

Abstract
The α1β1 integrin (VLA-1) is a major collagen/laminin receptor that regulates fibroblast proliferation and mesangial cell migration and cell contraction. We have examined the effect of an antibody to VLA-1 in crescentic glomerulonephritis. Nephrotoxic nephritis was induced in Wistar-Kyoto rats and rats were given monoclonal antibody to VLA-1 (Ha31/8), 2.5 mg/kg, on alternate days. Antibodies were given from day −1 to day 10 or from day 14 to day 28. Treatment from day −1 to day 10, during the early inflammatory phase of nephrotoxic nephritis, had no effect on albuminuria or glomerular crescent formation. In the delayed treatment experiment, all rats developed florid crescentic glomerulonephritis, and control rats showed marked glomerular and tubulointerstitial scarring at day 32. VLA-1 expression, by immunohistochemistry, was increased in glomeruli and around tubules. Proteinuria did not differ between groups. In anti-VLA-1-treated rats, serum creatinine was significantly lower at day 32 (P = 0.002) and renal survival was significantly better (P = 0.045). Both glomerular and interstitial scarring were significantly less at day 32 in rats given anti-VLA-1 (P = 0.002). Deposition of ED(A) fibronectin, a marker of new matrix synthesis, and of type IV collagen, were reduced in glomeruli and interstitium in anti-VLA-1-treated animals (P = 0.0006). Expression of α-smooth muscle actin, a marker of myofibroblasts, showed no significant difference. Expression of matrix metalloproteinase-9 was increased in the glomeruli of rats treated with anti-VLA-1. We conclude that VLA-1 mediates both glomerular and interstitial fibrosis in crescentic glomerulonephritis and that neutralization of VLA-1, which enhanced expression of matrix metalloproteinase-9, is a possible therapeutic strategy in progressive renal scarring.
Integrins are transmembrane receptors for extracellular proteins. They mediate cell attachment, proliferation, and motility in response to the extracellular environment. They are heterodimeric, noncovalently associated glycoprotein complexes consisting of an α and a β chain. The β1 integrins are the largest group and are composed of a β1 chain associated with 1 of 12 α chains. They function predominantly in cell-matrix adhesion. Five members of this family are known to be major receptors for collagen, α1β1, α2β1, α3β1, α10β1, and α11β1. α1β1 integrin, also known as VLA-1, has a preference for type IV collagen and laminin. Expression of α1 in the adult animal is predominantly mesenchymal. In the kidney α1β1 integrin is found on glomerular mesangial and endothelial cells and on tubular epithelium. 1,2
Experiments using gene-targeted mice deficient in α1 have shown that α1 expression is essential for fibroblasts to adhere to and migrate on collagen type IV, 3 and that absence of α1 markedly reduces fibroblast proliferation on collagenous substrata. 4 α1β1 also supports gel contraction by smooth muscle cells, 5 suggesting a role in remodeling of collagenous tissue during scar formation. The integrins are important in the regulation of matrix metalloproteinases (MMPs) that control turnover of extracellular matrix. In mice lacking α1 there is increased expression of MMPs, including MMP-13, 6 MMP-2, MMP-7, and MMP-9. 7 In glomerular mesangial cells, α1β1 integrin mediates collagen-dependent adhesion, migration, and gel contraction. 8,9 Mesangial cell expression of α1β1 integrin is increased by transforming growth factor (TGF)-β. 8 α1 integrin plays a role in the mesangial expansion that is seen in a murine model of Alport’s syndrome. 10 These data suggest a possible role for α1β1 integrin in renal scarring.
α1β1 may also play a role in the interaction of inflammatory cells with extracellular matrix and, in particular, their migration through matrix after extravasation. Administration of an antibody to α1 chain significantly inhibits effector phase inflammatory responses in delayed-type hypersensitivity, contact hypersensitivity, and arthritis models in the mouse. 11 We have therefore studied the effect of an antibody that blocks the function of the α1 chain on inflammation and scarring in a rat model of glomerulonephritis. The model we have studied is nephrotoxic nephritis (NTN) in the Wistar-Kyoto (WKY) rat. We have previously described the natural history of this model, 12 which is induced by intravenous injection of a small dose of rabbit anti-rat glomerular basement membrane serum. Within 6 days rats develop marked glomerular macrophage infiltration and focal and segmental glomerulosclerosis. By day 10 the majority of glomeruli contain cellular crescents. This is followed in the third week by progressive glomerular and tubulointerstitial scarring with the development of end-stage renal failure at 5 to 6 weeks. In the present study we have examined the effect of a blocking antibody to α1 integrin chain administered either during the phase of acute inflammation, from before induction of glomerulonephritis to day 10, or during the scarring phase from days 14 to 28.
Materials and Methods
Reagents
Nephrotoxic serum, a rabbit antiserum to rat glomerular basement membrane, was prepared as previously described. 13 Hamster monoclonal antibody, Ha31/8, to rat α1 integrin was supplied by Biogen Inc., Cambridge, MA. An isotype-matched antibody, Ha4/8 was used as a control.
