

Abstract
Senescence marker protein-30 (SMP30) is a calcium-binding protein that decreases in an androgen-independent manner with aging. To elucidate the physiological role of this protein, we introduced a null mutation of the SMP30 gene into the germ line of mice. Despite the complete lack of SMP30 (SMP30−/−), these mutant mice were indistinguishable from their wild-type (SMP30+/+) littermates in terms of development and fertilization capability. We then investigated the tissue susceptibility for apoptosis induced by cytokine using primary cultured hepatocytes, because SMP30 could rescue cells from cell death caused by calcium influx, using a calcium ionophore as previously described. SMP30−/− hepatocytes were found to be more susceptible to apoptosis induced by tumor necrosis factor-α (TNF-α) plus actinomycin D (ActD) than SMP30+/+ hepatocytes. In addition, the TNF-α/ActD-induced caspase-8 activity in SMP30−/− hepatocytes was twofold greater than that in SMP30+/+ hepatocytes. In contrast, no significant difference was observed in the TNF-α/ActD-induced nuclear factor-κB activation of SMP30+/+ versus SMP30−/− hepatocytes, indicating that SMP30 is not related to TNF-α/ActD-induced nuclear factor-κB activation itself. Moreover, deletion of the SMP30 gene enhanced liver injury after treatment in vivo with anti-Fas antibody and the SMP30+/− mice showed intermediate susceptibility to Fas-induced apoptosis. Collectively, these results demonstrate that SMP30 acts to protect cells from apoptosis.
The development and aging of tissues are governed by numerous factors that control their constituent proteins. During a survey of age-associated changes in soluble proteins of the rat liver, we discovered a novel age-associated protein designated senescence marker protein-30 (SMP30). 1 The amount of SMP30 significantly decreases with aging in an androgen-independent manner. 1-3 SMP30 is a 34-kd protein expressed mainly in hepatocytes and renal tubular epithelia. 1 By reverse transcriptase-polymerase chain reaction (PCR) analysis, SMP30 transcripts have been detected in multiple tissues including the liver, kidney, brain, lung, adrenal gland, stomach, ovary, uterus, testis, and epidermis (our unpublished results). The alignment of SMP30’s amino acid sequences deduced from a cloned cDNA sequence revealed a highly conserved structure among humans, rats, and mice. 2,4,5 According to a database search, SMP30 is a previously unknown and unique protein, wholly separate from any family of other proteins and without any functional domain in the structure, suggesting its distinctive and pivotal role in many organs. The SMP30 gene, which is located in the p11.3 to q11.2 segment of the X chromosome, 4 could be a candidate agent of X-linked diseases mapped to that region. Recently, a gene homologous to SMP30 was found during cold acclimation at a moderately low temperature (15°C) in Drosophila melanogaster and in the anterior fat body from the Sarcophaga peregrina (flesh fly). 6,7 Moreover, a Xenopus orthologue of SMP30 selectively expressed in the pronephric tubes from stage 32 onwards was identified by in situ hybridization. 8 These discoveries clearly support the important biological role of SMP30 in lower as well as higher animals.
After our discovery, another group reported a novel Ca2+-binding protein named “regucalcin,” which is identical to SMP30. 9 However, SMP30 does not have an EF-hand motif like the Ca2+-binding domain. We previously reported that SMP30 participates in Ca2+ efflux by activating the calmodulin-dependent Ca2+ pump in HepG2 cells and renal tubular epithelial cells and confers on these cells resistance to injury caused by high intracellular Ca2+ concentrations. 10,11 Studies performed in vitro suggest that SMP30 stimulates ATP-dependent 45Ca2+ uptake by isolated rat liver mitochondria and incites Ca2+ pump activity in renal cortex mitochondria. 12,13
According to proteomic analysis of differential protein expression in primary hepatocyte cultures, SMP30 content increased at 24 hours with tumor necrosis factor-α (TNF-α) present. 14 TNF-α routinely induces apoptosis in mammalian cells by increasing intracellular Ca2+ concentrations. 15,16 In our previous report, SMP30 rescued mammalian cells from death by enhancing plasma membrane Ca2+ pump activity. 10,11 These outcomes strongly suggest that SMP30 acts as a potent anti-apoptotic agent. To confirm this possibility, we generated SMP30 knockout (SMP30−/−) mice and examined the sensitivity to TNF-α- and Fas-induced apoptosis in vitro and in vivo, respectively. The results show that SMP30 knockout mouse livers are highly susceptible to apoptosis, indicating that SMP30 has a protective role in cell injury such as apoptosis and hypoxia.
Materials and Methods
Gene Targeting
To construct the targeting vector, we isolated one phage clone from a mouse genomic library of 129/Sv strain (Stratagene) using rat SMP30 cDNA. 2 An ∼6-kb ClaI-ClaI fragment and a 2-kb ClaI-SalI fragment including exons I, II, and III were isolated as the long arm and short arm, respectively, and used for constructing a targeting vector in pMCDT-A(A+T/pau) (Figure 1A) ▶ . A diphtheria toxin A gene cassette was included in the construct as a negative selection marker against random integration.
Figure 1.

