

Abstract
Platelet-derived growth factor (PDGF) is a potent stimulant of smooth muscle cell migration and proliferation in culture. To test the role of PDGF in the accumulation of smooth muscle cells in vivo, we evaluated ApoE −/− mice that develop complex lesions of atherosclerosis. Fetal liver cells from PDGF-B-deficient embryos were used to replace the circulating cells of lethally irradiated ApoE −/− mice. One month after transplant, all monocytes in PDGF-B −/− chimeras are of donor origin (lack PDGF), and no PDGF-BB is detected in circulating platelets, primary sources of PDGF in lesions. Although lesion volumes are comparable in the PDGF-B +/+ and −/− chimeras at 35 weeks, lesions in PDGF-B −/− chimeras contain mostly macrophages, appear less mature, and have a reduced frequency of fibrous cap formation as compared with PDGF-B +/+ chimeras. However, after 45 weeks, smooth muscle cell accumulation in fibrous caps is indistinguishable in the two groups. Comparison of elicited peritoneal macrophages by RNase protection assay shows an altered cytokine and cytokine receptor profile in PDGF-B −/− chimeras. ApoE −/− mice were also treated for up to 50 weeks with a PDGF receptor antagonist that blocks all three PDGF receptor dimers. Blockade of the PDGF receptors similarly delays, but does not prevent, accumulation of smooth muscle and fibrous cap formation. Thus, elimination of PDGF-B from circulating cells or blockade of PDGF receptors does not appear sufficient to prevent smooth muscle accumulation in advanced lesions of atherosclerosis.
Platelet-derived growth factor (PDGF) was first recognized, and later purified, based on its ability to stimulate the proliferation of a number of connective tissue cells, particularly smooth muscle cells (SMC). 1,2 PDGF is released from activated platelets and also synthesized by a number of other cells after activation or injury. Although it has not been possible to analyze the effect of targeted deletion of PDGF A- or PDGF B-chain or either of its receptors in adult animals because of embryonic lethality, 3-6 examination of mice from chimeric blastocysts (composed of a mixture of wild-type cells and cells with targeted inactivation of the PDGF β-receptor) demonstrated a role for the PDGF β-receptor in all muscle lineages. 7 Analysis of the genotypes in cells competing for representation in different cell lineages revealed that PDGF β-receptor −/− cells were reduced eight-fold in SMC-rich tissues relative to PDGF β-receptor +/+ cells.
PDGF is a family of disulfide-bonded homo- or heterodimers of four possible subunits (PDGF-A, PDGF-B, PDGF-C, and PDGF-D) which act on cells by binding to homo- or heterodimers of the two PDGF receptor proteins, PDGF α-receptor and β-receptor. PDGF-B is able to bind to both the PDGF α- and β-receptors, whereas PDGF-A can bind only to the PDGF α-receptor. 8 PDGF-C and PDGF-D have been identified recently, 9-12 and much less is known about their roles and cellular sources. PDGF-CC and PDGF-DD form only homodimers, with PDGF-CC binding to PDGF α-receptor homodimers or PDGF α/β-receptor heterodimers 13 and PDGF-DD binding to PDGF β-receptor homodimers or PDGF α/β-receptor heterodimers. 12 After vascular injury of a normal artery, increased levels of PDGF-A and PDGF β-receptor have been detected primarily in SMC neointima. 14 In vascular grafts 15 and atherosclerosis, 16 which have a significant inflammatory component, macrophages are a major source of PDGF-A and PDGF-B, whereas SMC in the neointima express both PDGF-A and PDGF-B to a lesser extent. Increased expression of the PDGF β-receptor is also observed. Although weak staining for PDGF-C has been observed in medial SMC of the normal arterial wall 11 and PDGF-D mRNA has been detected in circulating leukocytes, 10 changes in the expression of PDGF-C and PDGF-D have not been analyzed after injury or in atherosclerosis.
A role for PDGF in attracting SMC has been established in models of restenosis and vascular grafts, 17-22 in which the increase in SMC occurs within the first 2 to 4 weeks after injury or implant. However, SMC accumulation in atherosclerosis occurs over decades in humans and many months in animals placed on high cholesterol diets that accelerate lesion formation. 23 Protracted lesion formation requires a prolonged period of treatment and poses problems for experimental investigation, particularly with the administration of blocking antibodies. Studies in which endogenous neutralizing antibodies were induced in rabbits before initiation of the atherosclerotic diet 24,25 and PDGF receptor antibodies were administered long term in the ApoE null mouse model of atherosclerosis 26 have suggested the possibility that blockade of PDGF or the PDGF β-receptor can reduce lesion size. In this study, we have examined lesions in detail in ApoE −/− mice at extended time points and used two different approaches to test the role of PDGF in SMC accumulation in advanced lesions of atherosclerosis: hematopoietic chimeras lacking PDGF-B in their circulating cells and mice treated daily with a PDGF receptor antagonist. Although we demonstrate evidence that PDGF can transiently delay SMC accumulation in ApoE −/− mice at 35 weeks, neither method of blockade is able to prevent formation of advanced fibrous caps analyzed at 45 and 50 weeks.
Materials and Methods
Chemicals and Reagents
Antibodies used for immunostaining include: Mac-2 antibody for macrophages, 27 a rat anti-mouse monoclonal antibody (ATCC, Manassas, VA, monoclonal supernatant diluted 1:4), PGF-007, a mouse monoclonal antibody to PDGF-B16, and anti-α-actin (DAKO, Carpinteria, CA) for SMC.
Animals
Male ApoE −/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME) at the age of 5 weeks. This strain was produced by backcrossing the ApoEtmlUnc-null strain 10 times to C57BL/6J (G10). PDGF-B +/− mice that are 129 Ola/C57BL/6J 3 were backcrossed 6 times with ApoE −/− (G6) mice in the Department of Medical Biochemistry, University of Göteborg (Göteborg, Sweden). All experimental protocols were approved by the Animal Care Institutional Review Board (University of Washington) or the IACUC Review committee (COR Therapeutics, Inc., South San Francisco, CA). Details describing first generation PDGF-B null hematopoietic chimeras have been published. 28 In this study, male ApoE −/− recipients of 7 to 8 weeks were prepared by total body irradiation in a single 14-Gy fraction from dual-opposed Co-60 sources at an exposure rate of 20 cGy/min on the day before transplant. The next day, 5 × 106 to 10 × 106 viable marrow cells from a pool of first generation chimeras (repopulated with fetal liver cells from ApoE null/PDGF-B +/+ or −/− E16.5 fetal livers from littermate embryos) were injected into each recipient through the lateral tail vein.
Treatment with PDGF Receptor Antagonist
The PDGF receptor antagonist CT52923 29 was pulverized using a mortar and pestle and suspended in 0.5% methyl cellulose. The suspension of CT52923 or 0.5% methyl cellulose (vehicle) was orally administered to ApoE null mice at a dose of 60 mg/kg once daily from 8 weeks of age to the time of sacrifice. Plasma samples were obtained at various time points to measure cholesterol levels, drug concentration, and ability to inhibit ex vivo PDGF receptor tyrosine kinase phosphorylation.
Blood Analyses
Venous blood (100–200 μl) from chimeras was obtained from the orbital venous plexus without killing the mice or from the heart at the time of sacrifice. Peripheral blood cell counts were measured at the Phoenix Central Laboratory (Everett, WA). Plasma cholesterol levels were measured at the Northwest Lipid Research Laboratories (Seattle, WA). Plasma concentrations of the PDGF receptor antagonist CT52923 were determined by high pressure liquid chromatography (HPLC, detection limit 0.5 μg/ml), and the ability of the plasma to inhibit PDGF receptor phosphorylation was determined in an ex vivo receptor autophosphorylation assay. 29 For analysis of mouse platelets with a chain-specific PDGF ELISA, 30 blood was collected in sodium citrate (final concentration 0.37%), and the platelet-rich plasma was collected after centrifugation at 4°C for 15 minutes at 200 × g. Platelets were pelleted by centrifugation at 4°C for 15 minutes at 800 × g and then lysed for ELISA analysis.