Localization of VLA-1
Inhibition of rat α1 integrin is by injection of the monoclonal antibody Ha31/8. To demonstrate localization of this antibody two male WKY rats (Charles River, Margate, UK), were given a single dose of 2.5 mg or 5 mg of Ha31/8 intraperitoneally. Rats were killed 24 hours later and kidneys were snap-frozen. Frozen sections were used to detect the localization of the injected hamster anti-rat α1 integrin by immunofluorescence using an anti-Armenian and Syrian hamster IgG-PE-conjugated antibody (BD Pharmingen, Oxford, UK).
Nephrotoxic Nephritis
Male WKY rats, weighing 200 to 220 g, were given 0.1 ml of nephrotoxic serum intravenously. At intervals rats were bled from the tail vein under isoflurane anesthesia. Urine was collected by housing rats in metabolic cages for 24-hour periods with free access to food and water. Rats were killed under isoflurane anesthesia, and blood was collected from the abdominal aorta. Samples of kidney were fixed in 10% formal saline and processed into paraffin wax, or snap-frozen in isopentane cooled in liquid nitrogen. Urinary albumin was measured by rocket immunoelectrophoresis, 14 and proteinuria by the sulfosalicylic acid method. 15 Urine and serum creatinine were measured on an Olympus AU600 analyzer (Olympus, Eastleigh, UK).
Experimental Design
Experiment 1
Experiment 1 was designed to study the effect of antibody to α1 chain on the initial phase of NTN. NTN was induced in 10 rats. Five were treated with anti-α1 antibody, Ha31/8, 2.5 mg/kg intraperitoneally (i.p.) on alternate days, starting 24 hours before induction of glomerulonephritis. Five were treated with control antibody Ha4/8. All rats were killed at day 10.
Experiment 2
Experiment 2 was designed to test the effect of antibody to α1 on established glomerulonephritis during the phase of glomerular and tubulointerstitial scarring. NTN was induced in 14 rats. From day 14, the rats were treated with anti-α1 integrin antibody, Ha31/8, 2.5 mg/kg i.p. or with control antibody, on alternate days until day 28. Rats were killed on day 32 for assessment of glomerular scarring.
Experiment 3
Experiment 3 followed the same protocol as experiment 2 with treatment from day 14 to day 28. There were six rats in each group. At day 28 treatment was stopped and rats were followed until they had to be killed because of deteriorating renal function. In addition to these experiments, sections from our previous study of the natural history 12 of this model of glomerulonephritis were available for assessment of α1β1 integrin, collagen type IV, and fibronectin expression at other time points.
Histology and Immunohistochemistry
In experiment 1, numbers of crescents were counted in 100 consecutive glomeruli in each section. In experiment 2, an initial semiquantitative assessment of glomerular and tubulointerstitial scarring was performed on hematoxylin and eosin-stained sections using a scale from 0 (no scarring) to 4 (scarring involving more than 75% of glomerular or interstitial area). Further assessment of scarring was performed by ranking the intensity of Masson’s trichrome staining in glomeruli and interstitium, and by quantitative image analysis of sections stained by immunohistochemistry for fibronectin and collagen type IV.
Antibodies against the following were used for immunohistochemistry: monocytes/macrophages (monoclonal antibody ED1; Serotec, Oxford, UK), CD8 cells (monoclonal antibody OX8, Serotec), α1β1 integrin (Chemicon, Temecula, CA), collagen type IV (Southern Biotechnology Associates, Birmingham, AL), extra domain (ED) (A) fibronectin (IST-9; Harlan-Sera Lab, Loughborough, UK), Ki-67 (Novocastra, Newcastle on Tyne, UK), MMP-9 (Biocarta, Oxford, UK), and α-smooth muscle actin (1A4; DAKO, Cambridge, UK). Staining was performed on frozen sections for α1β1 integrin, MMP-9, and ED(A) fibronectin. The other antibodies were used on paraffin sections pretreated by microwaving in citrate buffer for 3 × 5 minutes. Concentrations of primary antibody were as follows: ED1, 1:500; OX8, 1:200; type IV collagen, 1:1000; α-smooth muscle actin, 1:1000; ED(A) fibronectin, 1:1000; α1β1 integrin, 1:100; Ki-67, 1:1000; and MMP-9, 1:100. Type IV collagen was detected with biotinylated goat anti-mouse immunoglobulin (DAKO) and the others with biotinylated rabbit anti-mouse immunoglobulin (DAKO) followed by streptavidin-biotin-peroxidase complexes (DAKO). Apoptotic cells were detected by staining of paraffin-embedded tissue using the ApopTag fluorescein in situ apoptosis detection kit (Appligene, Harefield, UK) following the manufacturer’s instructions. For quantitative assessment of staining of type IV collagen, ED(A) fibronectin, and α-smooth muscle actin images were captured using a color CoolView camera (Photonic Science, Robertsbridge, UK) and analyzed using ImagePro plus software (Media Cybernetics, Silver Spring, MD). Digital images were captured and color segmentation was performed to highlight the stained area. The software then calculated this as a percentage of the total defined area, eg, a glomerular cross-section. A minimum of 20 glomeruli or 20 interstitial areas was examined per section. Glomerular macrophages and CD8 cells were counted in at least 25 glomeruli per section. Interstitial macrophages were assessed by ranking sections for intensity of interstitial ED1 staining. MMP-9 staining was assessed by ranking sections. All histological assessment and image analysis was performed on coded, randomized sections by a blinded observer.
