

Abstract
Our previous studies demonstrated that enhanced epithelial cell proliferation is important for healing of experimental esophageal ulcers. However, the roles of angiogenesis, its major mediator, vascular endothelial growth factor (VEGF), and the mechanism(s) regulating VEGF expression during esophageal ulcer healing remain unknown. Esophageal ulcers were induced in rats by focal application of acetic acid. We studied expressions of hypoxia-inducible transcription factor-1α (HIF-1α), an activator of the VEGF gene, and VEGF by reverse transcriptase-polymerase chain reaction, Western blotting, and immunostaining. To determine the efficacy of VEGF gene therapy in esophageal ulcer healing, we studied whether a single local injection of plasmid cDNA encoding recombinant human VEGF165 affects ulcer healing and angiogenesis. Esophageal ulceration induced HIF-1α protein expression and VEGF gene activation reflected by increased VEGF mRNA (240%) and VEGF protein (310%) levels. HIF-1α protein was expressed in microvessels bordering necrosis where it co-localized with VEGF. Injection of cDNA encoding VEGF165 significantly enhanced angiogenesis and accelerated esophageal ulcer healing. These results: 1) suggest that HIF-1α may mediate esophageal ulceration-triggered VEGF gene activation, 2) indicate an essential role of VEGF and angiogenesis in esophageal ulcer healing, and 3) demonstrate the feasibility of gene therapy for the treatment of esophageal ulcers.
Although the cellular and molecular mechanisms of gastric ulcer healing have been extensively studied and well characterized, 1-5 the mechanisms involved in the healing of esophageal ulcers remain poorly understood. Our previous studies demonstrated that enhanced epithelial proliferation at the ulcer margin is an important component of experimental esophageal ulcer healing. 6,7 However, the role of other factors, such as angiogenesis and its mediators, in esophageal ulcer healing remains unknown. Angiogenesis—formation of new capillary blood vessels—in granulation tissue at the ulcer base is essential for gastric ulcer healing because it enables oxygen and nutrient delivery to the healing site. 8 There are however, major structural and functional differences between gastric and esophageal mucosa as well as differences in mediators of gastric and esophageal ulcer healing. Gastric mucosa consists of glands composed of a single layer of epithelial cells embedded into a richly vascularized lamina propria. 9 In contrast, esophageal mucosa is composed of a thick stratified squamous epithelium consisting of several layers of epithelial cells covering less well vascularized lamina propria. 9 Therefore, the healing of esophageal ulcers could be less dependent on angiogenesis than that of gastric ulcers. Epidermal growth factor and hepatocyte growth factor (but not keratinocyte growth factor) genes are induced by gastric ulceration and play an essential role in healing of gastric glandular epithelial structures. 1-3,10 In contrast, as demonstrated in our previous study, keratinocyte growth factor seems to be important for esophageal re-epithelialization and esophageal ulcer healing. 6 The role of angiogenic growth factors in esophageal ulcer healing has not been explored.
Vascular endothelial growth factor (VEGF), an endothelial cell-specific mitogen, is the most potent angiogenic growth factor. 11 Previously, we demonstrated that exogenous VEGF accelerates healing of ethanol-induced gastric erosions 12 and that VEGF gene activation is required to elicit the angiogenic response in acutely injured gastric mucosa. 13 VEGF has also been implicated in the angiogenic response to gastric ulceration 14 and a single local injection of a nonviral plasmid encoding recombinant human (rh) VEGF165 has been shown to stimulate angiogenesis and accelerate experimental gastric ulcer healing. 15 However, the roles of endogenous and exogenous VEGF in healing of esophageal ulcers remain unexplored. In addition, the mechanism(s) responsible for the induction of VEGF expression during esophageal and/or gastrointestinal ulcer healing are not known. Hypoxia is a potent stimulator of VEGF gene expression. 16,17 Hypoxia induces VEGF gene expression via the hypoxia-inducible factor (HIF)-1, 18,19 which is composed of two subunits: HIF-1α and HIF-1β. 20,21 Under normoxic conditions, HIF-1β protein is relatively stable, whereas, HIF-1α protein is continuously produced but rapidly degraded. 22 In contrast, hypoxia stabilizes the HIF-1α protein leading to its accumulation within the cell and formation of the active HIF-1 complex. 21,22 A recent study demonstrated that HIF-1α mRNA is induced during dermal wound healing, 23 but the expression of HIF-1α protein during healing of esophageal as well as other gastrointestinal ulcers has not been investigated.
This study was aimed to determine whether: 1) esophageal ulceration induces HIF-1α, 2) activates the VEGF gene, and 3) a single local injection of a nonviral plasmid encoding rhVEGF165 cDNA affects angiogenesis and healing of experimental esophageal ulcers.