Targeted disruption of the SMP30 gene. A: Partial restriction maps of the wild-type SMP30 locus, targeting vector, and mutant locus. To generate the mutated locus, exon III was disrupted by insertion of a positive selection marker (neo) and dual-negative selection markers (tk and Pr/DT-A). The sense and anti-sense PCR primers (TS3 and TS4) used for genomic PCR are indicated with arrows. B: PCR analysis of genomic DNA isolated from: lane 1, the wild-type allele (Y/+) from a male mouse; lane 2, male mouse with the targeted allele (Y/−); lane 3, female mouse homozygous for the wild-type allele (+/+); lane 4, female heterozygous mouse (+/−); lane 5, female mouse homozygous for the targeted allele (−/−). The wild-type and mutated genes gave 280-bp and 1363-bp PCR products, respectively. C: Western blotting of SMP30 (33 kd) showed the lack of SMP30 in livers of SMP30Y/− and SMP30−/− mice.
To generate SMP30−/− mutant mice, we electroporated into the targeting vector and selected 129/Sv E14 embryonic stem (ES) cells. To delete homologous recombinant ES clones, we used cell lysates of the G418-resistant clones as templates for PCR amplification with a SMP30 flanking primer (TS3, 5′-CTAGCCAT GGTGGATGAAGAT-3′; TA4, 5′-CAAGTAACTCTAGGTATGGAC-3′). Expected sizes for wild-type SMP30 and mutant SMP30 are 280 bp and 1363 bp, respectively. ES cells heterozygous for the targeted mutation were injected into C57BL/6 blastocysts to generate chimeras. The resulting male chimeras were mated with female C57BL/6 mice. Germ-line transmission of the injected ES was confirmed by inheritance of agouti coat color in the F1 animals, and all agouti offspring were tested for the mutated SMP30 allele by genomic PCR (Figure 1B) ▶ and Western blotting (Figure 1C) ▶ . Mice heterozygous for the trapped allele were backcrossed seven times (N7) to C57BL/6 mice and intercrossed eight times (F8). All subsequent analyses were performed with N7F8 generation and 4-month-old mice, and all animals received humane care in compliance with institutional guidelines.
Western Blotting Analysis
Equal amounts of protein (10 μg/lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on vertical slab gels (1 mm × 9 cm) containing 10% acrylamide and 0.25% N,N′-methylenebisacrylamide, by the method of Laemmli. 17 Proteins were then electrophoretically transferred from acrylamide gels onto a nitrocellulose membrane (BAS-85; Schleicher & Schuell) by the method of Towbin and colleagues. 18 The membrane was then incubated successively with rabbit anti-rat SMP30 antibody (1:5000) and horseradish peroxidase-labeled goat anti-rabbit IgG (Bio-Rad Laboratories). Chemiluminescence signals were detected on Kodak X-Omat AR films using Renaissance (DuPont NEN). Protein was quantified by the Bradford protein assay (Bio-Rad Laboratories) using bovine serum albumin as a standard.
Isolation and Culture of Mouse Hepatocytes
Mouse hepatocytes were isolated by the collagenase perfusion method as described before. 19 Briefly, each liver was perfused in situ through the vena cava with EGTA solution containing 0.5 mmol/L EGTA, 5 mmol/L glucose, 136 mmol/L NaCl, 5.3 mmol/L KCl, 0.3 mmol/L Na2HPO4, 0.4 mmol/L KH2PO4, and 10 mmol/L HEPES, pH 7.2, at a rate of 15 ml/minute. After that perfusion, the first solution was replaced with collagenase solution containing 0.5 mg/ml collagenase, 0.05 mg/ml trypsin inhibitor, 4.5 mmol/L CaCl2, 136 mmol/L NaCl2, 5.3 mmol/L KCl, 0.3 mmol/L NaH2PO4, 0.4 mmol/L KH2PO4, and 10 mmol/L HEPES, pH 7.4. After the second perfusion, the livers were removed and filtered through nylon mesh (100 μm) and then washed three times with Hanks’ balanced salt solution and twice with Williams medium E to remove nonparenchymal cells. Final preparations were suspended at 2.5 × 105 cells/ml in Williams medium E containing 0.1 mg/ml aprotinin and 10−9 mol/L dexamethasone supplemented with 2.5% fetal calf serum and then placed into culture plates coated with rat type I collagen (Sigma). Cells were cultured at 37°C under 5% CO2 in air for 3 hours to allow attachment to the wells, then the medium was replaced with serum-free Williams medium E containing 0.1 mg/ml of aprotinin and 10−9 mol/L dexamethasone. Cell viability was determined by trypan blue dye exclusion before plating.
Immunohistochemistry
For immunohistochemical staining, hepatocytes were cultured on Falcon culture slides for 20 hours, fixed for 15 minutes with 10% formalin in phosphate-buffered saline (PBS), and permeabilized for 3 minutes with 0.5% Triton X-100 in PBS. For immunofluorescence assay, cells were stained with rabbit anti-mouse SMP30 antibody (1:400) and rhodamine-phalloidin (1:500) (Molecular Probes) for detection of actin fiber 20,21 and then with fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (1:500) (Biosource International). Stained cells were examined with an LSM-510 laser-scanning microscope (Carl Zeiss).
Induction and Determination of Apoptosis
Apoptosis was induced by incubating the hepatocytes in a culture medium containing 20 ng/ml TNF-α and various concentrations of a transcription inhibitor actinomycin D (ActD) for various time points as specified in the figure legends.