Statistics
Results are presented as mean ± SE. Comparisons between groups are by Mann-Whitney U-test. Renal survival data were analyzed by Kaplan-Meier analysis.
Results
We have previously described, in detail, the natural history of this model of NTN in the WKY rat. 12 By day 4, rats have prominent glomerular hypercellularity. Segmental glomerular fibrinoid necrosis is seen by day 6 and by day 10 cellular crescents are present in the majority of the glomeruli. This is followed by progressive glomerular and tubulointerstitial scarring with renal failure by ∼6 weeks. Proteinuria is first apparent by day 4, increases during the first 2 weeks, and then declines as renal failure develops. We examined sections for the expression of α1β1 integrin. In normal kidneys there was staining for α1β1 in the mesangial regions of glomeruli and on Bowman’s capsule with very faint staining at the basal aspect of some tubular cells. In nephritic kidneys at day 14 mesangial staining was increased and many tubules showed prominent staining that was localized at the base of the tubular cells; this was more marked by day 35 (Figure 1) ▶ . After injection of a single dose of the hamster anti-rat α1 integrin monoclonal antibody, localization of this antibody could be detected on frozen sections in mesangial areas of glomeruli and around the tubules.
Figure 1.
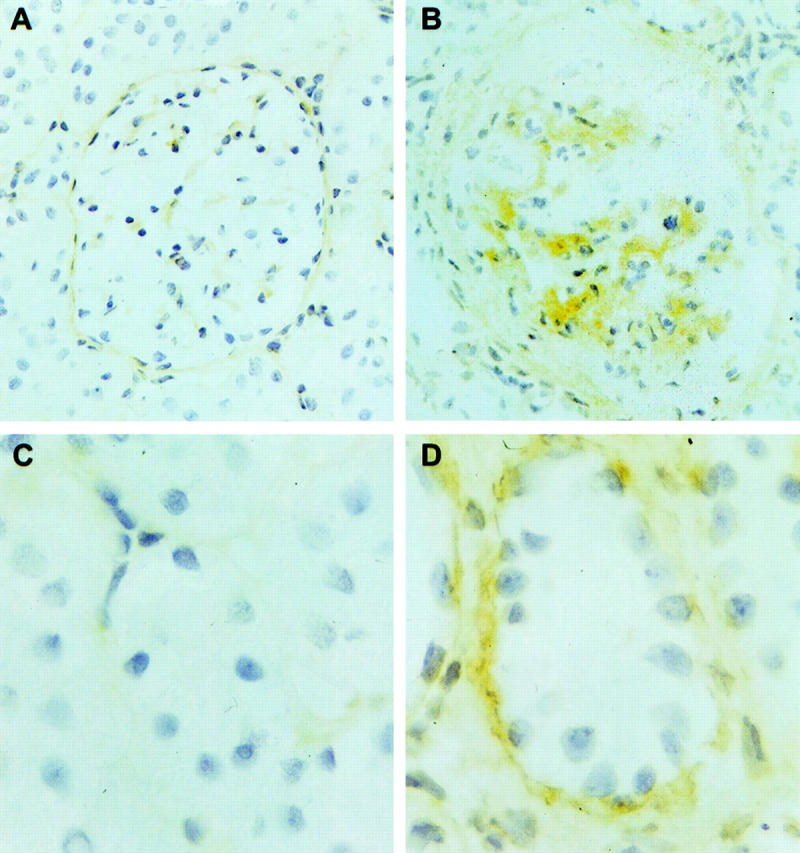
Immunohistochemical staining for α1β1 integrin. In normal kidneys there is faint staining in glomerular mesangial areas and in Bowman’s capsule (A); there is little peritubular staining (C). At day 35 of NTN there is increased staining in the mesangium (B) and on the basal aspect of renal tubules (D).
Experiment 1
Rats were treated with anti-α1 antibody or control antibody from the day before induction of glomerulonephritis until day 10. There was no significant effect on proteinuria at day 6 or day 10 or on glomerular injury at day 10 as assessed by glomerular crescent formation (Table 1) ▶ .
Table 1.