Materials and Methods
Induction of Esophageal Ulcers
This study was approved by the Subcommittee for Animal Studies of the Long Beach (CA) Department of Veterans Affairs Medical Center. Male Sprague-Dawley rats, weighing 225 to 250 g, were used in the experiments. Rats were kept individually in wire-bottom cages with free access to a standard rat chow (Rodent diet no.8504; Harlan Teklad, Madison, WI) and water. The animal room was illuminated on 12-hour light-dark cycles. Room temperature was kept at 18 to 22°C and humidity at 60 to 70%. Rats underwent laparotomy under ketamine-xylazine anesthesia (40 mg/kg body wt of ketamine and 5 mg/kg body wt of xylazine, i.p.). Esophageal ulcers were induced by modification of the method described by Tsuji and colleagues 24 In brief, 100% acetic acid (30 μL) was applied focally to the anterior wall of the intra-abdominal esophagus through a polyethylene tube (3-mm inner diameter) for 3 minutes. The area was then washed with isotonic saline and the abdomen was closed. Control rats underwent similar procedures except application of acetic acid (sham operation).
Effect of Ulceration on HIF-1α, HIF-1β, and VEGF Expression
Rats with esophageal ulcers and sham-operated rats were euthanized 1, 3, and 7 days after ulcer induction or sham operation. In each rat, a 1-cm-long segment of the esophagus was excised and cut longitudinally (through the center of the ulcer crater in rats with esophageal ulcers) into two portions. One half was snap-frozen in liquid nitrogen and stored at −80°C for RNA isolation and protein extraction and the other half was fixed in 10% formalin for immunostaining.
Effects of Injection of a Plasmid Encoding rhVEGF165 on Esophageal Ulcer Healing
In rats undergoing gene therapy, immediately after ulcer induction either 100 μg of plasmid DNA encoding the full-length cDNA of rhVEGF165 (VEGF group) or 100 μg of plasmid without cDNA insert (control group) was injected into the esophageal muscle layers around the area of ulcer induction. Three and 7 days after ulcer induction/injection, six rats from each group (control and VEGF) were euthanized. In each rat, the esophagus was excised, opened longitudinally, and the ulcer was photographed. The area of mucosal defect (ulcer area) was measured using a computerized video analysis system (Image 1/FL; Universal Imaging Corp., Westchester, PA), and a 1-cm-long segment of the esophagus (including ulcer) was excised and fixed in 10% formalin for immunohistochemical staining. In additional experiments, rats injected with plasmid encoding rhVEGF165 or with control plasmid were euthanized 7 and 14 days after ulcer induction.
Plasmid Preparation
The plasmid encoding the full-length cDNA of the rhVEGF165 gene fused at the C-terminus to the 6×His epitope tag was constructed essentially in the same way as previously described for the plasmid encoding rh angiopoietin-1. 25,26 The same plasmid used for VEGF gene construct, but without a cDNA insert, served as control. Plasmids were amplified in DH5a bacteria and were purified free of endotoxin using an endo-free plasmid maxi kit (Qiagen, Valencia, CA).
Determination of VEGF mRNA Expression by Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
RNA was isolated using RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. RT-PCR was performed as described previously. 13 The primers for VEGF were 5′-CCTGGTGGACATCTTCCAGGAGTACC-3′ (sense)and 5′-GAAGCTCATCTCTCCTATGTGCTGGC-3′ (anti-sense), and the size of the amplified fragment conserved in all of the variant spliced forms was 196 bp. PCR for β-actin was used as a positive control and as an internal standard. The specific primer set for rat β-actin was purchased from Clontech Laboratories, Inc., Palo Alto, CA. For the quantitative assessment of the PCR products, a computerized video analysis system (Image-1/FL, Universal Imaging Corp.) was used. The results are expressed as the VEGF/β-actin ratio.
Determination of HIF-1α, HIF-1β, VEGF, and 6×His-VEGF165 Protein Expression by Western Blotting
Protein extraction and Western blotting were performed as described previously. 13 Equal amounts of protein (150 μg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated with mouse anti-HIF-1α monoclonal antibody (Novus Biologicals, Littleton, CO), mouse anti-HIF-1β monoclonal antibody (Novus Biologicals), or mouse anti-VEGF monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature for 1 hour. The membranes were then washed and incubated with peroxidase-conjugated anti-mouse goat immunoglobulin (Transduction Laboratories, Lexington, KY) at room temperature for 1 hour. For determination of 6×His-VEGF165 fusion protein expression, the membranes were incubated with rabbit polyclonal anti-6×His antibody (Santa Cruz Biotechnology) and peroxidase-conjugated anti-rabbit goat immunoglobulin (Sigma Chemical Co., St. Louis, MO). Signal of bound antibodies was visualized using enhanced chemiluminescence Western blotting detection reagents (Amersham Life Science, Arlington Heights, IL). Protein levels were measured using a computerized video analysis system (Image-1/FL, Universal Imaging Corp.).
