

Abstract
Although argyrophilic grain disease is characterized histopathologically by τ-positive lesions known as argyrophilic grains located predominantly in limbic brain regions in the absence of other diagnostic neuropathologies, the biochemical correlates of argyrophilic grains in gray and white matter have not been reported. Thus, we analyzed insoluble (pathological) τ proteins in five argyrophilic grain disease brains in comparison with those seen in Alzheimer’s disease and Pick’s disease. Analyses of separately dissected gray and white matter samples from various cortical regions revealed that pathological τ in argyrophilic grain disease was confined primarily to mediotemporal neocortical gray and adjacent white matter, and also to the allocortex, amygdala, and hippocampus. The amounts of sarcosyl-insoluble τ in all five cases were substantially lower than in Alzheimer’s disease and Pick’s disease, but the amounts of sarcosyl-insoluble τ in white matter were higher or comparable to that detected in gray matter from the same region, which distinguishes argyrophilic grain disease from Alzheimer’s disease. The banding patterns of τ isoforms in argyrophilic grain disease varied: in three cases they were similar to Alzheimer’s disease, but in two other cases, 4 microtubule binding repeat (4R) τ predominated, which distinguishes argyrophilic grain disease from classical Pick’s disease. The differences between these three diseases were re-enforced by the predominance of straight τ filaments from argyrophilic grain disease brains. Thus, we conclude that argyrophilic grain disease is a distinct tauopathy characterized by prominent accumulation of argyrophilic grains in limbic brain regions in association with the characteristic τ biochemical and ultrastructural profile reported here.
Argyrophilic grain disease (AGD) is a recently described neurodegenerative disorder of the elderly that results in dementia and severe brain degeneration. 1-4 Prominent argyrophilic grains (AGs), ie, granular or punctuate deposits that are highly variable in size and shape, together with the presence of coiled bodies, are diagnostic hallmarks of AGD in the absence of additional neurodegenerative brain lesions sufficient to meet diagnostic criteria for Alzheimer’s disease (AD) or other dementing disorders. 5-8 AGs are most abundant in the limbic system, especially in the CA1 region of hippocampus, amygdala, and subcortical white matter as well as in deep cortical layers in mediotemporal and allocortical regions. 5 AGs are immunopositive for major phosphorylation-dependent anti-τ antibodies similarly to filamentous τ inclusions in AD 9,10 and other tauopathies. 11 The presence of argyrophilic glial cytoplasmic inclusions in AGD and the presence of AGs in other neurodegenerative disorders, including Pick’s disease (PiD), progressive supranuclear palsy, 12,13 corticobasal degeneration, multiple system atrophy, 14,15 and motoneuron disease 15-17 suggests that AGs may co-occur with other pathologies. 16,18 Nonetheless, in a small percentage of dementia patients (usually older than 65 years of age), AGs are the overwhelmingly predominant neuropathological lesions, thereby suggesting that AGD is a distinct neurodegenerative disorder. 5,16,19 However, the mechanism(s) underlying the formation of AGs is poorly understood, and biochemical analyses of the abnormal τ proteins gray and white matter of this tauopathy have not been reported.
Thus, we characterized pathological τ biochemically in frozen brain tissue from five AGD cases and compared the brain region-specific distribution of τ pathology and biochemical τ profile in AGD with those seen in AD and PiD.
Materials and Methods
Case Selection
Frozen brain samples from five patients with neuropathologically diagnosed AGD, two with AD, and two with PiD were obtained through the German Brain Bank Brain-Net (Munich Institute for Neuropathology) (cases 1 to 4) and The Center for Neurodegenerative Disease Research, University of Pennsylvania (cases 5 to 9). Demographic data and other information on these cases are summarized in Table 1 ▶ .
Table 1.