Effect of Treatment with Anti-α1 Antibody on NTN at Day 10
| Treatment group | Albuminuria (mg/day) | Glomerular crescents (%) | Creatinine clearance (ml/min) |
|---|---|---|---|
| Anti-α1 | 166 ± 24 | 55 ± 11 | 1.0 ± 0.1 |
| Control | 207 ± 19 | 50 ± 2 | 1.1 ± 0.01 |
Experiments 2 and 3
Rats with NTN were treated with anti-α1 antibody or control antibody from day 14 to day 28. There was no difference between the groups in levels of proteinuria (Figure 2) ▶ . At day 32, treated animals had a lower level of serum creatinine than control animals (Figure 3) ▶ . This was associated with reduced glomerular and tubulointerstitial scarring in rats killed at day 32 (Figure 4) ▶ . Semiquantitative assessment of glomerular scarring on a scale of 0 to 4 gave a score for control rats of 3.3 ± 0.2 and for treated rats 2.1 ± 0.1 (P = 0.0023); for tubulointerstitial scarring the semiquantitative scores were control 3.3 ± 0.2 and Ha31/8-treated rats 2 ± 0.2 (P = 0.0023). This reduction in glomerular and tubulointerstitial scarring was confirmed by ranking Masson’s trichrome stained sections (Figure 5) ▶ . Immunohistochemistry showed that by day 32 untreated rats had marked glomerular accumulation of ED(A) fibronectin (Figure 6) ▶ and cortical deposition of type IV collagen.
Figure 2.

Twenty-four-hour proteinuria in rats with NTN treated with anti-α1 antibody or control antibody. There was no difference between the groups.
Figure 3.

Serum creatinine at day 32 in rats with NTN treated with anti-α1 antibody or control antibody. Creatinine is significantly lower in anti-α1-treated animals.
Figure 4.
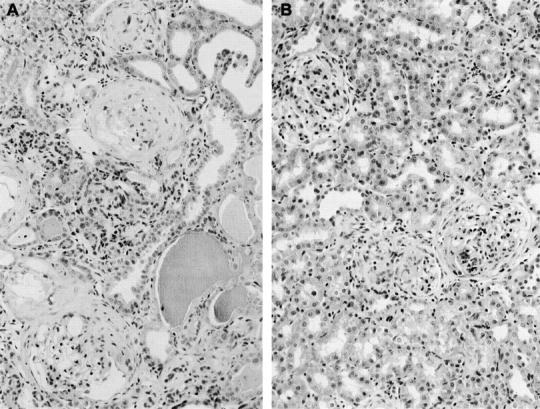
Histology of kidneys at day 32 in rats with NTN treated with anti-α1 antibody or control antibody. Control animals (A) showed marked glomerular scarring with fibrous crescents, together with tubular atrophy, tubular dilatation with cast formation, and expansion of the interstitium. All these changes are reduced in the anti-α1-treated animals (B).
Figure 5.
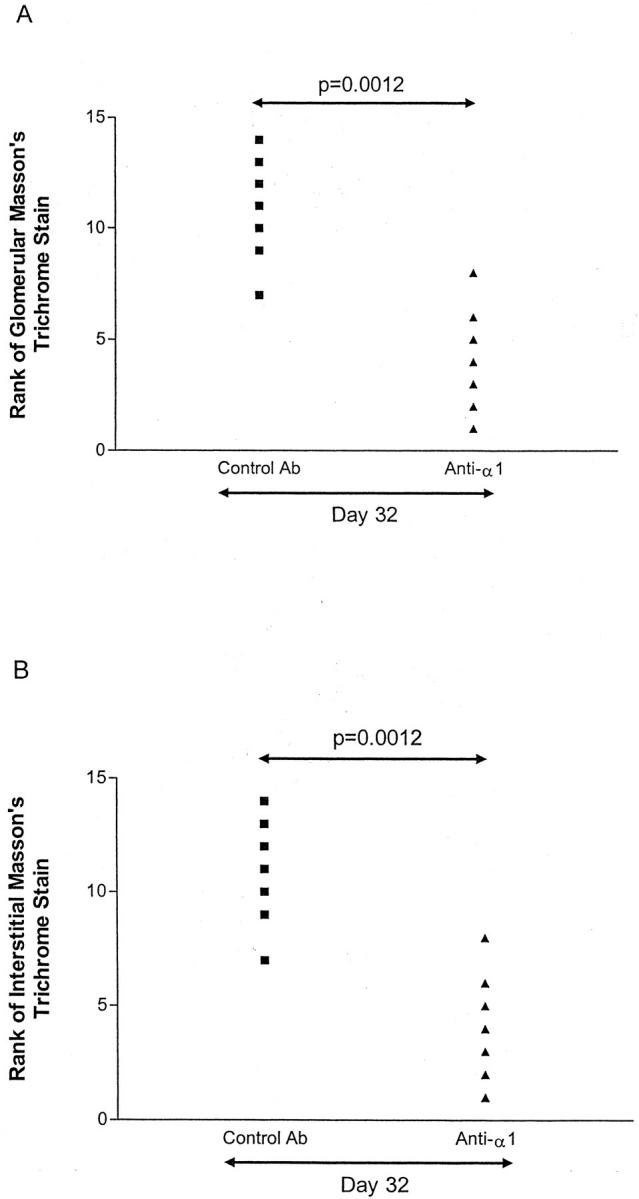
Graphs showing ranking of Masson’s trichrome staining in glomeruli (A) and interstitium (B). The intensity of staining of Masson’s trichrome-stained sections from rats with NTN at day 32 was ranked in randomized slides. There was significantly less staining in rats treated with anti-α1 antibody.