Determination of HIF-1α, VEGF, and 6×His-VEGF165 Protein Expression by Immunohistochemical Staining
Deparaffinized sections were incubated with either anti-HIF-1α antibody or anti-VEGF antibody overnight at 4°C. After washing with phosphate-buffered saline (PBS), the sections were incubated with multilink swine immunoglobulin (DAKO, Carpinteria, CA) followed by washing with PBS and incubation with StreptABComplex (DAKO). The color was developed with the AEC Substrate System (DAKO) and the sections were counterstained with Mayer’s hematoxylin (DAKO). For determination of 6×His-VEGF165 protein expression, the sections were incubated with anti-6×His antibody and corresponding fluorescein isothiocyanate-conjugated immunoglobulin (Sigma Chemical Co.). Immunofluorescence signal was evaluated using a Nikon Optiphot epifluorescence microscope (Nikon Inc., Garden City, NY) with an Omega filter fluorescein isothiocyanate/Tex Red.
Assessment of Angiogenesis
To identify microvessels, enhanced polymer one-step staining 27 with monoclonal mouse antibody against Factor VIII-related antigen (DAKO), which specifically detects endothelial cells, was used. Deparaffinized sections were incubated with antibody for 1 hour at room temperature. Color was developed with 3,3′-diaminobenzidine tetrahydrochloride (DAKO) and the sections were counterstained with Mayer’s hematoxylin (DAKO). Factor VIII-related antigen-positive microvessels were counted in two microscopic fields (×200 magnification) in granulation tissue immediately below the ulcer margin at each side. The results were expressed as a number of microvessels per mm2 (microvessel density). Coded sections were evaluated by two investigators unaware of the code. Two sections per each rat were evaluated and the mean was calculated.
Statistical Analysis
Results are expressed as the mean ± SD. Student’s t-test was used to determine statistical significance of differences between two treatment groups. Comparisons of data between multiple groups were performed with analysis of variance followed by Bonferroni correction. Pearson product moment correlation analysis was used to determine the significance of relationship between variables. A P value of <0.05 was considered statistically significant.
Results
HIF-1α and HIF-1β Protein Expression by Western Blotting
In normal, nonulcerated esophageal tissue of sham-operated rats HIF-1α protein expression was not detected by immunoblotting at all time points (Figure 1) ▶ . In contrast, HIF-1α protein expression was detected in ulcerated esophageal tissue as early as 1 day after ulcer induction and was significantly increased both 3 and 7 days versus 1 day after ulcer induction (Figure 1) ▶ . HIF-1β protein expression was not significantly affected by esophageal ulceration (Figure 1) ▶ .
Figure 1.

Western blot detection of HIF-1α and HIF-1β protein expression in ulcerated (UL) esophageal tissue versus nonulcerated esophageal tissue from sham-operated (SO) rats 1, 3, and 7 days after ulcer induction or sham operation. Top: Immunoblotting with anti-HIF-1α antibody detected specific ≈120-kd bands only in ulcerated, but not in nonulcerated esophageal tissue of sham-operated rats. Immunoblotting with anti-HIF-1β antibody detected specific ≈95-kd bands in both ulcerated and nonulcerated esophageal tissue of sham-operated rats. Bottom: Quantitative data for HIF-1α protein expression in ulcerated esophageal tissue. Data were obtained by a computerized video analysis of the Western blots. Values are expressed in intensity units and represent means ± SD. For each column, n = 6.
VEGF mRNA Expression by RT-PCR
VEGF mRNA levels in ulcerated esophageal tissue were significantly increased versus nonulcerated esophageal tissue 1 day after ulcer induction. Three and 7 days after ulcer induction, VEGF mRNA levels in ulcerated esophageal tissue were increased 240% and 210%, respectively, versus the corresponding nonulcerated esophageal tissue of sham-operated rats (Figure 2) ▶ .
Figure 2.

VEGF mRNA expression in ulcerated (UL) and nonulcerated esophageal tissue from sham-operated (SO) rats detected by RT-PCR. Tissues were obtained 1, 3, and 7 days after ulcer induction or sham operation. Top: RT-PCR products obtained with use of specific primers that recognize all four isoforms of VEGF mRNA (196 bp) and primers that recognize rat β-actin. Bottom: Quantitative data for VEGF mRNA expression. Data were obtained by computerized analysis of amplified PCR products. Each signal was normalized against the corresponding β-actin signal and the results are expressed as a ratio of VEGF/β-actin. Values are means ± SD. For each column, n = 6.