Demographic Information Summary
| Case no. | Age | Sex | PMI, hours | Braak stage | AG | Clinical information | 4R:3R* |
|---|---|---|---|---|---|---|---|
| 1 | 71 | F | 34 | I | +++ | Parkinson’s disease | 1.1 |
| 2 | 73 | M | 60 | II | ++ | Dementia | 0.9 |
| 3 | 85 | F | 48 | II | +++ | No neurologic or psychiatric symptoms | 1.2 |
| 4 | 78 | F | 39 | III | ++ | Dementia and parkinsonism | 2.4 |
| 5 | 54 | F | 12 | II | ++ | FTD w/motoneuron disease | 1.9 |
| 6 | 72 | M | 8 | I | − | PiD | 0.5 |
| 7 | 79 | M | 6 | II | − | PiD | 0.3 |
| 8 | 60 | F | 8 | V | − | AD | 0.9 |
| 9 | 79 | M | 5.5 | VII | − | AD | 1.0 |
PMI, postmortem interval; density of argyrophilic grains (AG) ++, moderate; +++, abundant.
*, see Results.
Immunocytochemistry
Fresh autopsy-derived brain tissue was fixed in neutral buffered formalin, paraffin-embedded, cut into 6-μm sections and processed as described elsewhere. 20 Additionally, frozen tissue from the area adjacent to the regions used for biochemical analysis was fixed in neutral buffered formalin and processed similarly to fresh-fixed material. Phosphorylation-dependent monoclonal antibodies (mAbs) PHF1 (1:500, a gift from Dr. P. Davies) were used for neuropathological evaluation.
Biochemical and Western Blot Analysis
Biochemical analyses of τ proteins in different brain regions were performed in five AGD cases and in one AD case using cortical gray and underlying white matter that were separately dissected and processed as described previously. 20-23 Frozen brain samples from frontal, occipital, temporal, parietal, occipital lobes, and cerebellum were used for the regional analysis of τ pathology. Also, tissue from the hippocampus, amygdala, and entorhinal allocortex was analyzed in cases 1, 3, and 4. Sarcosyl-insoluble τ samples were prepared, and the τ isoform profiles therein were examined after enzymatic dephosphorylation using Escherichia coli alkaline phosphatase (Sigma Chemical Co., St. Louis, MO) as described 22-24 and were resolved on 7.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. Six recombinant human brain τ isoforms were used as a standard for the Western blot studies including quantitative analysis of τ isoforms using a mixture of phosphorylation-independent τ mAb, Tau14 (1:3000; N-terminal) and Tau46 (1:1000; C-terminal). Mouse 125I-IgG was used as a secondary detection antibody. 22,23 Final data were presented as a ratio 4R:3R (Table 1) ▶ .
Electron Microscopy of Isolated Filaments
Dispersed τ filaments were prepared from separately dissected temporal cortical gray and white matter (case 2), adsorbed on carbon-coated 400-mesh grids, and stained with 0.4% uranyl acetate as described. 11,25 Electron photomicrographs were taken at a nominal magnification of ×120,000. Immunoelectron microscopy was performed on similar samples and probed with phosphorylation-dependent antibodies to τ (PHF1, 1:10). Goat anti-mouse antibodies conjugated to colloidal gold (10-nm diameter, 1:20; Amersham) were used as a secondary antibody. Negative staining with 1% uranyl acetate in 100% methanol was performed and grids were examined with a JEM1010 electron microscope at 80 kV.
Sequential Extraction of τ Proteins
Selected brain areas were used for the sequential extraction of τ proteins using buffers with increasing abilities to solubilize proteins. Each extraction step was repeated two times, samples were spun at 45 K for 30 minutes at 4°C, supernatants were collected and subjected to Western blot analysis. The following sequential series of solubilizing buffers with a mixture of protease inhibitors were used for extraction: 11 1) buffer A (0.75 mol/L NaCl, 50 mmol/L Tris buffer, pH 7.4, 2 mmol/L ethylenediaminetetraacetic acid; 2) 1% Triton in buffer A; 3) RIPA buffer (0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 2 mmol/L ethylenediaminetetraacetic acid, 150 mmol/L NaCl, 50 mmol/L Tris buffer, pH 8.0); 4) 2% SDS; 5) 70% formic acid. Because of differences in the amount of extraction buffer used in each step, protein concentrations were adjusted in samples prepared under similar conditions.