Figure 6.

Glomerular staining for ED(A) fibronectin at day 32. In control animals (A) there was marked deposition of fibronectin that was reduced in animals treated with anti-α1 antibody (B). Quantitation is shown in Figure 7 ▶ .
Figure 7 ▶ shows quantitation of glomerular ED(A) fibronectin in treated and untreated animals together with results for normal animals and day 14 NTN. Treatment with anti-α1 antibody significantly reduced glomerular ED(A) fibronectin deposition; in treated animals there was no significant difference from levels seen at day 14. Similar data are shown for cortical type IV collagen in Figure 8 ▶ . There was a significant reduction in deposition of type IV collagen in anti-α1-treated animals. It is possible that one mechanism by which α1 blockade prevents fibrosis is by preventing cells from acquiring a myofibroblastic phenotype. We therefore stained sections for the myofibroblast marker, α-smooth muscle actin. There was no significant difference in glomerular staining between the two groups (anti-α1-treated, 24 ± 2% of glomerular area stained; control antibody, 29 ± 3%).
Figure 7.

Glomerular staining for ED(A) fibronectin. Staining was analyzed by image analysis and expressed as percentage area of glomerulus stained as described in Materials and Methods. There was no staining in normal glomeruli. There was a slight increase by day 14 and a marked increase in control animals from day 14 to day 32. This was significantly inhibited by treatment with anti-α1 antibody.
Figure 8.

Cortical staining for type IV collagen. Staining was analyzed by image analysis and expressed as percentage of the cortical area stained. There was a marked increase in type IV collagen between days 14 and 32 that was significantly ameliorated in the rats treated with anti-α1 antibody.
Quantitation of glomerular inflammatory cells showed no significant difference in numbers of glomerular macrophages (ED1+) cells between treated and control groups, but CD8 cell numbers were higher in the treated group at day 32 (Figure 9) ▶ . Ranking of sections for interstitial ED1 cell infiltration showed no difference between the groups. We performed assessment of proliferating cells by Ki-67 immunostaining, and of apoptotic cells by terminal deoxynucleotidyl transferase end labeling. In the rats treated with anti-α1 antibody there were significantly more proliferating cells (10.5 ± 1.5 cells/glomerulus versus 2.0 ± 0.9; P = 0.0041) and apoptotic cells (0.86 ± 0.19 apoptotic bodies/glomerulus versus 0.35 ± 0.13; P = 0.0175) in glomeruli and it seems likely that this reflects the fact that there was much less glomerular scarring in the treated group.
Figure 9.
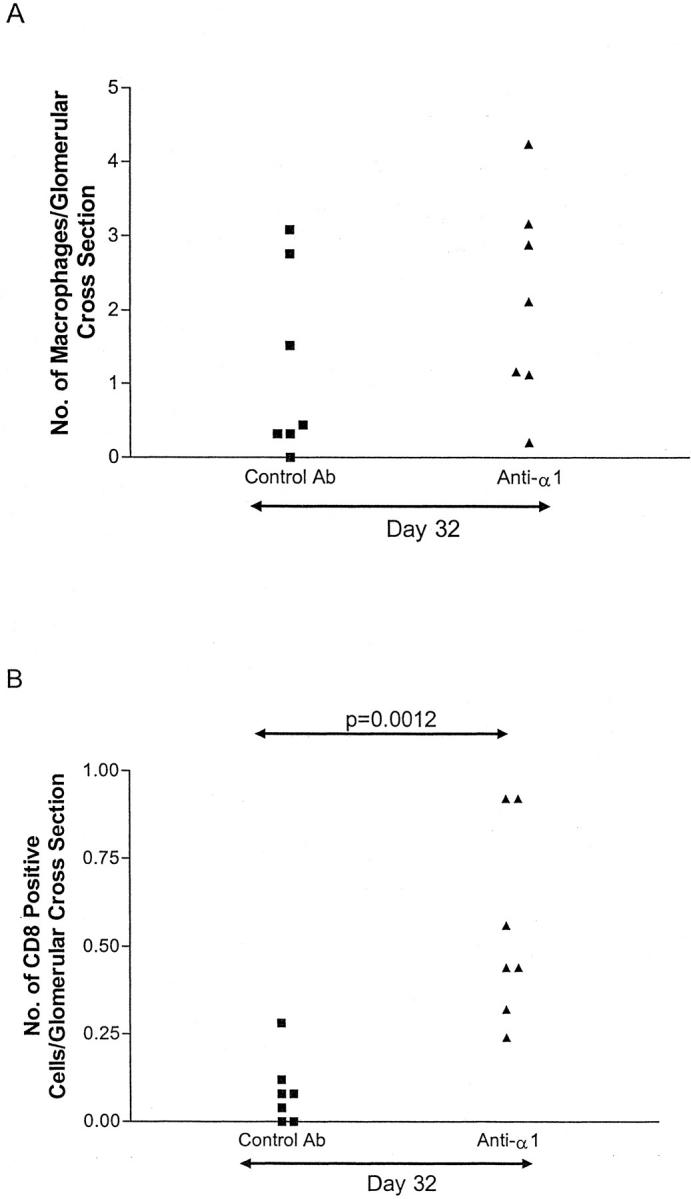
Glomerular infiltration by macrophages and CD8 cells at day 32. Immunohistochemistry was performed for macrophages (ED1+) (A) and CD8 cells (OX8+) (B). Glomerular CD8 cells were significantly higher in the rats treated with anti-α1 antibody. There was no difference in macrophage numbers.