Apoptosis was determined by assessing the following: nuclear changes indicative of apoptosis (ie, chromatin condensation, nuclear fragmentation) using the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) dihydrochloride; and externalization of phosphatidylserine residues using FITC-conjugated annexin V. Cells cultured on Falcon culture slides were washed with ice-cold PBS and placed in a custom-made chamber for viewing with an inverted fluorescence microscope. Cells were incubated in binding buffer (10 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2) containing FITC-labeled annexin V and DAPI for 15 minutes at room temperature in the dark. After incubation, cells were viewed by fluorescence microscopy using excitation and emission filters of 380 and 430 nm for DAPI, and 490 and 515 nm for FITC.
XTT Assay
Cell viability was determined by the XTT assay using the Cell Proliferation Kit II (XTT) (Roche Molecular Biochemicals). Briefly, cells in 12-well plates were incubated in the presence of 20 ng/ml of TNF-α and various concentrations of ActD in a final volume of 0.5 ml for 24 hours at 37°C. Thereafter, 0.5 ml of XTT solution (0.3 mg/ml in PBS) was added to each well. After a 4-hour incubation at 37°C, the OD at 492 nm were measured using a 12-well multiscanner autoreader, with the XTT solution as a blank.
Enzyme Activity Assay
Caspase-8 activity was determined with a Caspase-8 Colorimetric Assay Kit (BioVision Research Products). Briefly, the cells were washed with ice-cold PBS and harvested by scraping with a cell scraper in 500 μl of cell lysis buffer from the assay kit. The cytosolic fraction was obtained by centrifugation at 10,000 × g for 5 minutes. Caspase-8 activity was evaluated by measuring proteolytic cleavage of the chromogenic substrate, IETD-pNA,. The cell lysate (100 μg of protein) was added to 2× reaction buffer from the assay kit containing 200 μmol/L of IETD-pNA in a final volume of 105 μl. Finally, the reaction mixture was incubated at 37°C for 2 hours. The increase in absorbance of enzymatically released pNA was measured at 405 nm in a microplate reader.
Nuclear Extract Preparation
Cells were washed with 10 ml of ice-cold PBS and harvested by scraping with a cell scraper in 400 μl of buffer A (10 mmol/L HEPES, pH 7.8, 10 mmol/L KCl, 0.1 mmol/L ethylenediaminetetraacetic acid, 2 mmol/L dithiothreitol, 1 mmol/L phenylmethyl sulfonyl fluoride, 5 μg/ml aprotinin, 3 μg/ml pepstatin A, 5 μg/ml leupeptin) from plates to microtube. The cells were allowed to swell on ice for 15 minutes, after which 25 μl of 10% Nonidet P-40 was added, and the tube was vortexed vigorously for 10 seconds. The homogenate was centrifuged for 30 seconds in a microcentrifuge. The nuclear pellet was resuspended in buffer B (20 mmol/L HEPES, pH 7.8, 0.42 mol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 5 mmol/L dithiothreitol, 1 mmol/L phenylmethyl sulfonyl fluoride, 10% glycerol), and the tube was rocked gently at 4°C for 30 minutes on a shaking platform. The nuclear extract was centrifuged for 10 minutes in a microcentrifuge at 4°C, and the supernatant was frozen at −70°C in aliquots until the electrophoretic mobility shift assay (EMSA) was performed.
EMSA
For the EMSA, the nuclear factor-κB (NF-κB)-specific oligonucleotide was obtained from Santa Cruz Biotechnology, Inc. The oligonucleotide was end-labeled using T4 polynucleotide kinase and [γ-32P] ATP (Amersham). For competition studies, 3.5 pmol of unlabeled oligonucleotide was used. Nuclear extract without labeled oligonucleotide was preincubated for 15 minutes at 4°C followed by 20 minutes of incubation at room temperature, after the addition of labeled oligonucleotide. The binding reaction contained 10 μg of sample protein, 5 μl of 5× incubation buffer (20% glycerol, 5 mmol/L MgCl2, 5 mmol/L ethylenediaminetetraacetic acid, 5 mmol/L dithiothreitol, 500 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.5, and 0.4 mg/ml calf thymus DNA). In some of the binding reactions poly dIdC (Pharmacia) was added to a final concentration of 2 μg. The nuclear protein-[32P] oligonucleotide complex was separated from free [32P]-labeled oligonucleotide by electrophoresis through a 6% native polyacrylamide gel. The gels were dried and autoradiographed with Kodak X-Omat AR films. For supershift assay some of the binding reactions contained 200 μg of anti-p50 or anti-p65 antibody (Santa Cruz Biotechnology, Inc.) along with 2 mg of poly dIdC.
Anti-Fas Antibody Treatment of Mice
Mice were injected via the tail vein with anti-Fas antibody (Jo2, Pharmingen) diluted in pyrogen-free saline (3 μg/25 g mouse body weight). Six hours later, the blood sample and liver tissue were procured. The level of hepatocyte damage was assessed by the amount of alanine aminotransferase (ALT) released in serum using a standard clinical analyzer (Hitachi). The liver specimens were fixed in 10% neutral formalin overnight, and then embedded in paraffin and stained with hematoxylin and eosin (H&E). In situ apoptosis was detected by the method of terminal dUTP nick-end labeling using in situ apoptosis detection kit (Takara).
Statistical Methods
The results are expressed as the mean ± SEM. The probability of statistical differences between experimental groups was determined by a Student’s t-test, as indicated.