Results
The clinical diagnosis for all cases used in this study is summarized in Table 1 ▶ . Three of five AGD patients were presented with symptoms of dementia (cases 2, 4, and 5), one patient was diagnosed with Parkinson’s disease (case 1), and one patient (case 3) did not have any significant neurological or psychiatric symptoms that were identified during life. The two PiD cases used here were described elsewhere. 11
Immunocytochemical Findings
Neuropathological evaluation of five AGD cases revealed a variable distribution and abundance of AGs in selected brain areas. The highest density of AGs was found throughout CA1 of the hippocampus and in the amygdala, as well as in mediotemporal and entorhinal deep cortical areas and white matter (Figure 1) ▶ . Coiled bodies were also notably present in deep cortical layers and white matter. Mild AD pathology (eg, sparse tangles and neuropil threads) was present in all cases, thereby supporting the observations by others that AGs frequently occur with a low abundance of other τ lesions in AGD, and that cases of AGD with AGs in the complete absence of other brain lesions are very rare. 10,16,19 The distribution and the intensity of neuritic changes corresponded to Braak stages I to III. 2 Senile plaques were unremarkable in all AGD cases in densities resembling plaque distribution found in nondemented elderly controls rather than those in AD.
Figure 1.

Neuropathology of AGD cases. Sections of hippocampus, amygdala, entorhinal cortex, and temporal cortex of different AGD cases were stained with τ mAb PHF1 (1:3000). AGs are seen in amygdala (A, case 1, arrows show the punctate AGs distribution; B, case 5; C, case 2), hippocampus (D, case 1; F, case 3) and temporal cortex (C, case 4), whereas AD-type pathology and AGs are seen in entorhinal cortex (E, case 3). E: Glial inclusions are seen in temporal cortex (case 2). Original magnifications: ×2300 (A, C–F); ×7750 (B).
Biochemical Analysis of τ Proteins
To analyze the distribution of pathological τ in AGD and to assess the τ isoform composition in this tauopathy, we conducted brain region-specific analysis of sarcosyl-insoluble τ proteins in multiple brain regions in separately dissected gray and white matter samples. The amount of sarcosyl-insoluble τ was variable between the cases and different brain regions (Figure 2A) ▶ . Pathological τ in cases 1, 4, and 5 was confined primarily to the temporal lobe in both gray and white matter, whereas frontal, parietal, and occipital regions had relatively sparse of pathology. Interestingly, although the τ pathology in cases 2 and 3 was mild overall, it was restricted predominantly to the white matter. As expected, based on immunohistochemical analysis of the adjacent tissue, the amount of sarcosyl-insoluble τ in the amygdala, entorhinal allocortex, and hippocampus was substantially higher than in other cortical areas (cases 3 and 4). Insoluble τ proteins in low quantities also were detected in brain regions where sparse tangles and occasional treads, but not AGs, were found by neuropathological examination. The distribution and amount of sarcosyl-insoluble τ in all AGD cases were notably different from AD. Whereas in AGD pathological τ was detected only in selected brain regions, abundant sarcosyl-insoluble τ was present in gray matter of all cortical areas in AD cases.
Figure 2.