In view of the role of MMPs in controlling matrix synthesis and the fact that MMPs have been shown to be increased in α1 knockout mice we examined glomerular expression of MMP-9 by immunohistochemistry. We first stained sections from various time points of the model. We found no MMP-9 staining in normal glomeruli and a marked expression at day 7 which was maintained to day 14 (Figure 10) ▶ . In rats treated with anti-α1 antibody glomerular MMP-9 expression remained increased and significantly higher than in rats treated with control antibody, in which it was almost back at baseline at day 32 (P = 0.0041) (Figure 10) ▶ .
Figure 10.

Immunohistochemical staining for MMP-9. No glomerular staining was detectable in normal kidney (A). At day 7 of disease there was marked staining in glomeruli (B). By day 32 this had returned almost to baseline in animals treated with control antibody (C) but was significantly enhanced by treatment with anti-α1 antibody (D).
In the renal survival experiment, rats were treated from day 14 to day 28 and then followed until they died or had to be killed because of deteriorating clinical condition as assessed by an independent observer. The results of this renal survival cohort are shown in Figure 11 ▶ . Treatment with Ha31/8 significantly prolonged survival.
Figure 11.

Graph showing renal survival in rats with NTN treated with anti-α1 antibody or control antibody. There was significantly prolonged survival in the anti-α1-treated group.
Discussion
A major determinant of outcome in glomerular disease is progressive fibrosis. We have shown that treatment with an antibody to α1 integrin chain reduces glomerular and tubulointerstitial scarring and preserves renal function in a model of focal necrotizing glomerulonephritis. Histologically, this model shows focal and segmental necrosis followed by the development of crescents. The changes are very similar to those seen in human crescentic glomerulonephritis as seen, for example, in anti-neutrophil cytoplasm antibody-associated disease. 16 These human diseases are typically associated with progression to glomerular and interstitial scarring and therefore any treatment that can ameliorate this progression is potentially of considerable clinical importance.
In the first instance we examined the effect of anti-α1 on the inflammatory phase of the model. During the first 10 days of the model there is accumulation of macrophages in glomeruli in response to deposition of rabbit immunoglobulin. This is followed by segmental tuft necrosis and crescent formation. Blockade of α1 integrin has previously been shown to be effective in reducing inflammation in immunologically mediated models in the mouse delayed-type hypersensitivity, contact hypersensitivity, and anti-collagen antibody-induced arthritis. 11 α1β1 integrin is expressed on activated T cells, monocytes, and neutrophils. Therefore, possible mechanisms of the beneficial effects in these models of inflammation include disruption of leukocyte migration through extracellular matrix and an effect on leukocyte activation. However, in our model, we found no effect of treatment with anti-α1 on glomerular inflammation as assessed by either proteinuria or glomerular crescent formation indicating that α1β1 integrin is not critical in glomerular inflammation. This is in contrast to our previous study using anti-VLA-4 antibody, which reduced acute glomerular injury in this model. 17
There is considerable in vitro evidence that α1β1 integrin plays a role in remodeling of extracellular matrix. It is expressed on fibroblasts, mesangial cells, and tubular cells. It is a major receptor for type IV collagen, which is the main type of collagen found in the glomerulus and in tubular basement membranes, and is critical to adhesion and migration of fibroblasts and mesangial cells on type IV collagen and laminin. 3,5 In addition, collagen gel contraction, an in vitro model of collagenous tissue remodeling, is dependent on α1β1 integrin expression in both fibroblasts and mesangial cells. 5,8 We found, by immunohistochemistry, that there was increased expression of α1β1 integrin both in glomeruli and around tubules during the scarring phase of the model. One possible mediator of this increased expression is TGF-β, which has been shown in vitro to increase mesangial cell expression of α1β1. 8 Increased glomerular α1β1 expression was also found in a model of mesangial proliferative glomerulonephritis where it paralleled increased TGF-β synthesis. 18 In human glomerulonephritis, mesangial expression of α1β1 correlated with expression of type IV collagen, fibronectin, α-smooth muscle actin, and TGF-β. 1 We quantitated glomerular accumulation of extracellular matrix by measuring deposition of fibronectin, and specifically of the extra domain A, ED(A), form, which is almost undetectable in normal glomeruli and is said to be a marker of TGF-β-induced matrix accumulation. 19 We found that there was a modest increase in glomerular ED(A) fibronectin by 14 days and that blockade of α1 integrin from day 14 to 28 prevented most of the increase seen in the control rats during that period.