Quantitation of Atherosclerotic Lesions in the Brachiocephalic Trunk
After mice were killed, they were perfusion-fixed with methyl Carnoy’s fixative. The aorta was dissected and fixed in methyl Carnoy’s for 48 hours. The brachiocephalic trunk (innominate artery), from the bifurcation off the aortic arch to the branching point to the right subclavian artery and common carotid artery, was dissected and embedded in a sandwich cassette. Using a random start site from a random number table and within the first 75 μm, we serially sectioned the entire brachiocephalic trunk (5-μm sections), and every 75 μm a section was stained with hematoxylin and eosin (H & E). All of the H & E-stained images were captured with a microscope equipped with a Hamamatsu CCD camera (Bridgewater, NJ). Lesion area was quantitated with NIH Image 1.59 software. The volume of the brachiocephalic trunk lesion was calculated by the Cavalieri stereologic method [∑ (lesion area) × (distance; 75 μm)]. All analyses were done without knowledge of the tissue source.
Collagen Quantitation, Fibrous Cap Scoring, and Area Analysis
Sirius red was used to stain collagen fibrils 31 and quantitated using polarization microscopy. Images were captured with a Spot Insight digital camera (Diagnostic Instruments, Sterling Heights, MI), and collagen area was quantitated using a color threshold and Image-Pro Plus 4.5 Software (Media Cybernetics, Silver Spring, MD). Fibrous cap formation was evaluated in H & E-stained slides. All of the H & E-stained sections in the brachiocephalic trunk were examined randomly by a single observer without knowledge of the tissue source. The extent of fibrous cap formation was scored as four different levels: thick (greater than 4 elastic layers), intermediate (two to four elastic layers), thin (a single elastic layer), and no fibrous cap (foam cell lesion only with no fibrous cap). Elastic fibers in the fibrous cap identified in H & E-stained sections were verified by staining serial sections with Verhoeff-Van Gieson (VVG) and Gomori’s aldehyde fuchsin (GAF) elastin-specific stains. 32 Fibrous cap lesion area was also evaluated for the most advanced lesion in each mouse by using the VVG-stained slides. Only the elastin-stained area was selected using Photoshop (Adobe Systems Inc., San Jose, CA) and quantitated with NIH Image 1.59. The VVG-stained area was expressed as a percentage of total lesion area.
Immunohistochemistry
All immunohistochemical procedures were performed as previously described. 33 Endogenous peroxidase activity was blocked by incubating the tissue sections in 0.3% H2O2 with 1% sodium azide. Primary antibodies were incubated overnight at 4°C with the sections in 3% serum matched to the species of the secondary antibody. Biotinylated second antibodies were incubated for 30 minutes at room temp followed by 30 minutes with horseradish peroxidase-conjugated streptavidin (1/5000; ImmunoResearch Laboratories, Inc., West Grove, PA), and the antibody binding was visualized with diaminobenzidine (Sigma, St. Louis, MO). The percentage of lesion area occupied by staining for macrophages was quantitated as described for fibrous cap area analysis.
TaqMan Quantitative PCR
Peritoneal macrophages from PDGF-B chimeric ApoE −/− mice were collected four days after injection of 3% thioglycolate (BD Biosciences, San Diego, CA) into the peritoneal cavity. RNA was isolated with Trizol, followed by LiCl precipitation and RNeasy column (Qiagen, Inc., Valencia, CA) after removal of lymphocytes from the purified peritoneal macrophages with anti-CD2 selection. cDNA was primed by random hexamers and made from the extracted RNA by the use of the Superscript Preamplification System (Gibco/BRL, Rockville, MD). Transcript levels were quantitated by real-time PCR as previously described. 34 Standard 18 seconds primers and TaqMan probe and custom-made PDGF B-chain primers and TaqMan probe were obtained from Perkin Elmer Biosystems (Foster City, CA): PDGF B-chain forward primer, tccggagtcgagttggaaag; reverse primer, ggcgattacagcaggctctg; probe, FAM-tcgagggaggaggagccta. Thermocycling was performed on the GeneAmp 5700 Sequence Detection System (Perkin Elmer) using the following parameters: 50°C for 2 minutes, 95°C for 10 minutes, then alternating 40 times between 95°C for 20 seconds and 50°C for 60 seconds. Threshold (CT) values were calculated by the GeneAmp 5700 SDS Detector software. Each sample was analyzed in triplicate PCR reactions accompanied by a standard curve and two no-template control reactions.
Macrophage RNA Preparation and RNase Protection Assay
Total RNA was isolated from peritoneal macrophages using Trizol reagent (Gibco/BRL). Peritoneal macrophages were collected from PDGF-B +/+ and −/− chimeras at 24 weeks (n = 1), 30 weeks (n = 3), 35 weeks (n = 2), and 45 weeks (n = 2) on day 4 after intraperitoneal injection of aged, sterile 3% thioglycolate (Difco Laboratories, Becton Dickinson Microbiology, Sparks, MD). Multiprobe template sets for mouse chemokines, cytokines, and their receptors (mCK-1, mCK-2b, mDK-4, mCK-5, mCR-1, mCR-3, and mCR-5) were purchased from PharMingen International (San Diego, CA). RNA probes were synthesized with α-[32P]UTP (Amersham Pharmacia Biotech, Piscataway, NJ) and used within 2 days. RNase protection assays were performed according to the manufacturer’s instructions. Ten to 20 μg of total RNA was used in each hybridization reaction, and RNase-protected probe fragments were resolved on 0.4 mm 5% polyacrylamide gels containing 8 mol/L urea. After they were dried, the gels were quantitated by PhosphorImage analysis. L32 and GAPDH were used for normalization.
Statistical Analysis
Data are presented as means ± SEM. Statistical analyses used Wilcoxon rank sum (Mann-Whitney) test statistic with Instat 2.01 (Graphpad Software, San Diego, CA). A value of P < 0.05 indicates statistical significance.
Results
Generation of PDGF-B Hematological Chimeras to Test the Role of PDGF-B in Lesion Formation
Lesions of atherosclerosis result from focal accumulation within the arterial intima of mononuclear inflammatory cells from the circulation and SMC from the underlying media. PDGF is a potent stimulant of SMC migration and proliferation in culture, suggesting that PDGF may play a role in the accumulation of SMC in atherogenesis. We have directly tested the role of PDGF in the accumulation of SMC in vivo using ApoE −/− mice that develop complex lesions of atherosclerosis. Although targeted deletion of the PDGF A- or B-chain 3,5 or PDGF receptor genes 4,6 is embryonic lethal, we have developed chimeric mice in which fetal liver cells from PDGF-B-deficient embryos are used to replace the circulating cells of lethally irradiated ApoE −/− mice, and marrow from these chimeras were used to repopulate the marrow of lethally irradiated study mice (Figure 1) ▶ .
Figure 1.

Two different approaches were used to test the role of PDGF in the formation of lesions of atherosclerosis in chow-fed ApoE −/− mice. Lesion formation was analyzed in two separate sets of experiments. 1) Bone marrow of lethally irradiated ApoE −/− mice was repopulated by transplant at 7 weeks with the bone marrow of PDGF-B hematopoietic chimeras generated by repopulation of first generation chimeras with fetal liver cells from PDGF-B +/+ and −/− embryos (top). 2) ApoE −/− mice were treated with a PDGF receptor antagonist or a vehicle control starting at 8 weeks. In both cases, treatments to remove or inhibit PDGF preceded the initiation of foam cell lesion formation as indicated by the bars at the bottom summarizing the normal kinetics of lesion progression. Lesion analysis was performed between 25 and 50 weeks to focus on the period of time during which fibrous caps are formed by the infiltrating SMC.