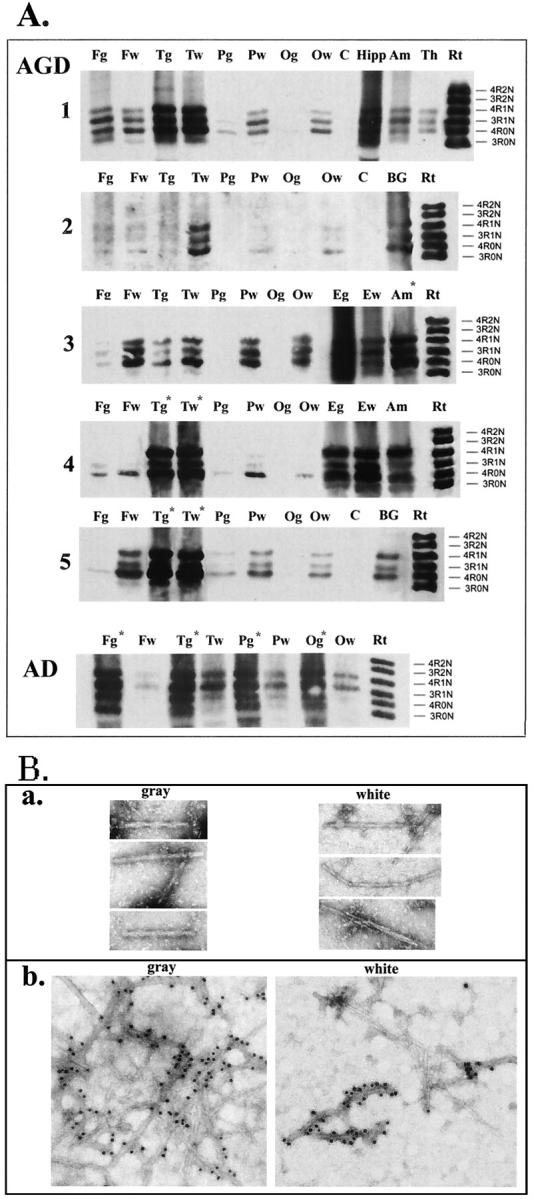
Biochemical and ultrastructural analysis of sarcosyl-insoluble τ. A: Dephosphorylated samples from different brain regions were resolved onto 7.5% SDS-polyacrylamide gel. Twenty percent of total sarcosyl-insoluble material from each sample was loaded in each lane, and nitrocellulose replicas were probed with a mixture of mAbs Tau14 and Tau46. A: Brain region-specific distribution of pathological τ. F, frontal; T, temporal; P, parietal; O, occipital cortex; C, cerebellum; Hipp, hippocampus; Amy, amygdala; Th, thalamus; E, entorhinal cortex; BG, basal ganglia; g, gray matter; w, white matter. A mixture of six recombinant human τ isoforms is shown on the right for the comparison (Rt). Case numbers are shown on the left and on the right. Asterisks indicate samples with 30% reduction of loaded material to avoid the saturation. B: Dispersed τ filaments were isolated from frozen temporal cortical gray and white matter (case 2) adjacent to tissue used for biochemical analysis. a: Electron microphotographs of τ filament stained with 0.4% uranyl acetate. Microphotographs were taken at a nominal original magnification of ×120,000. b: Immunogold labeling of τ filaments using PHF1 antibodies.
In dephosphorylated samples, τ isoforms were comprised of a mixture of 3R- and 4R-τ in all five AGD cases similar to AD (Figure 2A) ▶ . However, unlike AD in which 3R- and 4R-τ ratio is ∼1, 22 a preponderance of 4R-τ isoforms (4R-0N and 4R-1N) in the mediotemporal and entorhinal regions was evident in cases 3 (4R:3R = 1.2:1), 4 (2.2:1), and 5 (1.9:1).
