difference in the potency of the PDGF isoforms used with significantly more TGF-β1 generated after sequential stimulation by 25 mmol/L of d-glucose and PDGF-AA than 25 mmol/L of d-glucose and PDGF-BB. This is consistent with our previous observations in primary cultures of human proximal tubular epithelial cells in which sequential glucose- and PDGF-stimulated TGF-β1 secretion was inhibited by anti-PDGF α-receptor subunit antibody, suggesting that the effect of PDGF was mediated via the PDGF α-receptor subunit. 11
It is known that the long CG rich 5′UTR of TGF-β1 mRNA is involved in regulation of its translation. More specifically elements spanning bases +1 to +147 of the 5′UTR have previously been shown to be involved in the inhibition of TGF-β1 translation, 26 and the binding of a cytosolic factor may regulate this inhibitory effect. 27 Interestingly it has been shown that the inhibitory effect of the putative stem loop structure of the 5′UTR sequence of TGF-β1 spanning bases +1 to +147 was unrelated to rapid degradation of the mRNA. The presence of this sequence in fact increased the stability of the mRNA. 27 This suggests that an increase in TGF-β1 mRNA stability does not always result in an increase in its translation. We have also previously demonstrated that insulin directly alters the production of TGF-β1 by renal proximal tubular cells by a posttranscriptional mechanism. This increase in TGF-β1 translation was not associated with alteration in TGF-β1 mRNA stability as assessed by actinomycin D chase. This again suggests that mechanisms other than modification of mRNA stability stimulate TGF-β1 translation. The effect of insulin was related to increased binding of a cytoplasmic protein to the putative stem loop structure +1 to +147 in the 5′UTR of TGF-β1 mRNA. This suggests that under basal conditions translation may be suppressed by constitutively expressed cytoplasmic proteins, but that once stimulated the pattern of cytoplasmic protein binding to this region is altered thus overcoming the repression of TGF-β1 mRNA translation seen in the basal state. It is apparent therefore that facilitation of TGF-β1 translation by PDGF occurred via a different mechanism to that previously reported for insulin, and that these differ again from the mechanisms repressing TGF-β1 mRNA translation in a nonstimulated setting. It is clear therefore that although TGF-β1 synthesis may be regulated at the level of transcription, translation, secretion, and activation, numerous different mechanisms that are stimulus-specific, influence TGF-β1 mRNA translation.
In conclusion the synergistic effect of glucose and cytokines on TGF-β1 synthesis suggest that in diabetic nephropathy, the role of glucose is to lower the threshold at which a second stimulus such as PDGF or interleukin-1β, may result in the generation of a profibrotic response. Because not all patients with diabetes develop progressive renal disease, understanding how factors in combination with glucose initiate and perpetuate renal fibrosis is an important area for research. From our observations we suggests that the role of glucose is to prime the kidney for an injurious response to other stimuli.
Footnotes
Address reprint requests to Dr. A. O. Phillips, Institute of Nephrology, University of Wales College of Medicine, Heath Park, Cardiff, Wales, United Kingdom CF14 4XN. E-mail: phillipsao@cf.ac.uk.
Supported by a National Kidney Research Fund Training Fellowship (to D. F.), and a Glaxo Wellcome Advanced Fellowship (to A. O. P.).