The ApoE −/− recipients were transplanted at 7 to 8 weeks before monocyte adhesion and early lesion formation. 33 Complete repopulation by donor monocytes, granulocytes, and B cells was achieved within 30 days after transplant, while T-cell reconstitution required more than 100 days for both PDGF-B +/+ and −/− chimeras. 28 Peritoneal macrophages from PDGF-B −/− chimeric mice harvested at 24, 27, and 31 weeks do not express the PDGF-B gene as detected by real time PCR, whereas PDGF B-chain mRNA copy number of 90 per 106 18S copies is detected in peritoneal macrophages from PDGF-B +/+ chimeras (data not shown). Also PDGF-B was not detected in platelet extracts from PDGF-B −/− chimeras by PDGF-B-specific ELISA (data not shown). No significant differences in body weights, plasma cholesterol levels, lipoprotein profiles, red cell counts, platelet counts, or differential counts of granulocytes, lymphocytes, or monocytes are found between PDGF-B −/− and PDGF-B +/+ ApoE −/− chimeras (Table 1 ▶ and data not shown).
Table 1.
Body Weights and Blood Parameters of PDGF-B Chimeric ApoE−/− Mice
| Parameter | 35 weeks | 45 weeks | ||
|---|---|---|---|---|
| PDGF-B +/+ (n = 9) | PDGF-B −/− (n = 8) | PDGF-B +/+ (n = 12) | PDGF-B −/− (n = 13) | |
| Body weight (g) | 25.3 ± 1.5 | 26.0 ± 2.8 | 24.0 ± 1.7 | 22.9 ± 1.9 |
| RBC (×106/μl) | 8.0 ± 0.7 | 7.5 ± 9.0 | 7.6 ± 0.5 | 7.1 ± 0.2 |
| Hematocrit (%) | 37.4 ± 2.8 | 35.0 ± 3.5 | 27.8 ± 3.7 | 28.5 ± 2.2 |
| WBC (×103/μl) | 8.6 ± 2.8 | 10.5 ± 3.0 | ND | ND |
| PMN (×103/μl) | 2.1 ± 2.5 | 2.3 ± 2.0 | ND | ND |
| Lymphocyte (×103/μl) | 5.5 ± 2.2 | 7.0 ± 2.2 | ND | ND |
| Monocyte (×103/μl) | 0.6 ± 0.4 | 0.9 ± 0.5 | ND | ND |
| Platelet (×103/μl) | 490 ± 150 | 670 ± 280 | 772 ± 149 | 851 ± 111 |
| Plasma cholesterol (mg/dl) | 393 ± 67 | 417 ± 29 | 463 ± 71 | 489 ± 104 |
ND, not determined.
Elimination of PDGF-B from Circulating Cells of ApoE −/− Mice Does Not Affect Lesion Volume at 35 Weeks but Does Alter Lesion Characteristics
Blockade of PDGF ligands or infusion of PDGF in vivo in acute balloon injury models of the normal carotid artery has demonstrated a role for PDGF in the stimulation of SMC migration and proliferation responsible for SMC-rich neointimal formation. 17-19 Analysis of injury-induced lesion formation with blocking antibodies to the two PDGF receptors further supports an involvement of PDGF in the accumulation of SMC in the neointima. 20,22 In human atherosclerotic lesions and animal models of atherosclerosis, macrophages are a major source of PDGF-B. 16 Similarly, in lesions of ApoE −/− mice (data not shown) and PDGF-B +/+ chimeras (Figure 2) ▶ , many of the macrophages are positive for PDGF-B in foam cell-rich lesions (Figure 2A) ▶ , as well as more advanced lesions (Figure 2E) ▶ . Consistent with real-time PCR analysis of isolated cells, no staining for PDGF-B is observed in PDGF-B −/− chimeras (Figure 2D) ▶ .
Figure 2.
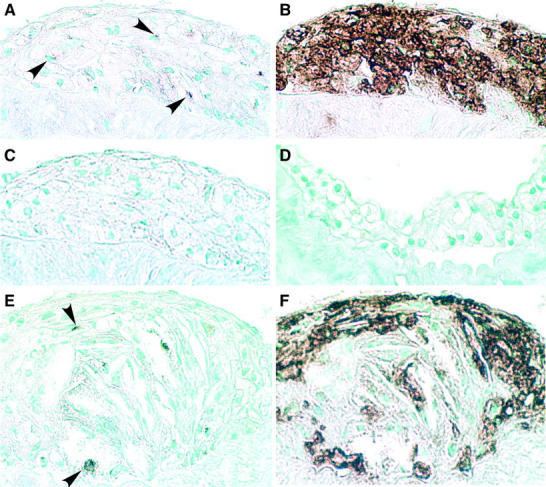
PDGF-B is primarily expressed in macrophages in developing lesions of ApoE −/− mice. To localize PDGF-B in lesions of 35-week ApoE −/−/PDGF-B chimeras, adjacent sections were stained with the PDGF-B monoclonal antibody PGF007 (A, D, and E) or with the macrophage-specific antibody Mac-2 (B and D) or normal mouse IgG1 (C). Fatty streaks (A–C) and advanced lesions (E and F) from PDGF-B +/+ chimeras contain primarily macrophages that stain positively for PDGF-B. No significant staining is observed with normal mouse IgG (C) or with the PDGF-B antibody in lesions from PDGF-B −/− chimeras (D). Arrows in A and E indicate some of the PDGF-B-positive cells. Magnification, ×40 for all panels. Lesions from chimeras at 35 weeks (+/+, n = 26; −/−, n = 29) and 45 weeks (+/+, n = 10; −/−, n = 10) showed similar immunostaining profiles.
To evaluate SMC accumulation in atherosclerotic lesions in the ApoE −/−/PDGF-B chimera mice, we initially examined mice at 35 weeks, a time point when consistent fibrous plaque formation is observed in non-irradiated ApoE −/− mice (Figure 1) ▶ . 33 To evaluate lesion volume, we examined the entire length of the brachiocephalic trunk (innominate artery) from a random start site within the initial 75-μm and determined lesion area at 75-μm intervals. As shown in Figure 3A ▶ , lesion volume varies significantly within an individual experimental group and is comparable for PDGF-B +/+ and −/− chimeras.
Figure 3.

At 35 weeks, no significant difference is observed between PDGF-B +/+ and −/− chimeras in atherosclerotic lesion volume in the brachiocephalic trunk, but lesion characteristics are altered. A: Mean lesion volume in PDGF-B +/+ and −/− chimeras was determined by evaluation of the entire length of the brachiocephalic trunk (innominate artery) at 75-μm intervals from a random start site within the first 75 μm of the vessel. The data shown are for 9 PDGF-B +/+ and 8 PDGF-B −/− chimeras and representative of an additional 17 PDGF-B +/+ and 16 PDGF-B −/− chimeras analyzed for all criteria shown in this figure. B: The frequency and extent (thin, intermediate, and thick) of fibrous cap formation was evaluated along the entire brachiocephalic trunk of all mice in this group (+/+, n = 9; −/−, n = 8) as described in A. The extent of fibrous cap formation was scored as thick (greater than 4 elastic layers), intermediate (2–4 elastic layers), and thin (a single elastic layer). The mean ± SEM is shown for 9 PDGF-B +/+ and 8 PDGF-B −/− chimeras, but P values are 0.2, considered not significant because of variability in fibrous cap formation. C: The occurrence of lesions with cholesterol clefts and necrotic cores was determined throughout the length of the brachiocephalic trunk at 75-μm intervals. The mean ± SEM is shown for 9 PDGF-B +/+ and 8 PDGF-B −/− chimeras (P = 0.5, considered not significant). D: The percentage of lesions occupied by macrophages (Mac-2-positive staining) was quantitated in lesions larger than 10,000 μm2 for PDGF-B +/+ (n = 14) and PDGF-B −/− (n = 18) chimeras (P = 0.07, considered not quite significant). E: The fibrous cap area was determined as a percentage of total lesion area for the most advanced lesion in each mouse. However, a significant number of animals in both group did not develop fibrous caps. F–I: Fibrous cap formation was evaluated by VVG staining. (F and H) PDGF-B +/+ chimeras show a more mature fibrous cap (F, thick; H, intermediate) than do (G and I) PDGF-B −/− chimeras (I, thin). Lesions in PDGF-B −/− chimeras are occupied primarily by macrophages (K) detected by staining with Mac-2 antibody, while lesions (J) in PDGF-B +/+ chimeras appear to have fewer macrophages and extensive deposits of extracellular matrix (F and H). Magnifications: F and G, ×40; H–K, ×20.