Results
Establishment of SMP30 Knockout Mice
SMP30 knockout mice was generated by inserting the targeting vector into the third exon of SMP30 gene (Figure 1A) ▶ . This targeting vector, which expressed the selective marker, neomycin transferase, was transfected into ES cells to create a mutated allele. Three of 502 recombinant ES cells were found to be SMP30+/−. We then crossed chimeric mice from one of the clones (no. 41) with C57BL/6 mice to produce SMP30+/− progeny. Mice homozygous for the mutation were identified by genomic PCR analysis (Figure 1B) ▶ . Intercross of heterozygous founder mice yielded wild-type, heterozygous, and homozygous progeny. Heterozygous male mice of this strain do not exist because of the X chromosome’s location in the SMP30 gene. Western blot analysis using rabbit anti-rat SMP30 antibody showed that the livers of SMP30Y/− and SMP30−/− mice completely lacked SMP30 (Figure 1C ▶ , lanes 2 and 5). We observed no obvious differences between SMP30+/+ and SMP30−/− mice with respect to gross physical appearance, organ morphology, or reproductive capability. SMP30 knockout mice were established as recombinant inbred strain after backcrossed seven times (N7) to C57BL/6 mice and intercrossed eight times (F8).
Expression of SMP30 in Primary Cultured Hepatocytes
To confirm SMP30 expression and to determine its intracellular distribution, we used primary cultured hepatocytes from female mice of three genotypes, SMP30+/+, SMP30+/−, and SMP30−/−. Subsequent cytoskeletal actin staining with rhodamine-phalloidin was clear in the hepatocytes of all three types (Figure 2; B, D, and F) ▶ . Rabbit anti-mouse SMP30 antibody recognized mouse SMP30 in the hepatocytes of SMP30+/+ mice (Figure 2A) ▶ as SMP30 proteins in both the cytoplasm and nuclei. SMP30 in cytoplasm was diffusely distributed but not unevenly distributed. In SMP30+/− hepatocytes, SMP30 was expressed in a mosaic manner because of X-chromosome inactivation. 22 However, SMP30-negative cells were present among the actin-positive cells (Figure 2, C and D) ▶ . Hepatocytes from SMP30−/− mice did not stain for SMP30 (Figure 2E) ▶ . Nevertheless, the shapes and sizes of hepatocytes from all three genotypes were virtually identical microscopically.
Figure 2.

Intracellular localization of SMP30. Primary cultured hepatocytes from SMP30+/+ (A, B), SMP30+/− (C, D), and SMP30−/− (E, F) mice were stained with rabbit anti-mouse SMP30 and FITC-labeled goat anti-rabbit IgG (A, C, E). Actin fibers were detected by using rhodamine-phalloidin (B, D, F). Fluorescence was imaged by laser-scanning confocal microscopy. A and B, C and D, and E and F indicate the same field.
Increased Susceptibility to TNF-α Induced Apoptosis in SMP30 Knockout Mice
TNF-α induces apoptosis in mammalian cells. 15,16 To investigate the effect of TNF-α-induced apoptosis, hepatocytes were treated with TNF-α plus ActD. The degree of apoptosis was determined by the externalization of phosphatidylserine residues using FITC-conjugated annexin V and the nuclear changes visible with the DNA-binding dye, DAPI (Figure 3) ▶ . On exposure to 20 ng/ml of TNF-α plus 10 ng/ml of ActD (TNF-α/ActD), almost all SMP30−/− hepatocytes became stained with annexin V; these SMP30-deficient cells thus exhibited chromatin condensation and nuclear fragmentation indicating copious apoptosis not seen in untreated control cells (Figure 3, B and D) ▶ . In SMP30+/+ mice, however, few cells underwent apoptosis, ie, little annexin V-staining or chromatin condensation was apparent after exposure to TNF-α/ActD in comparison with untreated control cells (Figure 3, A and C) ▶ . When SMP30+/+ and SMP30−/− hepatocytes were exposed to 20 ng/ml of TNF-α plus 100 ng/ml of ActD, almost all cells were stained with annexin V and detached from culture plates because of progressive cell death (Figure 3, E and F) ▶ . The degree of cell death when exposed to TNF-α plus various concentration of ActD was determined by XTT assay (Figure 4) ▶ . When SMP30+/+ and SMP30−/− hepatocytes were exposed to 20 ng/ml of TNF-α plus 10 ng/ml of ActD, cell death in SMP30−/− cells was ∼40% higher than SMP30+/+ cells. The degree of cell death as determined by the XTT assay was virtually identical to the DAPI and annexin V staining of hepatocytes (Figure 3) ▶ . In addition, neither TNF-α alone (100 ng/ml) nor ActD alone (200 ng/ml) induced apoptosis in SMP30+/+ or SMP30−/− hepatocytes (data not shown).
Figure 3.

Induction of apoptosis by TNF-α/ActD. SMP30+/+ (A, C, E) and SMP30−/− (B, D, F) hepatocytes were incubated for 24 hours in medium lacking (A, B) or containing 20 ng/ml of TNF-α plus 10 ng/ml of ActD (C, D) or 100 ng/ml of ActD (E, F). Apoptosis was detected by staining with both FITC-conjugated annexin V (lighter green than background) and DAPI (blue). Cells were considered apoptotic when positive for either annexin V staining, chromatin condensation, or nuclear fragmentation.
Figure 4.