References
- 1.Ritz E, Stefanski A: Diabetic nephropathy in type II diabetes. Am J Kidney Dis 1996, 27:167-194 [DOI] [PubMed] [Google Scholar]
- 2.Hasslacher C, Ritz E, Wahl P, Michael C: Similar risks of nephropathy in patients with type I or type II diabetes mellitus. Nephrol Dial Transplant 1989, 4:859-863 [DOI] [PubMed] [Google Scholar]
- 3.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin IR: Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes 1994, 43:1046-1051 [DOI] [PubMed] [Google Scholar]
- 4.Chihara J, Takegayashi S, Taguchi T, Yokatama K, Horada T, Naito S: Glomerulonephritis in diabetic patients and its effect on the prognosis. Nephron 1986, 43:45-49 [DOI] [PubMed] [Google Scholar]
- 5.Mauer SM, Steffes MW, Ellis EE, Sutherland DER, Brown DM, Goetz FC: Structural-functional relationships in diabetic nephropathy. J Clin Invest 1984, 74:1143-1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White KE, Bilous RW: Type 2 diabetic patients with nephropathy show structural-functional relationships that are similar to type 1 disease. J Am Soc Nephrol 2000, 11:1667-1673 [DOI] [PubMed] [Google Scholar]
- 7.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Muller GA: The pathogenesis of chronic renal failure in diabetic nephropathy. Path Res Pract 1991, 187:251-259 [DOI] [PubMed] [Google Scholar]
- 8.: The Diabetes Control and Complications Trial Research Group: The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin dependant diabetes mellitus. N Engl J Med 1993, 329:977-986 [DOI] [PubMed] [Google Scholar]
- 9.: The Diabetes Control and Complications Research Group: Effect of intensive therapy on the development and progression of diabetic nephropathy in the Diabetes Control and Complications Trial. Kidney Int 1995, 47:1703-1720 [DOI] [PubMed] [Google Scholar]
- 10.Ziyadeh FN, Sharma K, Ericksen M, Wolf G: Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor beta. J Clin Invest 1994, 93:536-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips AO, Steadman R, Topley N, Williams JD: Elevated D-glucose concentrations modulate TGF-β1 synthesis by human cultured renal proximal tubular cells: the permissive role of platelet-derived growth factor. Am J Pathol 1995, 147:362-374 [PMC free article] [PubMed] [Google Scholar]
- 12.Sassy-Pringent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, Bariety J, Bruneval P: Early glomerular macrophage recruitment in streptozotocin induced diabetic rats. Diabetes 2000, 49:466-475 [DOI] [PubMed] [Google Scholar]
- 13.Young BA, Johnson RJ, Alpers CE, Eng CE, Gordon K, Floege J, Couser W: Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int 1995, 47:935-944 [DOI] [PubMed] [Google Scholar]
- 14.Furuta T, Saito T, Ootaka T, Soma J, Obara K, Keishi A, Yohinaga K: The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis 1993, 21:480-485 [DOI] [PubMed] [Google Scholar]
- 15.Phillips AO, Topley N, Steadman R, Morrisey K, Williams J: Induction of TGF-β1 synthesis in D-glucose primed human proximal tubular cells: differential stimulation by the macrophage derived pro-inflammatory cytokines IL-1β and TNFα. Kidney Int 1996, 50:1546-1554 [DOI] [PubMed] [Google Scholar]
- 16.Phillips AO, Morrisey R, Steadman R, Williams JD: Polarity of stimulation and secretion of TGF-β1 by proximal tubular cells. Am J Pathol 1997, 150:1101-1111 [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA, Torok-Storb B: HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 1994, 45:48-57 [DOI] [PubMed] [Google Scholar]
- 18.Kim SJ, Jeang KT, Glick AB, Sporn MB, Roberts AB: Promoter sequences of the human transforming growth factor β1 gene responsive to transforming growth factor β1 auto-induction. J Biol Chem 1989, 264:7041-7045 [PubMed] [Google Scholar]
- 19.Peten EP, Striker LJ, Garcia-Perez A, Striker GE: Studies by competitive PCR of glomerulosclerosis in growth hormone transgenic mice. Kidney Int 1993, 43(Suppl 39):S55-S58 [PubMed] [Google Scholar]