VEGF Protein Expression by Western Blotting
Western blotting with specific anti-VEGF antibody demonstrated the presence of the secreted form of VEGF protein, VEGF165, in nonulcerated rat esophageal tissue (Figure 3) ▶ . One day after ulcer induction, VEGF165 protein levels in ulcerated esophageal tissue were not significantly different from those in nonulcerated esophageal tissue. However, 3 and 7 days after ulcer induction, VEGF165 protein levels in ulcerated tissue were increased 310% and 290%, respectively, versus the corresponding nonulcerated esophageal tissue from sham-operated rats (Figure 3) ▶ . The expression of the larger nonsecreted form of VEGF protein was affected by esophageal ulceration similarly to that of VEGF165 (data not shown).
Figure 3.

Western blot detection of VEGF protein expression in ulcerated (UL) esophageal tissue versus nonulcerated esophageal tissue from sham-operated (SO) rats 1, 3, and 7 days after ulcer induction or sham operation. Top: Immunoblotting with anti-VEGF antibody detected specific ≈26-kd bands for VEGF165. Bottom: Quantitative data for VEGF165 protein expression. Data were obtained by a computerized video analysis of the Western blots. Values are expressed in intensity units and represent means ± SD. For each column, n = 6.
HIF-1α and VEGF Protein Expression by Immunostaining
There was no positive staining for HIF-1α in sections of nonulcerated esophagus of sham-operated rats incubated with anti-HIF-1α antibody (Figure 4A) ▶ . In contrast, 3 days after ulcer induction, HIF-1α signal was present in endothelial cells of microvessels of the esophageal muscularis propria adjacent to the necrotic tissue (Figure 4B) ▶ and in submucosal vessels below the ulcer margin. In nonulcerated esophagus of sham-operated rats, VEGF signal was detected in some microvessels (endothelial cells and pericytes) of muscularis propria (Figure 4C) ▶ , in muscularis mucosa, in large submucosal vessels, and in basal epithelial cells. Three days after ulcer induction, VEGF signal was significantly enhanced and expanded to all microvessels of the muscularis propria adjacent to the ulcer bed (Figure 4D) ▶ . VEGF (but not HIF-1α) signal was also detected in esophageal epithelial cells at the ulcer margin (data not shown).
Figure 4.

Photomicrographs showing immunostaining for HIF-1α and VEGF in rat esophageal muscularis propria 3 days after sham-operation or ulcer induction. A: HIF-1α signal is absent in sections from sham-operated rats. B: HIF-1α expression in microvessels adjacent to the necrotic tissue of the ulcer bed. C: VEGF expression in microvessels of sham-operated rats. D: VEGF expression in microvessels adjacent to the necrotic tissue of the ulcer bed. Scale bars, 100 μm.
6×His-VEGF165 Protein Expression
We detected 6×His-VEGF165 fusion protein by Western blotting in ulcerated esophageal tissue obtained 7 days, but not at 14 days, after injection of plasmid encoding rhVEGF165 (Figure 5) ▶ . Seven days after ulcer induction, immunofluorescence staining of histological sections from ulcers injected with plasmid encoding rhVEGF165 revealed specific (green fluorescence) staining of vessels and microvessels in the granulation tissue of the ulcer bed (Figure 6B) ▶ . Specific (green fluorescence) staining was absent in sections of ulcers injected with control plasmid (Figure 6A) ▶ .
Figure 5.

6×His-VEGF165-fusion protein expression by Western blot analysis in ulcerated esophageal tissue 7 and 14 days after injection of either control plasmid (control) or plasmid encoding rhVEGF165 (VEGF).
Figure 6.

Photomicrographs showing expression by immunohistochemical staining of 6×His-VEGF165-fusion protein in granulation tissue of the ulcer bed 7 days after injection of plasmids. A: Control plasmid. Microvessels show absence of specific (green fluorescence) staining. B: Plasmid encoding rhVEGF165. Positive staining is present in numerous vessels and microvessels. Arrows indicate vessels. Scale bars, 50 μm.