Electron Microscopy of Isolated τ Filaments
The electron microscopy of tissue sections from AGD brains described by others 1,5,10,18 revealed that AGs consist of 9- to 19-nm straight τ filaments. To further characterize pathological τ in AGD, we isolated dispersed τ filaments from sarcosyl-insoluble fractions of the separately dissected temporal gray and white matter samples that were used for the biochemical studies of case 2 (Figure 2B) ▶ . In these preparations straight τ filaments were predominant structures (width, ∼9 to 22 nm), but paired helical filament (PHF)-like structures similar to those found in AD also were detected in low quantities only in the gray matter. The phosphorylation-dependent mAb PHF1 decorated the entire length of filaments isolated from both gray and white matter. A subset of PHF1-negative thin straight filaments (width, ∼9 to 10 nm) was also detected in the preparation from the white matter thereby suggesting heterogeneity of filaments in AGD.
Sequential Extraction of τ Proteins
Sarcosyl-insoluble τ proteins represent a substantial fraction of the total extractable τ pool and are commonly used to characterize the distribution of τ pathology throughout the brain. 11,23 To further characterize pathological τ proteins in AGD and to compare it with AD and PiD, we conducted studies of a wide spectrum of τ proteins ranging from soluble to very insoluble species using a graded sequential series of buffers containing detergents or acids with an increasing ability to solublize proteins. To ensure the completeness of extraction, each step was repeated twice. The amounts of soluble τ extracted from separately dissected samples of gray and white matter with 0.75 of mol/L NaCl (buffer A) and 1% Triton X-100 were comparable in different cases (Figure 3) ▶ . The more insoluble τ proteins (ie, those extracted with RIPA buffer) were detected only in two PiD cases, but these species were below the level of detection by Western blot methods in AD and AGD. Further extractions of samples with 2% SDS and 70% formic acid recovered the most highly insoluble τ proteins in variable amounts. Although the amount of SDS-soluble τ in all five AGD cases was variable and substantially lower than in AD and PiD, formic acid extracts contained comparable amounts of insoluble τ in all cases. Interestingly, in three (cases 2 to 5) of five AGD brains, highly insoluble τ species were detected in the formic acid extracts, and in cases 2 and 4, high-molecular weight τ-immunopositive aggregates were evident in the upper portion of the gels (arrows) consistent with a pool of formic acid resistant or insoluble τ. Also, a banding pattern of τ proteins in SDS-soluble fractions of AD brains was represented by a typical triplet of 69-, 64-, and 60-kd bands similar to a sarcosyl-insoluble τ pattern, whereas in PiD and AGD only two bands of 69 and 64 kd were evident in both SDS and formic acid extracts.
Figure 3.

Sequential extraction of τ proteins from AGD, AD, and PiD cases: Western blot analysis. Temporal cortex (AD, cases 8 and 9; PiD, cases 6 and 7; AGD, cases 1, 2, and 5) and entorhinal cortex and amygdala (AGD, case 4) with abundant τ pathology were studied after repeated extraction with buffers of increasing stringency as described in Materials and Methods. Lane 1: Buffer A (first extraction is shown); lane 2: 1% Triton X-100 in buffer A; lane 3: RIPA buffer; lane 4: 2% SDS; lane 5: 70% formic acid. A mixture of mAbs Tau14 and Tau46 were used for the Western blot analysis. An equal amount of material from each extraction step was loaded. Highly insoluble aggregated τ species were detected in AGD cases 2, 4, and 5 (arrowheads).
Discussion
Although AGD is well characterized neuropathologically based on the abundant presence of Gallyas and τ-positive AGs within the limbic system of elderly demented individuals, 2,5,6,26 biochemical analysis of τ proteins has been reported only recently in several AGD cases. 27 Analysis of τ proteins in five AGD brains reported here determined the pathological τ profiles in this tauopathy. Brain region-specific analyses of sarcosyl-insoluble fractions from separately dissected gray and white matter samples from various cortical regions revealed that pathological τ in AGD was confined primarily to mediotemporal cortical gray and adjacent white matter, and also to entorhinal cortex, amygdala, and hippocampus.