- 20.Garcia-Sanz JA, Mikulits W, Livingstone A, Lefkovits I, Mullner EW: Translational control: a general mechanism for gene regulation during T cell activation. FASEB J 1998, 12:299-306 [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA: Expression of transforming growth factor-β is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 1993, 90:1814-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrisey K, Evans RA, Wakefield L, Phillips AO: Translational regulation of renal proximal tubular epithelial cell transforming growth factor-β1 generation by insulin. Am J Pathol 2001, 159:1905-1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips AO, Topley N, Morrisey K, Williams JD, Steadman R: Basic fibroblast growth factor stimulates the release of pre-formed TGF-β1 from human proximal tubular cells in the absence of de-novo gene transcription or mRNA translation. Lab Invest 1997, 76:591-600 [PubMed] [Google Scholar]
- 24.Massague J: TGF-β signal transduction. Annu Rev Biochem 1998, 67:753-791 [DOI] [PubMed] [Google Scholar]
- 25.Sonenberg M: mRNA translation: influence of the 5′ and 3′ untranslated regions. Curr Opin Genet Dev 1994, 4:310-315 [DOI] [PubMed] [Google Scholar]
- 26.Kim SJ, Park K, Koellers D, Kim KY, Wakefield LM: Post-transcriptional regulation of the human transforming growth factor beta1 gene. J Biol Chem 1992, 267:13702-13707 [PubMed] [Google Scholar]
- 27.Romeo DS, Park K, Roberts AB, Sporn MB, Kim SJ: An element of the transforming growth factor-β 5′ untranslated region represses translation and specifically binds a cytosolic factor. Mol Endocrinol 1993, 7:759-766 [DOI] [PubMed] [Google Scholar]
- 28.Scotto L, Assoian RK: A GC-rich domain with bifunctional effects on mRNA and protein levels: implications for control of transforming growth factor β1 expression. Mol Cell Biol 1993, 13:3588-3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian SW, Kondaiah P, Casscells W, Roberts AB, Sporn MB: A second messenger RNA species of transforming growth factor β1 in infarcted rat heart. Cell Regul 1991, 2:241-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostrowski LE, Gray TE, Randell SH, Nettesheim P: Characterization of a third transforming growth factor β1 transcript in rat tracheal epithelial cells. Cell Growth Differ 1993, 4:985-991 [PubMed] [Google Scholar]
- 31.Romeo D, Allison RSH, Kondaiah P, Wakefield LM: Recharacterisation of the start sites for the major human transforming growth factor β1 mRNA. Gene 1997, 189:289-295 [DOI] [PubMed] [Google Scholar]
- 32.Arrick BA, Grendell RL, Griffin LA: Enhanced translational efficiency of a novel transforming growth factor β1 mRNA in human breast cancer cells. Mol Cell Biol 1994, 14:619-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson NL, Bazoberry F, Speir EH, Casscells W, Ferrans VJ, Flanders KC, Kondaiah P, Geiser A, Sporn MB: Transforming growth factor β1 in acute myocardial infarction in rats. Growth Factors 1988, 1:91-99 [DOI] [PubMed] [Google Scholar]
- 34.Allison RSH, Mumy ML, Wakefield LM: Translational control elements in the major human transforming growth factor β1 mRNA. Growth Factors 1998, 16:89-100 [DOI] [PubMed] [Google Scholar]
- 35.Fowlis DJ, Flanders KC, Duffie E, Balmain A, Akhurst RJ: Discordant transforming growth factor beta 1 RNA and protein localization during chemical carcinogenesis of the skin. Cell Growth Differ 1992, 3:81-91 [PubMed] [Google Scholar]
- 36.Lehnert SA, Akhurst RJ: Embryonic expression pattern of TGF beta type-1 RNA suggests both paracrine and autocrine mechanisms of action. Development 1988, 104:263-273 [DOI] [PubMed] [Google Scholar]
- 37.Floege J, Eng E, Young BA, Alpers CE, Barrett TB, Bowen-Pope DF, Johnson RJ: Infusion of platelet-derived growth factor or basic fibroblast growth factor induces selective glomerular mesangial cell proliferation and matrix accumulation in rats. J Clin Invest 1993, 92:2952-2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce GF, Mustoe TA, Lingelbach J, Masakowski VR, Griffin GL, Senior RM, Deuel TF: Platelet-derived growth factor and transforming growth factor beta enhance tissue repair activities by unique mechanisms. J Cell Biol 1989, 109:429-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierce GF, Vande Berg J, Rudolph R, Tarpley J, Mustoe TA: Platelet-derived growth factor-BB and transforming growth factor beta 1 selectively modulate glycosaminoglycans, collagen and myofibroblasts in excisional wounds. Am J Pathol 1991, 138:629-646 [PMC free article] [PubMed] [Google Scholar]