Injection of a Plasmid Encoding rhVEGF165 Accelerates Esophageal Ulcer Healing and Stimulates Angiogenesis
Three days after ulcer induction, no significant difference was detected in the ulcer size between the control and VEGF groups indicating that injection of plasmids did not affect the development of esophageal ulcers (data not shown). Seven days after ulcer induction, round or oval-shaped, clearly delineated deep mucosal defects (ulcers) were present in both groups, but the ulcers in the VEGF group were significantly smaller than in the control group (Figure 7) ▶ . The mean area of mucosal defect (ulcer area) was reduced by more than twofold from 3.1 mm2 in the control group to 1.46 mm2 in the VEGF group (see Figure 9 ▶ ). Three days after ulcer induction, only a few microvessels with lumen were detected in the ulcer bed (data not shown). Seven days after ulcer induction, granulation tissue was developed at the ulcer bed and contained well formed microvessels (Figure 8) ▶ . In rats injected with plasmid encoding rhVEGF165, the number of microvessels in granulation tissue was significantly increased compared with rats injected with control plasmid (Figure 8) ▶ . Quantitative assessment of the sections stained with antibody against Factor VIII-related antigen revealed that the microvessel density in granulation tissue of the ulcer bed was increased 2.5-fold in the VEGF group compared to the control group (Figure 9) ▶ . Seven days after ulcer induction/plasmid injection, a strong negative correlation was observed between the microvessel density and the ulcer area in either control (r = −0.992, P < 0.001) or rhVEGF165-injected (r = −0.978, P < 0.001) groups. Correlation analysis of pooled data from both groups is presented in Figure 10 ▶ .
Figure 7.

Macroscopic appearance of acetic acid-induced esophageal ulcers 7 days after injection of indicated plasmids. Esophagus was dissected and opened longitudinally. A: Treatment with control plasmid. B: Treatment with plasmid encoding rhVEGF165. Scale is marked in mm.
Figure 9.

Quantitative evaluation of ulcer area (left) and microvessel density in granulation tissue below the epithelium of the ulcer margin (right) in rats injected either with control plasmid (control) or plasmid encoding rhVEGF165 (VEGF) 7 days after ulcer induction/injection. Ulcer area (area of mucosal defect) was measured by a computerized video analysis system. Microvessel density was calculated as the number of microvessels per mm2 of granulation tissue section. Values are means ± SD. For each column, n = 6.
Figure 8.
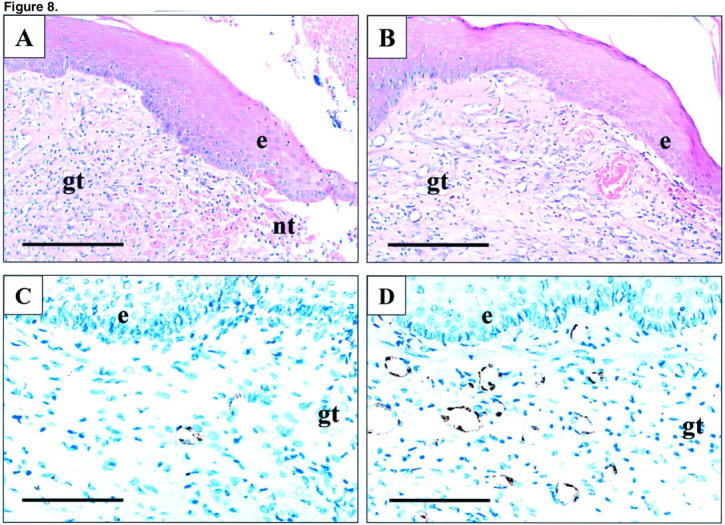
Photomicrographs of esophageal ulcer margin 7 days after injection of indicated plasmids. A and C: Control plasmid. B and D: Plasmid encoding rhVEGF165. A and B: H&E staining. C and D: Immunostaining for Factor VIII-related antigen. Factor VIII-related antigen expression (brown staining) is present in the cytoplasm of endothelial cells forming microvessels. e, epithelium; gt, granulation tissue; nt, necrotic tissue. Scale bars, 200 μm (A and B); 100 μm (C and D).
Figure 10.

Correlation between the ulcer area and the microvessel density (number of microvessels per mm2 of granulation tissue section) in rats treated with control plasmid and plasmid encoding rhVEGF165 7 days after ulcer induction (pooled data). r = −0.996, P < 0.001, n = 12.
Discussion
Vascular injury leading to ischemia is the major pathogenic factor in the development of acute and chronic tissue injury, including acetic acid-induced gastric ulcerations. 28 However, ischemia and the resultant reduction in tissue oxygen tension (hypoxia) also trigger the angiogenesis required to restore the microvascular network and blood supply and, thus, enable healing of damaged tissue. 29 In the present study, newly-formed microvessels were detected in granulation tissue of the esophageal ulcer bed indicating an intimate involvement of angiogenesis in the healing of esophageal ulcers. Furthermore, a strong negative correlation between the microvessel density in granulation tissue and the ulcer area in control rats indicates the important role of angiogenesis in spontaneous healing of esophageal ulcers.