To examine the extent of SMC accumulation and the frequency and extent of fibrous cap formation, we evaluated the same H & E-stained sections used to determine lesion volume. Adjacent sections stained with the elastin-specific stains, VVG and GAF, were also examined. The number of elastic layers present in fibrous caps was used to classify the fibrous caps as thin, intermediate, or thick. No difference is observed in the frequency of thin fibrous cap formation, whereas the frequency of intermediate and thick fibrous caps appears reduced in PDGF-B −/− chimeras (Figure 3B) ▶ . This is illustrated by a comparison of the fibrous caps in PDGF-B +/+ chimeras shown in Figure 3, F and H ▶ , as compared with PDGF-B −/− chimeras in Figure 3, G and I ▶ .
The maturity of the underlying lesion also appears different between the two chimeras. PDGF-B −/− chimeras appear to have a reduced frequency of cholesterol clefts and necrotic cores (Figure 3C) ▶ and an increase in the content of macrophages (Figure 3D) ▶ , as shown with an antibody to MAC-2 (compare Figure 3, J and K ▶ ). The PDGF-B −/− lesion is composed almost exclusively of lipid-laden macrophages (Figure 3, G, I, and K) ▶ , whereas the PDGF-B +/+ lesion contains significant amounts of accumulated extracellular matrix, varying numbers of macrophages, and abundant cholesterol crystals (Figure 3, F, H, and J) ▶ . However, the differences between PDGF-B +/+ and −/− chimeras do not quite reach statistical significance. This reflects the variability in the formation of any fibrous cap in both groups of chimeric mice (Figure 3E) ▶ .
By 45 Weeks, Smooth Muscle Accumulation in the Fibrous Caps of Lesions Is Indistinguishable in PDGF-B +/+ and −/− Chimeras
To ensure more consistent formation of fibrous caps in all chimeras, lesions were analyzed in two separate, matched sets of mice after 45 weeks. As observed at 35 weeks, lesion volume remains indistinguishable between PDGF-B +/+ and −/− chimeras (Figure 4A) ▶ . Although fibrous cap formation is more consistently observed in both groups of mice at 45 weeks, the frequency of intermediate and thick fibrous cap formation appears the same in PDGF-B −/− as compared with PDGF-B +/+ chimeras (Figure 4B) ▶ . This is well illustrated by representative sections histochemically stained with GAF to highlight connective tissue components deposited by SMC that migrate from the media into the neointima (Figure 4, C and D) ▶ . Thus, elimination of PDGF-B from circulating cells is not sufficient to prevent accumulation of SMC at late stages of atherosclerotic lesion formation.
Figure 4.

By 45 weeks, the fibrous caps of PDGF −/− chimeras are indistinguishable from those of PDGF-B +/+ chimeras. A: Mean lesion volume in PDGF-B +/+ and −/− chimeras was determined by evaluation of the entire length of the brachiocephalic trunk at 75-μm intervals using a random start site within the first 75 μm of the vessel. The mean ± SEM is shown for each PDGF-B +/+ and −/− chimera and representative of an additional 12 PDGF-B +/+ and 13 PDGF-B −/− chimeras analyzed for all criteria shown in this figure. B: The frequency and extent (thin and intermediate plus thick) of fibrous cap formation were evaluated along the entire brachiocephalic trunk of all mice in a second group (+/+, n = 10; −/−, n = 10) as described in A. The extracellular matrix content of the fibrous cap is highlighted for PDGF-B +/+ (C) and PDGF-B −/− (D) chimeras with GAF staining. Magnification, ×20.
Altered Macrophage Cytokine and Cytokine Receptor Gene Expression in ApoE −/−/ PDGF-B −/− Chimeras
To probe possible differences in cytokine gene expression between the PDGF-B +/+ and −/− chimeras that may contribute to the altered lesion characteristics of PDGF-B −/− chimeras, we purified total RNA from thioglycolate-elicited peritoneal macrophages from matched PDGF-B +/+ and −/− chimeras at 24 weeks (n = 1), 30 weeks (n = 3), 35 weeks (n = 2), and 45 weeks (n = 2). RNase protection assays were used to analyze expression of cytokines and cytokine receptors (Table 2) ▶ . In elicited peritoneal macrophages, interferon (IFN)-γ, interleukin (IL)-1α, and IL-15 are all increased in PDGF-B −/− chimeras (Table 2) ▶ , characteristic of activated macrophages. 35,36 In contrast, the potent chemokines—monocyte chemotactic protein (MCP)−1, macrophage inflammatory protein (MIP)−1α, and MIP-1β, MIP-2, and RANTES—are all decreased in macrophages (Table 2) ▶ . Two chemokine receptors (CCRs), CCR2 and CCR5, receptors for MCP-1 and RANTES, respectively, which are important in monocyte recruitment, 37,38 are both increased in PDGF-B −/− elicited macrophages (Table 2) ▶ .
Table 2.
Summary of Macrophage Gene Alterations Assayed by RNAse Protection Assay
| Genes | pMac* |
|---|---|
| Cytokines | |
| GM-CSF | 1 |
| IFN-γ | +1.7/0.5 (n = 3) |
| IL-1α | +1.7/0.2 (n = 1) |
| IL-10 | 1 |
| IL-15 | +1.97/0.7 (n = 3) |
| IL-18 | 1 |
| MCP-1 | −1.8/0.3 (n = 2) |
| MIP-1α | −3.2/0.2 (n = 2) |
| MIP-1β | −3.9/0.8 (n = 2) |
| MIP-2 | −2.0/0.3 (n = 2) |
| RANTES | −1.4/0.2 (n = 2) |
| Cytokine receptors | |
| CCR1 | 1 |
| CCR2 | +1.23/0.15 (n = 3) |
| CCR5 | +2.17/0.67 (n = 3) |
Data represent fold increase (+) or decrease (−) in gene expression levels in ApoE −/−/PDGF-B −/− chimeras compared with +/+ chimeras after normalization. Other genes included in the multiprobe sets that gave signals too low to allow interpretation were: IL-2, IL-4, IL-5, IL-6, IL-7, IL-13, CCR1b, CCR3, and CCR4. IL-4Rα, IL-6Rα, IL-12Rβ2, IL-13Rα, and IL-15Rα gave significant signals, but no difference was observed.
*/, standard deviation or differences in average; 1, no difference between PDGF-B −/− and +/+; n, number of samples analyzed.
Blockade of the PDGF Receptor Transiently Delays but Does Not Prevent Fibrous Cap Formation
Since it has been determined recently that an additional member of the PDGF gene family, PDGF-D, also binds to the PDGF β-receptor, 10,12 redundancy in the PDGF ligands may compensate for the absence of PDGF-B in macrophages that infiltrate the vessel wall to initiate lesion formation (Figure 5A) ▶ . Furthermore, elimination of PDGF-B from the circulating cells does not exclude other PDGF ligands made by endothelial cells and SMC of the vascular wall. Therefore, we administered a small molecule PDGF receptor antagonist (CT52923) that potently inhibits the kinase activity of both PDGF receptors 29 to analyze its effect on the progression of atherosclerosis in ApoE −/− mice (Figure 5A) ▶ . This antagonist blocks the kinase activity of the PDGF receptors and the highly related stem cell factor receptor (c-kit) with an IC50 of 100–200 nmol/L, while 45- to 200-fold higher concentrations of CT52923 are required to inhibit fms-like tyrosine kinase-3 and colony-stimulating factor-1 receptor, two other closely related PDGF receptor family kinases. 29 The efficacy of this drug in inhibiting the activity of PDGF β-receptor was analyzed by oral administration of the drug to ApoE −/− mice. At various time points after administration, plasma samples were collected and evaluated by ex vivo phosphorylation assay (Figure 5B) ▶ . At the concentration used to study lesion progression, the inhibitory effect of the antagonist was lost 12 hours after administration.