The amounts of sarcosyl-insoluble τ in all five AGD cases were substantially lower than those in AD and PiD. The abundance of τ-positive AGs usually correlates with advancing age and disease severity, 2,28 and there is some evidence that AGs can occur without other neurodegenerative changes or neuropsychological impairments. 16,19 In our study we found no obvious correlation between the amount of τ pathology and progression of dementia in all AGD cases. Interestingly, the most severe burden of AGs in our AGD cases was found in a 54-year-old patient (case 5) with clinical symptoms of frontotemporal dementia with motor neuron disease. Although the overall τ pathology in AGD was lower than in AD and PiD, the amount of sarcosyl-insoluble τ in AGD white matter was higher or comparable to that detected in gray matter from the same region. This distinguishes AGD from AD (wherein there is little or no white matter τ pathology), and these data support the notion that τ-positive glial inclusions are major neuropathological lesions in AGD subcortical white matter.
Notably, similar to AGD, white matter τ lesions also distinguish PiD, progressive supranuclear palsy, and corticalbasal degeneration from AD. 11,29-31 Although the banding patterns of τ isoforms in AGD varied, in some cases they were similar to AD, whereas in other cases or brain regions, 4R-0N and 4R-1N τ isoforms were overrepresented.
Ultrastructural analyses of τ filaments have been widely used to characterize τ lesions in tauopathies such as FTDP-17, 23,32-35 progressive supranuclear palsy, corticalbasal degeneration, 36,37 and PiD. 11,38-40 Isolated dispersed τ filaments from the AGD brain described here were straight with only occasional PHF-like structures present. Moreover, we used a sequential extraction approach 24 to analyze the profile of abnormal τ proteins in AGD, and showed that there was no significant difference between the banding pattern of normal τ in soluble fractions among cases, although the amount of τ protein was variable as described in other tauopathies. 20
Although the amount of SDS-extractable τ from gray and white matter was low in AGD, the amount of highly insoluble τ species in formic acid extracts of four of five AGD cases was comparable to that in AD and PiD cases. Interestingly, variable amounts of high-molecular weight τ-positive aggregates were evident in formic acid fractions mostly in the white matter samples from three AGD cases, and this is in concordance with our observation that sarcosyl-insoluble τ was also found predominantly in the white matter of different brain regions. The differential τ solubility reported for the first time in this study of AGD might provide additional information for understanding the mechanisms that lead to the formation of heterogeneous τ inclusions in different tauopathies, but further studies are needed to elucidate the role of abnormal τ and AGs in the pathogenesis of AGD.
Acknowledgments
We thank the families of our patients who made this research possible, Dr. M. Neuman for her help in collecting patient information, and Mr. D. Martinez for technical assistance.
Footnotes
Address reprint requests to Virginia M.-Y. Lee, Ph.D., Center for Neurodegenerative Disease Research, Maloney 3, HUP, 3600 Spruce St., Philadelphia, PA 19104-4283. E-mail: vmylee@mail.med.upenn.edu.
Supported by the National Institute on Aging (grants AG17586 to V. M.-Y. L. and AG 10124 to J. Q. T.).