TNF-α/ActD-induced cell death. SMP30+/+ and SMP30−/− hepatocytes were incubated for 24 hours in medium containing 20 ng/ml of TNF-α plus 1 ng/ml, 10 ng/ml, or 100 ng/ml of ActD. Percentage of survival was calculated relative to untreated controls by XTT assay. Values are expressed as average percentage of survival ± SEM of four independent experiments. Data were compared using Student’s t-test. *, P < 0.05, SMP30+/+ versus SMP30−/−.
Caspase-8 Activation as a Proapoptotic Pathway
Caspase-8 is a cysteine protease that plays a pivotal role in the proapoptotic pathway, and its activation initiates or is an early event of apoptosis. 23,24 To determine whether increased susceptibility to TNF-α-induced apoptosis in SMP30−/− hepatocytes is associated with caspase-8 activation, hepatocytes were treated with TNF-α/ActD for 10 hours and then lysed and assayed for caspase-8. In SMP30+/+ and SMP30−/− hepatocytes, caspase-8 was activated fourfold and eightfold, respectively, compared with that in untreated control cells (Figure 5) ▶ . Thus caspase-8 activation by TNF-α/ActD treatment is twofold higher in SMP30-deficient hepatocytes than in SMP30-containing hepatocytes.
Figure 5.

Induction of caspase-8 enzyme activity by TNF-α/ActD treatment. Apoptosis was induced in SMP30+/+ and SMP30−/− hepatocytes by treatment with 20 ng/ml of TNF-α plus 10 ng/ml of ActD for 10 hours. Data are expressed as percent activity of caspase-8 enzyme relative to basal levels (ie, pretreatment = 100% activity) in each experiment. Values are expressed as average percentage of activity ± SEM of four independent experiments. Data were compared using Student’s t-test. *, P < 0.01, SMP30+/+ versus SMP30−/−.
NF-κB Activation as Anti-Apoptotic Pathway
We next examined the influence of NF-κB activation on TNF-α signaling as an anti-apoptotic pathway in our SMP30 model. NF-κB DNA-binding activity was assessed by EMSA using the NF-κB binding site as a probe. TNF-α/ActD treatment for 60 minutes induced an increase of NF-κB DNA binding in both SMP30+/+ and SMP30−/− hepatocytes (Figure 6 ▶ , lanes 4 and 10). However, even without TNF-α/ActD, slight NF-κB-binding activity occurred (Figure 6 ▶ , lanes 1 and 7), although to a lesser extent than after TNF-α/ActD treatment. The NF-κB complex activated by TNF-α/ActD treatment of SMP30+/+ and SMP30−/− hepatocytes was composed of p50-p65 dimers, as determined by supershifts (Figure 6 ▶ ; lanes 5, 6, 11, and 12). Yet untreated (no TNF-α/ActD) SMP30+/+ and SMP30−/− hepatocytes also contained some p50-p65 dimers (Figure 6 ▶ ; lanes 2, 3, 8, and 9). This TNF-α/ActD-activated NF-κB presence in both SMP30+/+ and SMP30−/− hepatocytes without a significant difference in NF-κB-binding activity or composition of p50-p65 dimers indicates that a decline of NF-κB activation does not cause TNF-α/ActD-induced apoptosis in SMP30−/− hepatocytes.
Figure 6.

TNF-α/ActD-induced NF-κB activation. SMP30+/+ and SMP30−/− hepatocytes were treated with 20 ng/ml of TNF-α and 10 ng/ml of ActD for 60 minutes. Nuclear extracts were prepared as described in Materials and Methods and used in an EMSA to measure NF-κB DNA-binding activity using a NF-κB-specific oligo-nucleotide sequence (lanes 1 to 12). EMSA was performed after incubation of protein extracts with anti-p50 (lanes 2, 5, 8, and 11) or anti-p65 (lanes 3, 6, 9, and 12) antibodies to examine the protein composition of NF-κB and to demonstrate the specificity of DNA binding. A bold arrow indicates the NF-κB-DNA complex, and a thin arrow denotes supershifted bands produced by anti-p50 and anti-p65 antibodies.
Increased Susceptibility to Fas Induced Liver Damage in Vivo in SMP30 Knockout Mice
Intravenous administration of the anti-Fas antibody, Jo2, at high doses (>5 μg/25 g mouse body weight) induces death of the animal, SMP30+/+, SMP30+/−, and SMP30−/− mice, in 6 hours by fulminant hepatitis. At 6 hours after nonlethal doses (3 μg/25 g mouse body weight) of anti-Fas antibody treatment, serum ALT concentrations in SMP30−/− mice were significantly higher than in SMP30+/+ mice (Figure 7A) ▶ . In addition, serum ALT concentrations in SMP30+/− mice were higher than in SMP30+/+ mice and lower than SMP30−/− mice. Livers from the SMP30−/− and the SMP30+/− mice displayed extensive hemorrhagic lesions by H&E staining and numerous clusters of apoptotic cells by terminal dUTP nick-end labeling analysis (Figure 7B) ▶ . In contrast, liver specimens from the SMP30+/+ mice showed minimal damage. These results indicated that SMP30−/− and SMP30+/− mice were more susceptible to liver injury after anti-Fas antibody treatment and SMP30+/− mice showed intermediate susceptibility to Fas-induced apoptosis.
Figure 7.