In the interstitium, we quantitated type IV collagen because this is reported to be a sensitive marker of interstitial scarring. We found that α1 blockade markedly reduced the deposition of type IV collagen in the interstitium from days 14 to 28. These changes in glomerular and tubulointerstitial scarring were accompanied by preservation of renal function as determined by serum creatinine measurements. We found no difference in proteinuria between the two groups suggesting that this beneficial effect was not because of a reduction in inflammation. Consistent with this we found no difference in glomerular or interstitial macrophage numbers although we did find higher numbers of CD8 cells in the glomeruli of the anti-α1-treated animals. The significance of this is not clear although it most likely reflects the reduction in scarring with preservation of open capillary loops in the treated animals. We also found that there were increased proliferating and apoptotic cells in the treated animals and again, we think this merely reflects the reduction in scarring. The fact that interstitial macrophage numbers are similar in the treated and control groups suggests that α1 blockade is not acting by reducing macrophage migration in the interstitium, but we cannot exclude an effect on activation of interstitial macrophages.
Deposition of extracellular matrix in glomeruli is thought to follow the transformation of glomerular cells to a myofibroblastic phenotype. We therefore stained for the myofibroblast marker, α-smooth muscle actin, and found no difference between treated and control groups in glomerular expression. It therefore seems that myofibroblasts are present in the treated animals but that deposition of matrix by these cells is blocked. This suggests that the acquisition of the myofibroblastic phenotype is not dependent on stimulation through α1β1 integrin but that TGF-β stimulates the expression of both α-smooth muscle actin and α1β1 independently.
Extracellular matrix can be degraded by a number of MMPs and increased activity of MMPs would be expected to reduce matrix accumulation. Previous reports have shown increased MMP expression in mice genetically engineered to lack the integrin α1 chain 6,7 and we therefore hypothesized that the reduced scarring in rats given anti-α1 antibody might be because of increased synthesis of MMPs with consequent extracellular matrix degradation. We examined expression of MMP-9 also known as gelatinase B because, together with MMP-2, this is important in the degradation of type IV collagen, which is the major collagen found in the glomerulus. Fibronectin is also a substrate for MMP-9. In contrast to MMP-2, MMP-9 is known to be inducible in mesangial cells in response to proinflammatory stimuli. 20 Consistent with this we found a marked increase in glomerular MMP-9 during the inflammatory phase of disease at day 7. In rats treated with control antibody this had returned almost to baseline by day 32, whereas treatment with anti-α1 antibody led to increased and persistent glomerular MMP-9 expression. We suggest that this enhancement of MMP-9 expression is one way in which blockade of α1β1 integrin reduces glomerular scarring.
The role of α1β1 integrin has recently been examined in a mouse model of Alport’s syndrome, a basement membrane disorder resulting from mutations in type IV collagen genes. 10 Mice with genetic deletion of the collagen α3(IV) gene show progressive renal failure with marked increase in mesangial matrix and prominent deposition of fibronectin in the mesangium. When these mice were crossed with mice lacking the integrin α1 chain, the double knockouts showed significant amelioration of mesangial expansion and fibronectin deposition. Blockade of TGF-β1 had an additive protective effect in this model and it would be of interest in our model of glomerulonephritis to look for possible synergy between blockade of TGF-β1 and anti-α1 treatment.
In conclusion, we have shown that during the progressive glomerular and tubulointerstitial scarring that follows acute crescentic glomerulonephritis, there is increased expression of α1β1 integrin in glomeruli and around tubules. There is also increased glomerular and interstitial deposition of fibronectin and type IV collagen associated with deterioration in renal function. Blockade of α1β1 integrin using a monoclonal antibody markedly reduces fibronectin and collagen deposition and preserves renal function. This beneficial effect does not seem to be through a reduction in interstitial inflammation or through inhibition of myofibroblast transformation. It therefore probably reflects a disruption of the normal interaction of the myofibroblast with extracellular matrix, which limits the deposition of collagen and other matrix components. At least in part it may be through enhanced expression of MMP-9 leading to increased degradation of type IV collagen and fibronectin. Our results are particularly relevant to human disease because α1 blockade was effective even when started at a relatively late stage of disease when crescents were well established. This corresponds to the stage at which most patients will be diagnosed. This approach may, therefore, be worth investigating in the progressive scarring phase of human glomerulonephritis.
Acknowledgments
We thank Dr. Philip Gotwals and Dr. Victor Koteliansky for invaluable help and advice.
Footnotes
Address reprint requests to Dr. H. T. Cook, Department of Histopathology, Imperial College, Du Cane Rd., London W12 0NN, UK. E-mail: t.cook@ic.ac.uk.