- 40.Guillausseau PJ, Dupuy E, Bryckaert MC, Timsit J, Chanson P, Tobelem G, Caen JP, Lubetzki J: Platelet-derived growth factor in type 1 diabetes mellitus. Eur J Clin Invest 1989, 19:172-175 [DOI] [PubMed] [Google Scholar]
- 41.Mizutani M, Okuda Y, Yamaoka T, Tsukahara K, Isaka M, Bannai C, Yamashita K: High glucose and hyperosmolarity increase platelet-derived growth factor mRNA levels in cultured human vascular endothelial cells. Biochem Biophys Res Commun 1992, 187:664-669 [DOI] [PubMed] [Google Scholar]
- 42.Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R: TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990, 63:515-524 [DOI] [PubMed] [Google Scholar]
- 43.Ishikawa O, LeRot EC, Trojanowska M: Mitogenic effect of transforming growth factor beta1 on human fibroblasts involves the induction of platelet derived growth factor alpha receptors. J Cell Physiol 1990, 145:181-186 [DOI] [PubMed] [Google Scholar]
- 44.Haberstroh U, Zahner G, Disser M, Thaiss F, Wolf G, Stahl RAK: TGF-beta stimulates rat mesangial cell proliferation in culture: role of PDGF beta-receptor expression. Am J Physiol 1993, 264:F199-F205 [DOI] [PubMed] [Google Scholar]
- 45.Soma Y, Grotendorst GR: TGF-β stimulates primary human skin fibroblast DNA synthesis via an autocrine production of PDGF-related peptides. J Cell Physiol 1989, 140:246-253 [DOI] [PubMed] [Google Scholar]
- 46.Jakowlew SB, Cubert J, Danielpour D, Sporn MB, Roberts AB: Differential regulation of the expression of transforming growth factor beta mRNAs by growth factors and retinoic acid in chicken embryo chondrocytes, myocytes, and fibroblasts. J Cell Physiol 1992, 150:377-385 [DOI] [PubMed] [Google Scholar]
- 47.Villeger PM, Lotz M: Differential expression of TGF-beta isoforms by human articular chondrocytes in response to growth factors. J Cell Physiol 1992, 151:318-325 [DOI] [PubMed] [Google Scholar]
- 48.Sherer TB, Neff PS, Hankins GR, Tuttle JB: Mechanisms of increased NGF production in vascular smooth muscle of the spontaneously hypertensive rat. Exp Cell Res 1998, 241:186-193 [DOI] [PubMed] [Google Scholar]
- 49.Lyons RM, Gentry LE, Purchio AF, Moses HL: Mechanism of activation of latent recombinant transforming growth factor beta1 by plasmin. J Cell Biol 1990, 110:1361-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyons EM, Keski-Oja J, Moses HL: Proteolytic activation of latent transforming growth factor beta from fibroblast conditioned medium. J Cell Biol 1988, 106:1659-1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB: Characterisation of the activation of latent TGF-β by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self regulating system. J Cell Biol 1990, 111:757-763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribeiro SMF, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE: The activation sequence of thrombospondin 1 interacts with the latency associated peptide to regulate activation of latent transforming growth factor β. J Biol Chem 1999, 274:13586-13593 [DOI] [PubMed] [Google Scholar]
- 53.Schultz-Cherry S, Murphy-Ullrich JE: Thrombospondin causes activation of latent transforming growth factor β secreted by endothelial cells by a novel mechanism. J Cell Biol 1993, 122:923-932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schultz-Cherry S, Riberio S, Gentry L, Murphy-Ullrich JE: Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem 1994, 269:26775-26782 [PubMed] [Google Scholar]
- 55.Sato Y, Rifkin DB: Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor beta 1 like molecule by plasmin during co-culture. J Cell Biol 1989, 109:309-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato Y, Okada M, Seguchi T, Kuwano M, Sato S, Furuya A, Hanai N, Tamaoki T: The mechanism for the activation of latent TGF-beta during co-culture of endothelial cells and smooth muscle cells: cell-type specific targeting of latent TGF-beta to smooth muscle cells. J Cell Biol 1993, 123:1249-1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ross R, Raines EW, Bowen-Pope DF: The biology of platelet-derived growth factor. Cell 1986, 46:155-169 [DOI] [PubMed] [Google Scholar]