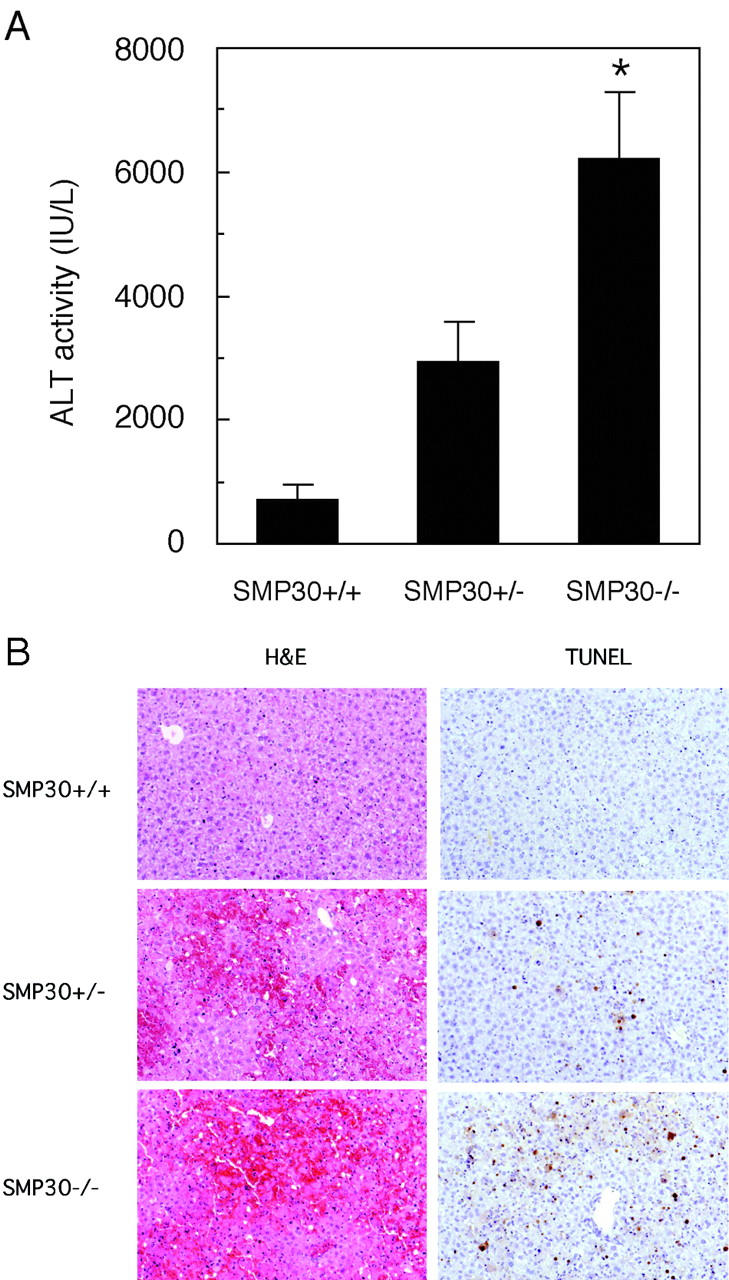
Anti-Fas antibody treatment in vivo. SMP30+/+, SMP30+/−, and SMP30−/− mice were injected via tail vein with anti-Fas antibody (3 μg/25 g mouse body weight). Mice were sacrificed after a 6-hour treatment with anti-Fas antibody. A: Serum ALT levels were measured. ALT values in control samples were less than 40 IU/ml. Values are expressed as an average of ALT level ± SEM of four animals. *, P < 0.05, SMP30+/+ versus SMP30−/−. B: H&E staining and terminal dUTP nick-end labeling analysis of liver specimens from SMP30+/+, SMP30+/−, and SMP30−/− mice injected with anti-Fas antibody. Original magnifications, ×100.
Discussion
SMP30 is a calcium-binding protein that decreases with aging independent of androgen. 1-3 We characterized SMP30 by generating a null mutation of the SMP30 gene in mice and comparing them to their normal counterparts. For that purpose, we used primary cultured hepatocytes from both groups, because SMP30 proteins are present most abundantly (<2% of total soluble protein) in the liver. 25 After cultivation, microscopic examination, and cytoskeletal actin staining, no difference was obvious in SMP30+/+ and SMP30−/− hepatocytes. Although SMP30 proteins are present in nuclei, there is no leader sequence for known nuclear translocation in the amino acid sequence of SMP30, and no biological significance has been found for such translocation of SMP30. In a preliminary experiment, almost half of the SMP30+/− hepatocytes expressed SMP30, indicating that the SMP30 deficiency had no effect on cell division or growth.
Our finding that SMP30−/− hepatocytes were far more susceptible to TNF-α-induced apoptosis in the presence of ActD than SMP30+/+ hepatocytes (Figures 3 and 4) ▶ ▶ . To confirm the increased susceptibility to apoptosis in vivo, we developed an in vivo apoptosis induction system using nonlethal doses of anti-Fas antibody. 26,27 As we expected, SMP30−/− and SMP30+/− mice were more susceptible to liver injury after anti-Fas antibody treatment in vivo (Figure 7) ▶ . Interestingly, SMP30+/− mice showed intermediate susceptibility to Fas-induced apoptosis. These results indicate that SMP30 prevents apoptosis and strongly suggests that the lack of SMP30 in SMP30 knockout mice heightened their apoptotic response.