Supported by an Action Research Training Fellowship (to A. A.).
References
- 1.Kuhara T, Kagami S, Kuroda Y: Expression of β1-integrins on activated mesangial cells in human glomerulonephritis. J Am Soc Nephrol 1997, 8:1679-1687 [DOI] [PubMed] [Google Scholar]
- 2.Simon EE, McDonald JA: Extracellular matrix receptors in the kidney cortex. Am J Physiol 1990, 259:F783-F792 [DOI] [PubMed] [Google Scholar]
- 3.Gardner H, Kreidberg J, Koteliansky V, Jaenisch R: Deletion of integrin α1 by homologous recombination permits normal murine development but gives rise to a specific defect in cell adhesion. Dev Biol 1996, 175:301-313 [DOI] [PubMed] [Google Scholar]
- 4.Pozzi A, Wary KK, Giancotti FG, Gardner HA: Integrin α1β1 mediates a unique collagen-dependent proliferation in vivo. J Cell Biol 1998, 142:587-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gotwals PJ, Chi-Rosso G, Lindner V, Yang J, Ling L, Fawell SE, Koteliansky VE: The α1β1 integrin is expressed during neointima formation in rat arteries and mediates collagen matrix reorganisation. J Clin Invest 1996, 97:2469-2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner H, Brosberg A, Pozzi A, Laato M, Heino J: Absence of integrin α1β1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci 1999, 112:263-272 [DOI] [PubMed] [Google Scholar]
- 7.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA: Elevated matrix metalloprotease and angiostatin levels in integrin α1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci USA 2000, 97:2202-2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kagami S, Kondo S, Loster K, Reutter W, Kuhara T, Yasutomo K, Kuroda Y: α1β1 integrin-mediated collagen matrix remodeling by rat mesangial cells is differentially regulated by transforming growth factor-β and platelet derived growth factor-BB. J Am Soc Nephrol 1999, 10:779-789 [DOI] [PubMed] [Google Scholar]
- 9.Kagami S, Kondo S, Urushihara M, Loster K, Reutter W, Saijo T, Kitamura A, Kobayashi S, Kuroda Y: Overexpression of α1β1 integrin directly affects rat mesangial cell behavior. Kidney Int 2000, 58:1088-1097 [DOI] [PubMed] [Google Scholar]
- 10.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Koteliansky V, Gotwals P, Amatucci A, Kalluri R: Integrin α1β1 and TGF-β1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol 2000, 157:1649-1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Fougerolles AR, Sprague AG, Nickerson-Nutter CL, Chi-Rosso G, Rennert PD, Gardner H, Gotwals PJ, Lobb RR, Koteliansky VE: Regulation of inflammation by collagen-binding integrins α1β1 and α2β1 in models of hypersensitivity and arthritis. J Clin Invest 2000, 105:721-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam FWK, Smith J, Morel D, Karkar AM, Thompson EM, Cook HT, Pusey CD: Development of scarring and renal failure in a rat model of crescentic glomerulonephritis. Nephrol Dial Transplant 1999, 14:1658-1666 [DOI] [PubMed] [Google Scholar]
- 13.Bhan AK, Schneeberger EE, Collins AB, McCluskey RT: Evidence for a pathogenic role of a cell-mediated immune mechanism in experimental glomerulonephritis. J Exp Med 1978, 148:246-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds J, Mavromatidis K, Cashman SJ, Evans DJ, Pusey CD: Experimental autoimmune glomerulonephritis (EAG) induced by homologous and heterologous glomerular basement membrane in two substrains of Wistar-Kyoto rat. Nephrol Dial Transplant 1998, 13:44-52 [DOI] [PubMed] [Google Scholar]
- 15.Baker FJ, Silverton RE, Luckcock ED: An Introduction to Medical Laboratory Technology. 1966. Butterworth, London
- 16.Cattell V, Cook HT: Pathology of glomerulonephritis. Pusey CD eds. The Treatment of Glomerulonephritis. 1999:pp 15-37 Kluwer, Dordrecht
- 17.Allen AR, McHale J, Smith J, Cook HT, Karkar A, Haskard DO, Lobb RR, Pusey CD: Endothelial expression of VCAM-1 in experimental crescentic nephritis and effect of antibodies to very late antigen-4 or VCAM-1 on glomerular injury. J Immunol 1999, 162:5519-5527 [PubMed] [Google Scholar]
- 18.Kagami S, Border WA, Ruoslahti E, Noble NA: Coordinated expression of β1 integrins and transforming growth factor-β-induced matrix proteins in glomerulonephritis. Lab Invest 1993, 69:68-76 [PubMed] [Google Scholar]
- 19.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E: Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360:361-364 [DOI] [PubMed] [Google Scholar]
- 20.Suto TS, Fine LG, Shimizu F, Kitamura M: In vivo transfer of engineered macrophages into the glomerulus. J Immunol 1997, 159:2476-2483 [PubMed] [Google Scholar]