Expression of constitutively active HIF-1α in skin of transgenic mice induces dermal hypervascularity and dramatically increases VEGF expression demonstrating the importance of HIF-1α for in vivo angiogenesis and VEGF expression. 30 However, the role of HIF-1α for angiogenesis and VEGF expression associated with healing of esophageal and/or gastrointestinal ulcers remains unclear. Previous studies have shown that hypoxia induces VEGF expression in pulmonary artery endothelial cells. 19 Our recent study demonstrated that hypoxia leads to accumulation of HIF-1α and the induction of VEGF expression and angiogenesis in rat gastric microvascular endothelial cells in vitro. 31 Here, we demonstrate that HIF-1α protein is not expressed in normal (nonischemic) esophageal tissue, but strongly expressed in ulcerated (ischemic) esophageal tissue. HIF-1α protein expression was localized to microvessels adjacent to the necrotic tissue of the ulcer bed. HIF-1α protein was expressed in ulcerated esophageal tissue at 1 day after ulcer induction preceding induction of VEGF protein expression. Moreover, HIF-1α signal was detected in endothelial cells of microvessels where it co-localized with that of VEGF as demonstrated by our immunohistochemical studies. Together, these results suggest that induction of HIF-1α protein expression may be involved in VEGF gene activation in regenerating microvessels during esophageal ulcer healing.
In situ hybridization studies revealed that VEGF mRNA is expressed by keratinocytes at the skin wound edge, identifying them as an important source of VEGF during wound healing. 32,33 HIF-1α mRNA expression was also detected by in situ hybridization in basal keratinocytes at the skin wound edge. 23 Our immunohistochemical studies revealed that VEGF protein, but not HIF-1α protein is expressed in esophageal epithelial cells at the ulcer margin. The earlier study 23 evaluated HIF-1α mRNA expression by in situ hybridization, whereas we determined HIF-1α protein expression by immunostaining. As mentioned in the introduction, HIF-1α is a constitutively synthesized protein that rapidly degrades under normoxic conditions. Hypoxia stabilizes HIF-1α leading to its intracellular accumulation. Therefore, it is possible that HIF-1α mRNA may also be expressed in esophageal epithelial cells, but this does not necessarily lead to HIF-1α protein accumulation which was what we evaluated in our study by the immunostaining technique.
VEGF gene transfection performed in the present study demonstrated the essential role of VEGF-induced angiogenesis in esophageal ulcer healing as reflected by a strong correlation between increased microvessel density and accelerated ulcer healing. The ulcers in the rhVEGF165-injected group were very small and similar in size explaining a slightly decreased correlation coefficient in the rhVEGF165-injected group compared to that in the control group. The expression of VEGF protein from the transgene was localized to regenerating microvessels of the ulcer bed indicating that the gene encoding rhVEGF165 was successfully transfected and was functionally active. Several clinical trials evaluating efficacy and safety of gene therapy with angiogenic growth factors reported no serious side effects associated with the angiogenesis agent. 34 Although the present study did not specifically address systemic reactions and side effects of the VEGF gene therapy, we did not observe any signs of distress, change in behavior, neovascularization of nontargeted tissues or malignancy, and there were no animal deaths related to gene transfection. In our previous study, we demonstrated that rhVEGF165 mRNA is expressed in ulcerated gastric tissue only transiently and disappeared from 7 days after plasmid injection. 15 Therefore, the respective protein could not be synthesized beyond this time point, whereas already synthesized protein might be degraded. This can explain our finding that rhVEGF165 protein was expressed only at day 7, but not at day 14, after plasmid injection. Because VEGF acts both as a mitogen and a survival factor for endothelial cells, 35,36 it is unlikely that discontinuation of the plasmid-specific VEGF protein expression was because of the increased cell turnover. In addition, expression of plasmid-specific VEGF protein was restricted to the granulation tissue of the ulcer bed. Therefore, transient local expression of VEGF from a transgene may represent a preferable new therapeutic approach in the treatment of esophageal ulcers.
This study was performed in an animal model of esophageal ulceration caused by serosal application of acetic acid. In humans, esophageal ulcers usually are presented as a complication of reflux esophagitis. In patients with reflux esophagitis and esophageal ulcers, frequent exposure of the ulcer base to gastric contents may adversely affect the outcome of VEGF gene therapy. Therefore, our findings cannot be directly translated into clinical esophageal ulcers. It should be pointed, however, that the morphological features of acetic acid-induced esophageal ulcers in rats are very similar to those of human esophageal ulcers, 6 which suggests that, regardless of the cause, once the ulcer develops, it undergoes similar common stages of repair and healing.