TNF-α treatment increases intracellular Ca2+ concentration leading to cell death in astroglial cells and L929 cells. 16,28 Our previous investigations have demonstrated that heightened expression of SMP30 rescued both HepG2 cells and renal tubular epithelial cells from death caused by Ca2+ influx, using a calcium ionophore. 10,11 TNF-α-induced apoptosis is mediated by the tumor necrosis factor receptor-1 (TNFR-1). The binding of TNF-α to a TNFR-1 induces the recruitment of cellular components to form the death-inducing signaling complex that then results in the autoactivation of caspase-8. 24,29 In the present study, increased susceptibility to TNF-α/ActD-induced apoptosis in SMP30−/− hepatocytes coincided with caspase-8 activation (Figure 5) ▶ , but the possible relationship between this event and increased intracellular Ca2+ concentration is not clear. If elevation of intracellular Ca2+ concentration is an early event that initiates or promotes TNF-α-induced apoptosis after caspase-8 activation, endogenous SMP30 may function as an anti-apoptotic factor to prevent a sudden elevation of intracellular Ca2+.
TNF-α binding to the TNFR-1 potentially both initiates and activates NF-κB, which suppresses apoptosis by induction of NF-κB-responsive protective genes. 30,31 Activation of NF-κB occurs via phosphorylation of inhibitory κB (IκB) at Ser32 and Ser36, resulting in the dissociation and subsequent nuclear localization of active NF-κB. 32 Recently, Chevalier and colleagues 14 reported that TNF-α induced SMP30 expression in primary cultured rat hepatocytes tested by using two-dimensional gel electrophoresis and mass spectrometry. That result strongly underscores the probability that SMP30 is an anti-apoptotic factor whose modulation of gene expression is mediated by NF-κB activation. Moreover, SMP30 may directly affect activation of NF-κB. To confirm the association of SMP30 with NF-κB activation, we examined the NF-κB activation by EMSA and supershift assay. However, because we noted no significant difference in NF-κB activity of SMP30+/+ versus SMP30−/− hepatocytes (Figure 6) ▶ , SMP30 may be an anti-apoptotic factor induced by TNF-α-mediated NF-κB activation but having no direct effect on NF-κB activation itself.
Our results using SMP30 knockout mouse strongly suggest that the age-associated decrease of SMP30 may increase the tissue’s susceptibility to such harmful responses as apoptosis and hypoxia. In fact, in our preliminary study, cells taken from aged SMP30 knockout mice and analyzed by electron microscopy suffered far more marked injury than those from their wild-type counterparts. The down-regulation of SMP30 may contribute to hepatic deterioration of cellular functions during aging.
Acknowledgments
We thank Ms. S. Kubo, Mr. Y. Fujita, and Ms. E. Moriizumi for technical assistance; Dr. Y. Kasahara for discussion and applying a patent; and Ms. P. Minick for excellent editorial assistance.
Footnotes
Address reprint requests to Akihito Ishigami, Ph.D., Department of Molecular Pathology, Tokyo Metropolitan Institute of Gerontology, 35-2 Sakae-cho, Itabashi-ku, Tokyo 173-0015, Japan. E-mail: ishigami@tmig.or.jp.
Supported by a grant-in-aid for Scientific Research from the Ministry of Education, Science, and Culture, Japan; and a grant from the Health Science Research Grants for Comprehensive Research on Aging and Health supported by Ministry of Health Labor and Welfare, Japan.
A. I. and T. F. both contributed equally to this work.
References
- 1.Fujita T, Uchida K, Maruyama N: Purification of senescence marker protein-30 (SMP30) and its androgen-independent decrease with age in the rat liver. Biochim Biophys Acta 1992, 1116:122-128 [DOI] [PubMed] [Google Scholar]
- 2.Fujita T, Shirasawa T, Uchida K, Maruyama N: Isolation of cDNA clone encoding rat senescence marker protein-30 (SMP30) and its tissue distribution. Biochim Biophys Acta 1992, 1132:297-305 [DOI] [PubMed] [Google Scholar]
- 3.Fujita T, Shirasawa T, Uchida K, Maruyama N: Gene regulation of senescence marker protein-30 (SMP30): coordinated up-regulation with tissue maturation and gradual down-regulation with aging. Mech Ageing Dev 1996, 87:219-229 [DOI] [PubMed] [Google Scholar]
- 4.Fujita T, Mandel JL, Shirasawa T, Hino O, Shirai T, Maruyama N: Isolation of cDNA clone encoding human homologue of senescence marker protein-30 (SMP30) and its location on the X chromosome. Biochim Biophys Acta 1995, 1263:249-252 [DOI] [PubMed] [Google Scholar]
- 5.Fujita T, Shirasawa T, Maruyama N: Isolation and characterization of genomic and cDNA clones encoding mouse senescence marker protein-30 (SMP30). Biochim Biophys Acta 1996, 1308:49-57 [DOI] [PubMed] [Google Scholar]
- 6.Goto SG: Expression of Drosophila homologue of senescence marker protein-30 during cold acclimation. J Insect Physiol 2000, 46:1111-1120 [DOI] [PubMed] [Google Scholar]
- 7.Nakajima Y, Natori S: Identification and characterization of an anterior fat body protein in an insect. J Biochem (Tokyo) 2000, 127:901-908 [DOI] [PubMed] [Google Scholar]