V. Y.-M. L. is the John H. Ware, III, Professor of Alzheimer’s Disease Research at the University of Pennsylvania.
References
- 1.Tolnay M, Spillantini MG, Goedert M, Ulrich J, Langui D, Probst A: Argyrophilic grain disease: widespread hypophosphorylation of tau protein in limbic neurons. Acta Neuropathol 1997, 93:477-484 [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E: Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm 1998, 105:801-819 [DOI] [PubMed] [Google Scholar]
- 3.Tolnay M, Probst A: Review: tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol 1999, 25:171-187 [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Bohl J, Bratzke H, Braak E: Pathological changes in the parahippocampal region in select non-Alzheimer’s dementias. Ann NY Acad Sci 2000, 911:221-239 [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989, 15:13-26 [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Braak E: Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 1987, 76:124-127 [DOI] [PubMed] [Google Scholar]
- 7.Itagaki S, McGeer PL, Akiyama H, Beattie BL, Walker DG, Moor DRW, McGeer EG: A case of adult-onset dementia with argyrophilic grains. Ann Neurol 1989, 26:685-689 [DOI] [PubMed] [Google Scholar]
- 8.Cras P, Perry G: Dementia with argyrophilic grains. Ann Neurol 1991, 30:853-854 [DOI] [PubMed] [Google Scholar]
- 9.Tolnay M, Ipsen S, Misti C, Probst A: Argyrophilic grain disease: occurrence of grains inside dendritic branches of neurons containing hyperphosphorylated tau protein. Brain Pathol 1997, 7:1176 [Google Scholar]
- 10.Jellinger KA: Dementia with grains (argyrophilic grain disease). Brain Pathol 1998, 8:377-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller B, Hulette CM, Feinstein SC, Trojanowski JQ, Lee VM-Y: Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol 2002, 51:730-739 [DOI] [PubMed] [Google Scholar]
- 12.Masliah E, Hansen LA, Quijata S, De Teresa R, Alford M, Kauss J, Terry R: Late onset dementia with argyrophilic grains and subcortical tangles or atypical progressive supranuclear palsy? Ann Neurol 1991, 29:389-396 [DOI] [PubMed] [Google Scholar]
- 13.Togo T, Dickson DW: Ballooned neurons in progressive supranuclear palsy are usually due to concurrent argyrophilic grain disease. Acta Neuropathol 2002, 104:53-56 [DOI] [PubMed] [Google Scholar]
- 14.Arima K, Muriyama S, Mukoyama M, Inose T: Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. Acta Neuropathol 1992, 83:455-460 [DOI] [PubMed] [Google Scholar]
- 15.Wakabayashi K, Kawachi I, Toyoshima Y, Takahashi H: Occurrence of argyrophilic grains in multiple system atrophy: histopathological examination of 26 autopsy cases. Brain Nerve 1999, 51:433-437 [PubMed] [Google Scholar]
- 16.Martinez-Lage P, Munoz DG: Prevalence of diseases associated with argyrophilic grains of Braak. Neuropathol Exp Neurol 1997, 56:157-164 [DOI] [PubMed] [Google Scholar]
- 17.Tolnay M, Monsch AU, Probst A: Argyrophilic grain disease: a frequent dementing disorder in aged patients. Adv Exp Med Biol 2001, 487:39-58 [PubMed] [Google Scholar]
- 18.Ikeda K, Akiyama H, Kondo H, Haga C: A study of dementia with argyrophilic grains. Possible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. Acta Neuropathol 1995, 89:409-414 [DOI] [PubMed] [Google Scholar]
- 19.Tolnay M, Schwietert M, Monsch AU, Staehelin H, Langui D, Probst A: Argyrophilic grain disease: distribution of grains in patients with and without dementia. Acta Neuropathol 1997, 94:353-358 [DOI] [PubMed] [Google Scholar]
- 20.Zhukareva V, Vogelsberg-Ragaglia V, Van Deerlin VMD, Bruce J, Shuck T, Grossman M, Clark CM, Arnold SE, Masliah E, Galasko D, Trojanowski JQ, Lee VM-Y: Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol 2001, 49:165-175 [DOI] [PubMed] [Google Scholar]
- 21.Lee VM-Y, Wang J, Trojanowski JQ: Purification of paired helical filament tau and normal tau from human brain tissue. Methods Enzymol 1999, 309:81-89 [DOI] [PubMed] [Google Scholar]