The results of the present study indicate that esophageal ulceration triggers induction of HIF-1α protein expression and activation of the VEGF gene and that angiogenesis is an essential component of esophageal ulcer healing. Our demonstration that VEGF gene therapy dramatically accelerates healing of experimental esophageal ulcers may provide a rationale for future clinical studies aimed at evaluating the efficacy of gene therapy with angiogenic growth factors for the treatment of esophageal ulcers.
Footnotes
Address reprint requests to A. S. Tarnawski, M.D., D.Sc., Professor of Medicine, Chief, Division of Gastroenterology, University of California, VA Medical Center, 5901 East Seventh St., Long Beach, CA 90822. E-mail: andrzej.tarnawski@med.va.gov.
Supported by the Department of Veterans Affairs Medical Research Service: Merit Reviews (to A.S.T. and M.K.J.) and the Research Enhancement Award Program (to A.S.T.).
KT was a visiting scientist from the Department of Surgery II, Kyushu University, Fukuoka, Japan; and WSM was a visiting scientist from the Department of Pathology, Chonbuk National University, Chonbuk, Korea.
References
- 1.Konturek SJ, Dembinski A, Warzecha Z, Brzozowski T, Gregory H: Role of epidermal growth factor in healing of chronic gastroduodenal ulcers in rats. Gastroenterology 1988, 94:1300-1307 [DOI] [PubMed] [Google Scholar]
- 2.Tarnawski A, Stachura J, Durbin T, Sarfeh IJ, Gergely H: Increased expression of epidermal growth factor receptor during gastric ulcer healing in rats. Gastroenterology 1992, 102:695-698 [DOI] [PubMed] [Google Scholar]
- 3.Schmassmann A, Stettler C, Poulsom R, Tarasova N, Hirschi C, Flogerzi B, Matsumoto K, Nakamura T, Halter F: Roles of hepatocyte growth factor and its receptor met during gastric ulcer healing in rats. Gastroenterology 1997, 113:1858-1872 [DOI] [PubMed] [Google Scholar]
- 4.Pai R, Ohta M, Itani RM, Sarfeh IJ, Tarnawski AS: Induction of mitogen-activated protein kinase signal transduction pathway during gastric ulcer healing in rats. Gastroenterology 1998, 114:706-713 [DOI] [PubMed] [Google Scholar]
- 5.Pai R, Jones MK, Tomikawa M, Tarnawski AS: Activation of Raf-1 during experimental gastric ulcer healing is Ras-mediated and protein kinase C-independent. Am J Pathol 1999, 155:1759-1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baatar D, Kawanaka H, Szabo IL, Pai R, Jones MK, Kitano S, Tarnawski AS: Esophageal ulceration activates keratinocyte growth factor and its receptor in rats: implications for ulcer healing. Gastroenterology 2002, 122:458-468 [DOI] [PubMed] [Google Scholar]
- 7.Baatar D, Jones MK, Pai R, Kawanaka H, Szabo IL, Moon WS, Kitano S, Tarnawski AS: Selective cyclooxygenase blocker delays healing of esophageal ulcers in rats and inhibits ulceration-triggered c-Met/hepatocyte growth factor receptor induction and extracellular signal-regulated kinase 2 activation. Am J Pathol 2002, 160:963-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarnawski A, Hollander D, Stachura J, Gergely H, Krause WJ, Sarfeh IJ: Role of angiogenesis in healing of experimental gastric ulcer. Halter F Garner A Tytgat GNJ eds. Mechanisms of Peptic Ulcer Healing. 1991:pp 165-171 Kluwer Academic Publishers, Dordrecht
- 9.Neutra MN, Padykula HA: The gastrointestinal tract. Weiss L eds. Modern Concepts of Gastrointestinal Histology. 1984:pp 658-706 Elsevier Science Publishing Co., New York
- 10.Kinoshita Y, Nakata H, Hassan S, Asahara M, Kawanami C, Matsushima Y, Naribayashi-Inomoto Y, Ping CY, Min D, Nakamura A, Chiba T: Gene expression of keratinocyte and hepatocyte growth factors during the healing of rat gastric mucosal lesions. Gastroenterology 1995, 109:1068-1077 [DOI] [PubMed] [Google Scholar]
- 11.Ferrara N: Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol 2001, 280:C1358-C1366 [DOI] [PubMed] [Google Scholar]
- 12.Tarnawski A, Sekhon S, Ichikawa Y, Sarfeh IJ: Vascular endothelial growth factor (VEGF) enhances angiogenesis in injured gastric mucosa and accelerates healing of ethanol-induced erosions. Gastroenterology 1998, 114:A307 [Google Scholar]
- 13.Jones MK, Itani RM, Wang H, Tomikawa M, Sarfeh IJ, Szabo S, Tarnawski S: Activation of VEGF and Ras genes in gastric mucosa during angiogenic response to ethanol injury. Am J Physiol 1999, 276:G1345-G1355 [DOI] [PubMed] [Google Scholar]