Figure 5.

The PDGF receptor antagonist inhibits all three combinations of receptor dimers and thus all possible ligand dimers. A: The lines drawn between the different PDGF ligand pairs and the PDGF receptor dimers indicate the demonstrated binding of the ligand pairs to the different receptor combinations. Only PDGF-BB is a universal ligand able to bind to all three receptor combinations, whereas the other ligand pairs are more restricted. Thus, in the PDGF-B null chimeras, the universal PDGF ligand has been deleted, but other forms of PDGF may be able to compensate for its absence. In contrast, the PDGF receptor antagonist blocks all three receptor combinations. B: Plasma concentration of CT52923 was determined by HPLC (detection limit 0.5 μg/ml), and inhibitory activity in an ex vivo phosphorylation assay was determined after a single oral dose of 60 mg/kg at each of the indicated time points for three ApoE −/− mice at each time point.
PDGF receptor antagonist CT52923 was administered to chow-fed ApoE −/− mice daily by oral gavage from 8 weeks until the time of sacrifice at 25, 35, and 50 weeks (Figure 1) ▶ . Over the course of treatment, changes in the clearance of the inhibitor are not observed (data not shown). Plasma cholesterol levels among unmanipulated, vehicle, and antagonist-treated groups are comparable at all time points examined (Table 3) ▶ . Body weight of the unmanipulated mice is greater than that of vehicle and antagonist-treated mice, most likely reflecting the stress of daily oral gavage (Table 3) ▶ . However, the body weights of the vehicle and PDGF antagonist groups are comparable. Lesion volume in the brachiocephalic trunk was measured using the same approach as described for the PDGF-B chimeras. Lesion volume increases with time in the unmanipulated (control), vehicle, and PDGF receptor antagonist-treated mice, but lesion volume among the groups is comparable (Figure 6A) ▶ .
Table 3.
Body Weights and Plasma Cholesterol Levels of ApoE −/− Mice Treated with the PDGF Receptor Antagonist
| Age | Type | Body weight (g) | Plasma cholesterol (mg/dl) |
|---|---|---|---|
| 25 weeks | Unmanipulated (n = 8) | 35.6 ± 3.5 | 545 ± 121 |
| Vehicle (n = 10) | 31.2 ± 2.8 | 521 ± 115 | |
| PDGF-βR antagonist (n = 15) | 32.1 ± 3.2 | 602 ± 147 | |
| 35 weeks | Unmanipulated (n = 10) | 34.6 ± 4.3 | 604 ± 169 |
| Vehicle (n = 10) | 31.2 ± 2.8 | 689 ± 87 | |
| PDGF-βR antagonist (n = 15) | 32.5 ± 3.1 | 651 ± 108 | |
| 50 weeks | Unmanipulated (n = 8) | 32.6 ± 2.4 | 636 ± 169 |
| Vehicle (n = 10) | 32.5 ± 3.8 | 639 ± 79 | |
| PDGF-βR antagonist (n = 15) | 32.2 ± 2.4 | 640 ± 91 |
Figure 6.
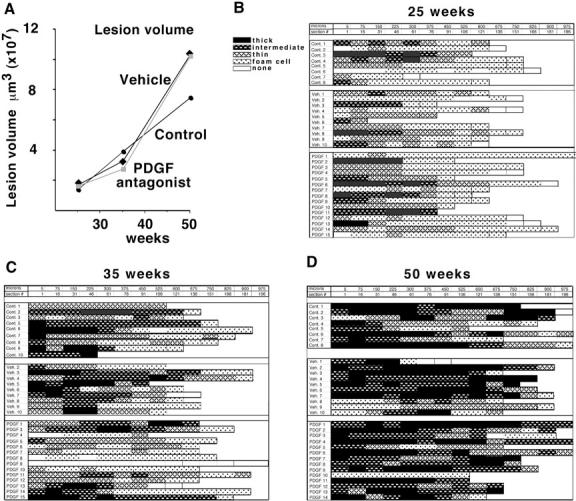
Daily administration of PDGF receptor antagonist CT52923 does not alter atherosclerotic lesion volume in the brachiocephalic trunk in ApoE −/− mice. A: Mean lesion volume in ApoE −/− mice that were untreated (control) or gavaged daily with vehicle or with the PDGF receptor antagonist CT52923 was determined by evaluation of the entire length of the brachiocephalic trunk at 75-μm intervals from a random start site. The number of mice evaluated at 25 weeks was: control, 8; vehicle, 10; CT52923, 15. At 35 weeks: control, = 10; vehicle, 10; CT52923, 14. At 50 weeks: control, 8; vehicle, 10; CT52923, 14. B–D: The lesion profile for each animal at 25 weeks (B), 35 weeks (C), and 45 weeks (D) is shown with the black and cross-hatched bars indicating varying extents of fibrous cap formation. As indicated in the key, the dotted box signifies foam cell lesions without any evidence of smooth muscle cells, and a white box has no evident lesion. Each box was scored from an H & E-stained section evaluated at 75-μm intervals using a random start site within the first 75 μm of the vessel.
To evaluate SMC accumulation at the three time points, the presence and extent of fibrous cap formation was determined along the full length of the brachiocephalic trunk for all animals (Figure 6, B, C, and D) ▶ . Even at 25 weeks, only two mice fail to show any evidence of fibrous cap formation in the brachiocephalic trunk (Figure 6B) ▶ . Fibrous caps also are not observed in two mice at 35 weeks, but fibrous caps containing lesions are observed in the brachiocephalic trunks of all mice at 50 weeks (Figure 6, C and D) ▶ . Analysis of the frequency of fibrous cap formation shows no difference at 25 weeks in thin or intermediate and thick fibrous caps among the three treatment groups (Figure 7, A and B) ▶ . However, at 35 weeks, the frequency of advanced (intermediate plus thick) fibrous cap-containing lesions is less in the antagonist-treated group than in the vehicle or unmanipulated group (P < 0.05), while the frequency of thin fibrous cap-containing lesions is similar in the three groups (Figure 7, A and B) ▶ . The decreased deposition of connective tissue at 35 weeks is further illustrated by the Masson’s trichrome staining of the PDGF receptor antagonist-treated mice (Figure 7D) ▶ compared with the vehicle-treated mice (Figure 7C) ▶ . Examination of Sirius red stained sections from the mice at 35 weeks by polarized microscopy demonstrated collagen birefringency in all of the lesions of vehicle-treated mice, but only in 5 of 14 mice treated with the PDGF receptor antagonist. At 50 weeks, the frequency of advanced (intermediate plus thick) fibrous cap-containing lesions is indistinguishable among the three groups (Figure 7, A and B) ▶ , and the change in the antagonist-treated group between 35 and 50 weeks is also significant (P < 0.001), implying that the lesions in the antagonist-treated mice “caught up” between 35 and 50 weeks. Extensive deposition of connective tissue is observed in both groups at 50 weeks (Figure 7, E and F) ▶ .
Figure 7.