- 8.Sato A, Asashima M, Yokota T, Nishinakamura R: Cloning and expression pattern of a Xenopus pronephros-specific gene, XSMP-30. Mech Dev 2000, 92:273-275 [DOI] [PubMed] [Google Scholar]
- 9.Shimokawa N, Yamaguchi M: Expression of hepatic calcium-binding protein regucalcin mRNA is mediated through Ca2+/calmodulin in rat liver. FEBS Lett 1993, 316:79-84 [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Inoue H, Kitamura T, Sato N, Shimosawa T, Maruyama N: Senescence marker protein-30 (SMP30) rescues cell death by enhancing plasma membrane Ca2+-pumping activity in Hep G2 cells. Biochem Biophys Res Commun 1998, 250:374-380 [DOI] [PubMed] [Google Scholar]
- 11.Inoue H, Fujita T, Kitamura T, Shimosawa T, Nagasawa R, Inoue R, Maruyama N, Nagasawa T: Senescence marker protein-30 (SMP30) enhances the calcium efflux from renal tubular epithelial cells. Clin Exp Nephrol 1999, 3:261-267 [Google Scholar]
- 12.Takahashi H, Yamaguchi M: Stimulatory effect of regucalcin on ATP-dependent Ca2+ uptake activity in rat liver mitochondria. J Cell Biochem 2000, 78:121-130 [PubMed] [Google Scholar]
- 13.Xue JH, Takahashi H, Yamaguchi M: Stimulatory effect of regucalcin on mitochondrial ATP-dependent calcium uptake activity in rat kidney cortex. J Cell Biochem 2000, 80:285-292 [DOI] [PubMed] [Google Scholar]
- 14.Chevalier S, Macdonald N, Tonge R, Rayner S, Rowlinson R, Shaw J, Young J, Davison M, Roberts RA: Proteomic analysis of differential protein expression in primary hepatocytes induced by EGF, tumour necrosis factor α or the peroxisome proliferator nafenopin. Eur J Biochem 2000, 267:4624-4634 [DOI] [PubMed] [Google Scholar]
- 15.Bellomo G, Perotti M, Taddei F, Mirabelli F, Finardi G, Nicotera P, Orrenius S: Tumor necrosis factor α induces apoptosis in mammary adenocarcinoma cells by an increase in intranuclear free Ca2+ concentration and DNA fragmentation. Cancer Res 1992, 52:1342-1346 [PubMed] [Google Scholar]
- 16.Kong SK, Fung KP, Choy YM, Lee CY: Slow increase in intranuclear and cytosolic free calcium concentrations in L929 cells is important in tumour necrosis factor-alpha-mediated cell death. Oncology 1997, 54:55-62 [DOI] [PubMed] [Google Scholar]
- 17.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 18.Towbin H, Staehelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979, 76:4350-4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishigami A, Roth GS: Age-related changes in DNA synthesis stimulated by epinephrine and isoproterenol in primary cultured rat hepatocytes. J Cell Physiol 1994, 158:231-236 [DOI] [PubMed] [Google Scholar]
- 20.Prentki M, Crettaz M, Jeanrenaud B: A possible complementary role of actin microfilaments and microtubules in triacylglycerol secretion by isolated rat hepatocytes. Biochim Biophys Acta 1980, 627:262-269 [DOI] [PubMed] [Google Scholar]
- 21.Prochniewicz-Nakayama E, Yanagida T, Oosawa F: Studies on conformation of F-actin in muscle fibers in the relaxed state, rigor, and during contraction using fluorescent phalloidin. J Cell Biol 1983, 97:1663-1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riggs AD, Pfeifer GP: X-chromosome inactivation and cell memory. Trends Genet 1992, 8:169-174 [DOI] [PubMed] [Google Scholar]
- 23.Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ: Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 2000, 106:1127-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashkenazi A, Dixit VM: Death receptors: signaling and modulation. Science 1998, 281:1305-1308 [DOI] [PubMed] [Google Scholar]
- 25.Fujita T, Shirasawa T, Maruyama N: Expression and structure of senescence marker protein-30 (SMP30) and its biological significance. Mech Ageing Dev 1999, 107:271-280 [DOI] [PubMed] [Google Scholar]
- 26.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itho N, Suda T, Nagata S: Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364:806-809 [DOI] [PubMed] [Google Scholar]
- 27.Guidotti JE, Mallet VO, Parlier D, Mitchell C, Fabre M, Jaffray P, Lambert M, Kahn A, Gilgenkrantz H: Fas/CD95 pathway induces mouse liver regeneration and allows for highly efficient retrovirus-mediated gene transfer. Hepatology 2001, 33:10-15 [DOI] [PubMed] [Google Scholar]
- 28.Koller H, Thiem K, Siebler M: Tumour necrosis factor-α increases intracellular Ca2+ and induces a depolarization in cultured astroglial cells. Brain 1996, 119:2021-2027 [DOI] [PubMed] [Google Scholar]
- 29.Baker SJ, Reddy EP: Modulation of life and death by the TNF receptor superfamily. Oncogene 1998, 17:3261-3270 [DOI] [PubMed] [Google Scholar]
- 30.Yuan J: Transducing signals of life and death. Curr Opin Cell Biol 1997, 9:247-251 [DOI] [PubMed] [Google Scholar]
- 31.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, Czaja MJ: NF-κB inactivation converts a hepatocyte cell line TNF-α response from proliferation to apoptosis. Am J Physiol 1998, 275:C1058-C1066 [DOI] [PubMed] [Google Scholar]
- 32.Clarkson RW, Heeley JL, Chapman R, Aillet F, Hay RT, Wyllie A, Watson CJ: NF-κB inhibits apoptosis in murine mammary epithelia. J Biol Chem 2000, 275:12737-12742 [DOI] [PubMed] [Google Scholar]