- 14.Takahashi M, Kawabe T, Ogura K, Maeda S, Mikami Y, Kaneko N, Terano A, Omata M: Expression of vascular endothelial growth factor at the human gastric ulcer margin and in cultured gastric fibroblasts: a new angiogenic factor for gastric ulcer healing. Biochem Biophys Res Commun 1997, 234:493-498 [DOI] [PubMed] [Google Scholar]
- 15.Jones MK, Kawanaka H, Baatar D, Szabo IL, Tsugawa K, Pai R, Koh GY, Kim I, Sarfeh IJ, Tarnawski AS: Gene therapy for gastric ulcers with single local injection of naked DNA encoding VEGF and angiopoietin-1. Gastroenterology 2001, 121:1040-1047 [DOI] [PubMed] [Google Scholar]
- 16.Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359:843-845 [DOI] [PubMed] [Google Scholar]
- 17.Minchenko A, Bauer T, Salceda S, Caro J: Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab Invest 1994, 71:374-379 [PubMed] [Google Scholar]
- 18.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL: Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996, 16:4604-4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Cox SR, Morita T, Kourembanas S: Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ Res 1995, 77:638-643 [DOI] [PubMed] [Google Scholar]
- 20.Wang GL, Semenza GL: Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995, 270:1230-1237 [DOI] [PubMed] [Google Scholar]
- 21.Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995, 92:5510-5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salceda S, Caro J: Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997, 272:22642-22647 [DOI] [PubMed] [Google Scholar]
- 23.Elson DA, Ryan HE, Snow JW, Johnson R, Arbeit JM: Coordinate up-regulation of hypoxia inducible factor (HIF)-1alpha and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res 2000, 60:6189-6195 [PubMed] [Google Scholar]
- 24.Tsuji H, Fuse Y, Kawamoto K, Fujino H, Kodama T: Healing process of experimental esophageal ulcers induced by acetic acid in rats. Scand J Gastroenterol 1989, 24(Suppl 162):6-10 [DOI] [PubMed] [Google Scholar]
- 25.Kim I, Moon SO, Koh KN, Kim H, Uhm CS, Kwak HJ, Kim NG, Koh GY: Molecular cloning, expression, and characterization of angiopoietin-related protein. J Biol Chem 1999, 274:26523-26528 [DOI] [PubMed] [Google Scholar]
- 26.Kwak HJ, So JN, Lee SJ, Kim I, Koh GY: Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS Lett 1999, 448:249-253 [DOI] [PubMed] [Google Scholar]
- 27.Sano K, Sekine J, Inokuchi T, Pe MB, Ma G: Enhanced polymer one-step staining for proliferating cell nuclear antigen in squamous cell carcinoma of the tongue. Biotech Histochem 1996, 71:273-237 [DOI] [PubMed] [Google Scholar]
- 28.Tarnawski A, Hollander D, Stachura J, Krause WJ, Eltorai M, Dabros W, Gergely HJ: Vascular and microvascular changes—key factors in the development of acetic acid-induced gastric ulcers in rats. Clin Gastroenterol 1990, 12(Suppl 1):S148-S157 [DOI] [PubMed] [Google Scholar]
- 29.Battegay EJ: Angiogenesis: mechanistic insights, neovascular diseases, and therapeutic prospects. J Mol Med 1995, 73:333-346 [DOI] [PubMed] [Google Scholar]
- 30.Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald DM, Johnson RS, Arbeit JM: Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev 2001, 15:2520-2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones MK, Szabo IL, Kawanaka H, Husain SS, Tarnawski AS: Von Hippel Lindau tumor suppressor and HIF-1alpha: new targets of NSAIDs inhibition of hypoxia-induced angiogenesis. EMBO J 2002, 16:264-266 [DOI] [PubMed] [Google Scholar]
- 32.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, van de Water LJ: Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. Exp Med 1992, 176:1375-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank S, Hubner G, Breier G, Longaker MT, Greenhalgh DG, Werner S: Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J Biol Chem 1995, 270:12607-12613 [DOI] [PubMed] [Google Scholar]
- 34.Epstein SE, Fuchs S, Zhou YF, Baffour R, Kornowski R: Therapeutic interventions for enhancing collateral development by administration of growth factors: basic principles, early results and potential hazards. Cardiovasc Res 2001, 49:532-542 [DOI] [PubMed] [Google Scholar]
- 35.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 36.Nor JE, Christensen J, Mooney DJ, Polverini PJ: Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol 1999, 154:375-384 [DOI] [PMC free article] [PubMed] [Google Scholar]