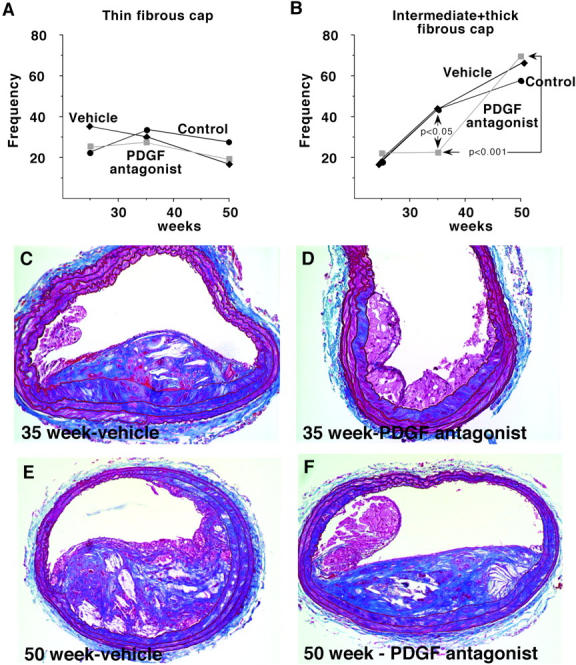
At 35 weeks, in PDGF receptor antagonist-treated ApoE −/− mice, advanced fibrous cap formation is delayed but at 50 weeks is indistinguishable from vehicle and control mice. The frequency and extent (thin in A and intermediate plus thick in B) of fibrous cap formation were evaluated along the entire brachiocephalic trunk of all mice analyzed in Figure 6, B, C, and D ▶ . Comparisons between groups were made using Mann-Whitney test and P values are indicated on the graph for statistically significant differences. C–F: Fibrous cap formation was evaluated by Masson’s trichrome staining, and representative sections are shown. At 35 weeks, PDGF receptor antagonist-treated mice show less mature fibrous caps (D) than vehicle-treated (C) mice. Magnification, ×20. However, by 50 weeks, there is no difference in the complexity or extent of the fibrous caps of the vehicle-treated mice (E) and the PDGF receptor antagonist-treated mice (F). Magnification, ×10.
Discussion
In Two Different Experimental Systems, Blockade of PDGF Appears to Delay but Does Not Prevent Fibrous Cap Formation in ApoE Null Mice
Although elimination of PDGF-B from circulating cells and blockade of both PDGF receptors appear to delay the formation of advanced lesions of atherosclerosis at 35 weeks, neither treatment was able to prevent formation of advanced fibrous caps at 45–50 weeks. The two approaches asked different questions, each with their own limitations. PDGF-B chimeric mice generated in this study express no detectable PDGF-B in their circulating cells by sensitive analysis of mRNA and protein. However, PDGF-B could still be made by endothelial cells and SMC within the vessel wall, and other chains of PDGF (PDGF-AA, PDGF-CC, and PDGF-DD) can be made by circulating and other cells in the vessel wall (Figure 4A) ▶ . In contrast, our studies with the PDGF receptor antagonist should block all forms of PDGF. However, a limitation of the receptor antagonist study is that the extended dosing of these animals restricted our administration of the antagonist to once daily, a dose that provided blockade of PDGF receptors for 8 to 10 hours. The PDGF receptor antagonist used in our study (CT52923) shows a high degree of specificity for PDGF. 29 Although other PDGF receptor antagonists have been described, they either have not been tested against a broad panel of kinases, 39-41 or they have been shown to inhibit other kinases, (eg, STI1571, which also blocks Abl kinase). 42 CT52923 further demonstrates specificity for PDGF in its inhibition of PDGF-stimulated proliferation and migration, whereas 50- to 100-fold higher concentrations are required to alter FGF-stimulated effects. 29 Thus, the two approaches that we used to block PDGF are highly specific but fail to prevent advanced fibrous cap formation.
Our results contrast with recent reports from two groups that blockade of PDGF can decrease the size of lesions of atherosclerosis. 24-26 In the rabbit studies, in which endogenous antibodies were induced by immunization with either PDGF-AA 25 or PDGF-BB, 24 lesions were evaluated 12 to 18 weeks after initiation of a 1% cholesterol diet. Lesion area evaluated by oil red O staining of the entire aorta shows a decrease of approximately 30%, and cross-sectional quantitation of aortic lesion-media ratios at a single site suggests up to 95% reduction. However, from the level of hypercholesterolemia achieved in these studies (800–1000 mg/dl), lesions at 12 to 18 weeks would be primarily foam cell-containing lesions with minimal or no SMC involvement. 43 A more recent examination of ApoE −/− mice fed a high fat diet, together with administration of anti-PDGF β-receptor antibodies between 12 to 18 weeks, indicates a 67% reduction in oil red O-stained lesion area in the aortic sinus. 26 The reduction in lesion area is associated with an 80% reduction in SMC counts in the limited number of animals (4 animals per group) and sections examined. The use of the lipid stain oil red O to quantitate lesion area in both studies favors lipid-rich lesions and would not detect lesions rich in connective tissue, characteristic of advanced fibrous cap-containing lesions. Therefore, it is difficult to conclude the true extent and nature of the reported reduction of lesion formation in these other studies. Neither of these studies examined extended time points.
Analysis of Advanced Lesions of Atherosclerosis Highlights the Difficulty in Assessing Changes in the Formation of Very Advanced Lesions of Atherosclerosis
The discrepancy between our study and those recently published emphasizes the many obstacles in evaluating advanced lesions of atherosclerosis. Unlike acute injury of a vessel, such as balloon injury of the rat carotid artery, in which mechanical injury of the media results in an immediate proliferative and migratory response of SMC at the site of injury, the accumulation of SMC in advanced lesions of atherosclerosis proceeds over many weeks in the ApoE −/− animals. 33 Although particular sites of lesion formation, such as the brachiocephalic trunk, are reproducible, 33 the exact location of the most advanced lesion within that site varies between animals. The most advanced and largest lesions in the brachiocephalic trunk frequently are found in the first segment that represents the area immediately adjacent to the aorta (Figure 6C) ▶ . However, very advanced lesions are also observed in the middle of the brachiocephalic trunk in the absence of extensive lesion development in the first segment adjacent to the aorta (Figure 6, B, C, and D) ▶ . Thus, studies in which lesions have been quantitated at a particular distance from a bifurcation or other anatomical reference may not be evaluating the site of maximal lesion formation.
Our analysis of advanced lesion formation in the brachiocephalic trunk highlights the variability in the formation of fibrous cap-containing lesions among the mice, especially in the PDGF-B chimeras. We avoided use of a high fat/high cholesterol diet that is often used to accelerate lesion formation because the very high cholesterol levels in these mice lead to even more variability in the extent of lesion formation (unpublished observations). 33 We also avoided analysis of the root of the aorta, a frequently reported site of lesion evaluation, because of the unusual anatomy of this site and the absence of lesions at this site in humans. 44 Further, our unpublished observations agree with suggestions that aortic root atherosclerosis may not reflect atherosclerosis in other regions of the vascular tree. 45,46 Potential differences in lesion progression at the brachiocephalic trunk versus the aortic root may partially explain the differences between our observations and those of Sano. 26
The clinical manifestations of atherosclerosis appear to result primarily from thromboses formed after physical rupture of the advanced atherosclerotic plaque. 47-51 Studies focused on late stage lesion development have quantitated changes in plaque characteristics, including content of macrophages, SMC, T-cells, collagen, lipid core, cholesterol clefts, and matrix metalloproteinases (this report). 52-56 Characterization of these features after genetic manipulation is critical to understanding the susceptibility to plaque rupture, which has been highly correlated with lesion characteristics rather than lesion size. 51,57-59
Does the Absence of a Prolonged Effect of PDGF Blockade on Fibrous Cap Formation Suggest Redundancy in Signals? How Might PDGF Blockade Enhance Lesion Formation?
The inability of either of our two approaches of PDGF blockade to induce a prolonged inhibition of fibrous cap formation in ApoE −/− mice suggests that other growth factors may be sufficient to promote SMC accumulation and connective tissue deposition. Consistent with this possibility, we recently reported that in PDGF-B hematopoietic chimeras in C57BL/6 mice, the absence of PDGF-B in circulating cells does not alter granulation tissue formation after sponge implantation despite an observed increase in vascularization in the PDGF-B −/− chimeras. 34 However, in contrast, analysis of mice derived from chimeric blastocysts composed of a mixture of wild-type cells and cells with targeted inactivation of the PDGF β-receptor demonstrates a selective advantage for fibroblasts expressing PDGF β-receptors in granulation tissue formation. 60 Even the 50% reduction in PDGF β-receptor gene dosage in PDGF β-receptor +/− cells reduces fibroblast participation by 85%. However, analysis of mice derived from chimeric blastocysts for the PDGF β-receptor is more sensitive and will reflect a selective advantage of wild-type cells over mutant cells rather than a requirement for the receptor. The kinetics and the release of specific factors that promote SMC migration and proliferation also may differ between the foreign body response in the sponge model and in the more slowly progressing, chronic inflammatory response in atherosclerosis. Similarly, although fibroblasts with the PDGF β-receptor have a selective advantage in wound healing, they do not have one during development. 7 Thus, our studies suggest that PDGF is not required for SMC accumulation in advanced fibrous caps and that other growth factors may compensate for its absence or blockade.