- 22.Hong M, Zhukareva V, Vogelsberg-Ragalia V, Wzolek Z, Reed L, Miller B, Geschwind DH, Bird TD, McKeel D, Goate A, Morris J, Wihlemsen KC, Schellenberg GD, Trojanowski JQ, Lee VM-Y: Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282:1914-1917 [DOI] [PubMed] [Google Scholar]
- 23.Lippa CF, Zhukareva VA, Kawarai T, Uryu K, Shafig M, Nee LE, Grafman J, Liang Y, George-Hyslop PH, Trojanowski JQ, Lee VM-Y: Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol 2000, 48:850-858 [PubMed] [Google Scholar]
- 24.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y: Age-dependent emergence and progression of tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 1999, 24:751-762 [DOI] [PubMed] [Google Scholar]
- 25.Ksiezak-Reading H, Wall JS: Mass and physical dimensions of two distinct populations of paired helical filaments. Neurobiol Aging 1994, 15:11-19 [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Braak E: On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol 1985, 68:325-332 [DOI] [PubMed] [Google Scholar]
- 27.Togo T, Sahara N, Yen S-H, Cookson N, Ishizawa T, Hutton M, De Silva R, Lees A, Dickson DW: Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 2002, 61:547-556 [DOI] [PubMed] [Google Scholar]
- 28.Davis DG, Ross GW, Petrovitch H, White LR, Hardman JM, Nelson JS, Thiessen P, Wang HZ, Patel E, Markesbery WR: Quantitation of argyrophilic grains in hippocampal CA-1 of aged Japanese-American men. J Neuropathol Exp Neurol 1997, 56:587Abstract [Google Scholar]
- 29.Ikeda K, Akiyama H, Haga C, Kondo H, Arima K, Oda T: Argyrophilic thread-like structures in cortical basal degeneration and supranuclear palsy. Neurosci Lett 1994, 174:157-159 [DOI] [PubMed] [Google Scholar]
- 30.Feany MB, Dickson DW: Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol 1996, 40:139-148 [DOI] [PubMed] [Google Scholar]
- 31.Chin S-M, Goldman JE: Glial inclusions in CNS degenerative disorders. J Neurol Exp Pathol 1996, 55:499-508 [DOI] [PubMed] [Google Scholar]
- 32.Goedert M, Spillantini MG, Crowter RA, Chen SC, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Peteresen RB, Gambetty PL: Tau gene mutation in familial progressive subcortical gliosis. Nat Med 1999, 5:454-457 [DOI] [PubMed] [Google Scholar]
- 33.Murrell JR, Spillantini MG, Smoth MJ, Hasegava M, Redi F, Crowter RA, Pietrini P, Getti B, Goedert M: Tau gene mutation G389R causes a tauopathy with abundant Pick body-like inclusions and axonal deposits. J Neuropath Exp Neurol 2000, 58:1207-1226 [DOI] [PubMed] [Google Scholar]
- 34.Spillantini MG, Growter RA, Kamphorst W, Heutink P, van Swieten JC: Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol 1998, 153:1359-1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasuda S, Takamatzu J, D’Souza I, Crowter RA, Kawamata T, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg GD, Tanaka C: A novel mutation at position +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto). Ann Neurol 2000, 47:422-429 [PubMed] [Google Scholar]
- 36.Crowter RA, Goedert M: Abnormal tau-containing filaments in neurodegenerative disorders. J Struct Biol 2000, 130:271-279 [DOI] [PubMed] [Google Scholar]
- 37.Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, Wiedenheim K, Dickson DW: Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol 1994, 145:1496-1508 [PMC free article] [PubMed] [Google Scholar]
- 38.Murayama S, Mori H, Ihara Y, Tomonaga M: Immunocytochemical and ultrastructural studies of Pick’s disease. Ann Neurol 1990, 27:394-405 [DOI] [PubMed] [Google Scholar]
- 39.King M, Ghoshal N, Wall JS, Binder LI, Ksiezak-Reding H: Structural analysis of Pick’s disease-derived and in-vitro assembled tau filaments. Am J Pathol 2001, 158:1481-1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee VM-Y, Balin BJ, Otvos L, Trojanowski JQ: A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 1991, 251:675-678 [DOI] [PubMed] [Google Scholar]