Blockade of PDGF may also alter expression of other gene(s), which could promote SMC accumulation via alternative mediators. PDGF is known to regulate a number of genes, 61 and the absence of some of these regulatory molecules could subsequently alter lesion progression. We probed peritoneal macrophages from PDGF-B +/+ and −/− chimeras and observed a significant shift in the expression of several cytokines and cytokine receptors (Table 2) ▶ . Monocytes are the first cells to accumulate in developing lesions of atherosclerosis, differentiate into macrophages within lesions, and are present throughout lesion progression. 58 In vitro studies have suggested that expression of the PDGF β-receptor increases with macrophage differentiation. 62 Thus, the absence of PDGF-B in circulating cells appears sufficient to modulate macrophage gene expression.
The genes that increase in the absence of PDGF-B include several pro-inflammatory genes characteristic of activated macrophages, 35,36 specifically IFN-γ, IL-1α, and IL-15. Increased levels of these genes would be predicted to promote atherosclerosis in the ApoE null mouse. 63-66 The associated increase in the chemokine receptors CCR2 and CCR5 may be due to reduced levels of their ligands, MCP-1 and RANTES. These ligands were originally identified as immediate early genes in mesenchymal cells stimulated by PDGF. 61,67,68 Reduced expression of MCP-1 and RANTES in peritoneal macrophages in the absence of endogenous PDGF-B suggests that they are PDGF-regulated early genes in macrophages as well. The decrease in macrophage expression of these potent monocyte chemoattractants does not prevent monocyte infiltration into lesions in PDGF-B −/− chimeras, and may be explained by the fact that other stimulants in atherosclerosis, such as oxidized LDL, 69 are sufficient to induce expression of MCP-1 and other chemokines in the vessel wall. Overexpression of CCR5 has been shown to increase the migratory rate of T cells toward RANTES and MIP-1α. 70 Thus, higher levels of CCR2 and CCR5 expression could enhance monocyte and T cell accumulation in PDGF-B −/− chimeras and further promote lesion progression.
The data on altered cytokine and cytokine gene expression in macrophages from PDGF-B −/− chimeras suggest a shift toward a proinflammatory phenotype that could further enhance lesion development. Although our current data do not allow us to determine whether the gene alterations are linked to changes in lesion progression, they suggest that elimination of a potent “gene switch,” such as PDGF, can significantly affect multiple components of the chronic inflammatory response responsible for the progression of lesions of atherosclerosis.
Acknowledgments
We thank Roderick Browne, Kelli McIntyre, Li-Chuan Huang, Bonnie Ashleman, Francis Deguzman, and Jing Chen for expert technical assistance, and Barbara Droker for editorial assistance.
Footnotes
Address reprint requests to Elaine W. Raines, Harborview Medical Center, 325 9th Avenue, Box 359675, Seattle, WA 98104-2499. E-mail: ewraines@u.washington.edu.
Supported by National Institutes of Health grants HL18645 (to E.W.R. and R.R.), HL55257 (to P.J.M.), and HL07828 (training grant to J.T.); the Swedish Medical Research Council, Cancer Foundation, Inga-Britt and Anne Lundberg Foundation, and a grant from the Novo Nordisk Foundation (to C.B.); and Deutsche Forschungsgemeinschaft grant Ka1078/1 (to W.E.K.).
Current address of Koichi Kozaki: Department of Geriatric Medicine, University of Tokyo Hospital, Tokyo, Japan.
References
- 1.Ross R, Raines EW, Bowen-Pope DF: The biology of platelet-derived growth factor. Cell 1986, 46:155-169 [DOI] [PubMed] [Google Scholar]
- 2.Raines EW, Ross R: Smooth muscle cells and the pathogenesis of the lesions of atherosclerosis. Br Heart J 1993, 69:S30-S37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C: Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev 1994, 8:1875-1887 [DOI] [PubMed] [Google Scholar]
- 4.Soriano P: Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes Dev 1994, 8:1888-1896 [DOI] [PubMed] [Google Scholar]
- 5.Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, Pekna M, Hellstrom M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Tornell J, Heath JK, Betsholtz C: PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 1996, 85:863-873 [DOI] [PubMed] [Google Scholar]
- 6.Soriano P: The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 1997, 124:2691-2700 [DOI] [PubMed] [Google Scholar]
- 7.Crosby JR, Seifert RA, Soriano P, Bowen-Pope DF: Chimaeric analysis reveals role of Pdgf receptors in all muscle lineages. Nat Genet 1998, 18:385-388 [DOI] [PubMed] [Google Scholar]
- 8.Seifert RA, Hart CE, Phillips PE, Forstrom JW, Ross R, Murray MJ, Bowen-Pope DF: Two different subunits associate to create isoform-specific platelet-derived growth factor receptors. J Biol Chem 1989, 264:8771-8778 [PubMed] [Google Scholar]
- 9.Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U: PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat Cell Biol 2000, 2:302-309 [DOI] [PubMed] [Google Scholar]
- 10.Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U: PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat Cell Biol 2001, 3:512-516 [DOI] [PubMed] [Google Scholar]
- 11.Uutela M, Lauren J, Bergsten E, Li X, Horelli-Kuitunen N, Eriksson U, Alitalo K: Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFC genes. Circulation 2001, 103:2242-2247 [DOI] [PubMed] [Google Scholar]
- 12.LaRochelle WJ, Jeffers M, McDonald WF, Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M, Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets J, Shimkets RA, Rothberg JM, Lichenstein HS: PDGF-D, a new protease-activated growth factor. Nat Cell Biol 2001, 3:517-521 [DOI] [PubMed] [Google Scholar]
- 13.Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL, Humes JM, Palmer TE, Hart CE: Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF α and β receptor. J Biol Chem 2001, 276:27406-27414 [DOI] [PubMed] [Google Scholar]
- 14.Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM: PDGF ligand and receptor gene expression during repair of arterial injury. J Cell Biol 1990, 111:2149-2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraiss LW, Raines EW, Wilcox JN, Seifert RA, Barrett TB, Kirkman TR, Hart CE, Bowen-Pope DF, Ross R, Clowes AW: Regional expression of the platelet-derived growth factor and its receptors in a primate graft model of vessel wall assembly. J Clin Invest 1993, 92:338-348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross R, Masuda J, Raines EW, Gown AM, Katsuda S, Sasahara M, Malden LT, Masuko H, Sato H: Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science 1990, 248:1009-1012 [DOI] [PubMed] [Google Scholar]
- 17.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R: Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science 1991, 253:1129-1132 [DOI] [PubMed] [Google Scholar]
- 18.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW: Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest 1992, 89:507-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson CL, Raines EW, Ross R, Reidy MA: Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler Thromb 1993, 13:1218-1226 [DOI] [PubMed] [Google Scholar]
- 20.Giese NA, Marijianowski MM, McCook O, Hancock A, Ramakrishnan V, Fretto LJ, Chen C, Kelly AB, Koziol JA, Wilcox JN, Hanson SR: The role of α and β platelet-derived growth factor receptor in the vascular response to injury in nonhuman primates. Arterioscler Thromb Vasc Biol 1999, 19:900-909 [DOI] [PubMed] [Google Scholar]
- 21.Hart CE, Kraiss LW, Vergel S, Gilbertson D, Kenagy R, Kirkman T, Crandall DL, Tickle S, Finney H, Yarranton G, Clowes AW: PDGFβ receptor blockade inhibits intimal hyperplasia in the baboon. Circulation 1999, 99:564-569 [DOI] [PubMed] [Google Scholar]
- 22.Davies MG, Owens EL, Mason DP, Lea H, Tran PK, Vergel S, Hawkins SA, Hart CE, Clowes AW: Effect of platelet-derived growth factor receptor-α and -β blockade on flow-induced neointimal formation in endothelialized baboon vascular grafts. Circ Res 2000, 86:779-786 [DOI] [PubMed] [Google Scholar]
- 23.Faggiotto A, Ross R: Studies of hypercholesterolemia in the nonhuman primate: II. fatty streak conversion to fibrous plaque. Arteriosclerosis 1984, 4:341-356 [DOI] [PubMed] [Google Scholar]
- 24.Rutherford C, Martin W, Carrier M, Anggard EE, Ferns GA: Endogenously elicited antibodies to platelet derived growth factor-BB and platelet cytosolic protein inhibit aortic lesion development in the cholesterol-fed rabbit. Int J Exp Pathol 1997, 78:21-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamb DJ, Avades TY, Ferns GA: Endogenous neutralizing antibodies against platelet-derived growth factor-aa inhibit atherogenesis in the cholesterol-fed rabbit. Arterioscler Thromb Vasc Biol 2001, 21:997-1003 [DOI] [PubMed] [Google Scholar]
- 26.Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, Nishikawa S, Nishikawa SI, Kita T: Functional blockade of platelet-derived growth factor receptor-β but not of receptor-α prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation 2001, 103:2955-2960 [DOI] [PubMed] [Google Scholar]
- 27.Ho MK, Springer TA: Mac-2, a novel 32, 000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J Immunol 1982, 128:1221-1228 [PubMed] [Google Scholar]
- 28.Kaminski WE, Lindahl P, Lin NL, Broudy VC, Crosby JR, Hellstrom M, Swolin B, Bowen-Pope DF, Martin PJ, Ross R, Betsholtz C, Raines EW: Basis of hematopoietic defects in platelet-derived growth factor (PDGF)-B and PDGF β-receptor null mice. Blood 2001, 97:1990-1998 [DOI] [PubMed] [Google Scholar]
- 29.Yu JC, Lokker NA, Hollenbach S, Apatira M, Li J, Betz A, Sedlock D, Oda S, Nomoto Y, Matsuno K, Ide S, Tsukuda E, Giese NA: Efficacy of the novel selective platelet-derived growth factor receptor antagonist ct52923 on cellular proliferation, migration, and suppression of neointima following vascular injury. J Pharmacol Exp Ther 2001, 298:1172-1178 [PubMed] [Google Scholar]
- 30.Raines EW, Ross R: Compartmentalization of PDGF on extracellular binding sites dependent on exon-6-encoded sequences. J Cell Biol 1992, 116:533-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Junqueira LC, Cossermelli W, Brentani R: Differential staining of collagens type I, II, and III by Sirius Red and polarization microscopy Arch Histol Jpn 1978, 41:267-274 [DOI] [PubMed] [Google Scholar]
- 32.Learn DB, Moloney SJ, Giddens LD: Quantitative determination of murine dermal elastic fibers by color image analysis: comparison of three staining methods. Biotech Histochem 1992, 67:125-130 [DOI] [PubMed] [Google Scholar]
- 33.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R: ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994, 14:133-140 [DOI] [PubMed] [Google Scholar]
- 34.Buetow BS, Crosby JR, Kaminski WE, Ramachandran RK, Lindahl P, Martin P, Betsholtz C, Seifert RA, Raines EW, Bowen-Pope DF: Platelet-derived growth factor B-chain of hematopoietic origin is not necessary for granulation tissue formation and its absence enhances vascularization. Am J Pathol 2001, 159:1869-1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhing RJ, Adams DO: Molecular events in the activation of murine macrophages. Agents Actions 1989, 26:9-14 [DOI] [PubMed] [Google Scholar]
- 36.Doherty TM, Seder RA, Sher A: Induction and regulation of IL-15 expression in murine macrophages. J Immunol 1996, 156:735-741 [PubMed] [Google Scholar]
- 37.Andres PG, Beck PL, Mizoguchi E, Mizoguchi A, Bhan AK, Dawson T, Kuziel WA, Maeda N, MacDermott RP, Podolsky DK, Reinecker HC: Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated Th2-type immune response in the intestine. J Immunol 2000, 164:6303-6312 [DOI] [PubMed] [Google Scholar]
- 38.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF: Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 1997, 100:2552-2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yagi M, Kato S, Kobayashi Y, Kubo K, Oyama S, Shimizu T, Nishitoba T, Isoe T, Nakamura K, Ohashi H, Kobayashi N, Iinuma N, Osawa T, Onose R, Osada H: Selective inhibition of platelet-derived growth factor (PDGF) receptor autophosphorylation and PDGF-mediated cellular events by a quinoline derivative. Exp Cell Res 1997, 234:285-292 [DOI] [PubMed] [Google Scholar]
- 40.Spacey GD, Uings IJ, Slater M, Hirst S, Bonser RW: Indolocarbazoles: potent and selective inhibitors of platelet-derived growth factor receptor autophosphorylation. Biochem Pharmacol 1998, 55:261-271 [DOI] [PubMed] [Google Scholar]
- 41.Bilder G, Wentz T, Leadley R, Amin D, Byan L, O’Conner B, Needle S, Galczenski H, Bostwick J, Kasiewski C, Myers M, Spada A, Merkel L, Ly C, Persons P, Page K, Perrone M, Dunwiddie C: Restenosis following angioplasty in the swine coronary artery is inhibited by an orally active PDGF-receptor tyrosine kinase inhibitor, RPR101511A. Circulation 1999, 99:3292-3299 [DOI] [PubMed] [Google Scholar]
- 42.Carroll M, Ohno-Jones S, Tamura S, Buchdunger E, Zimmermann J, Lydon NB, Gilliland DG, Druker BJ: CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins Blood 1997, 90:4947-4952 [PubMed] [Google Scholar]
- 43.Rosenfeld ME, Tsukada T, Chait A, Bierman EL, Gown AM, Ross R: Fatty streak expansion and maturation in Watanabe heritable hyperlipemic and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis 1987, 7:24-34 [DOI] [PubMed] [Google Scholar]
- 44.Reardon CA, Blachowicz L, White T, Cabana V, Wang Y, Lukens J, Bluestone J, Getz GS: Effect of immune deficiency on lipoproteins and atherosclerosis in male apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2001, 21:1011-1016 [DOI] [PubMed] [Google Scholar]
- 45.Witting PK, Pettersson K, Letters J, Stocker R: Site-specific antiatherogenic effect of probucol in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2000, 20:E26-E33 [DOI] [PubMed] [Google Scholar]
- 46.Reardon CA, Getz GS: Mouse models of atherosclerosis. Curr Opin Lipidol 2001, 12:167-173 [DOI] [PubMed] [Google Scholar]
- 47.Falk E: Why do plaques rupture? Circulation 1992, 86:III30-III42 [PubMed] [Google Scholar]
- 48.Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J: Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J 1993, 69:377-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross R: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362:801-809 [DOI] [PubMed] [Google Scholar]
- 50.Libby P: Molecular bases of the acute coronary syndromes. Circulation 1995, 91:2844-2850 [DOI] [PubMed] [Google Scholar]
- 51.Lee RT, Libby P: The unstable atheroma. Arterioscler Thromb Vasc Biol 1997, 17:1859-1867 [DOI] [PubMed] [Google Scholar]
- 52.Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA: Requirement for CD154 in the progression of atherosclerosis. Nat Med 1999, 5:1313-1316 [DOI] [PubMed] [Google Scholar]
- 53.Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ: Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci USA 2000, 97:7464-7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Jr, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM: Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest 2001, 107:675-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A: Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res 2001, 89:E41-E45 [DOI] [PubMed] [Google Scholar]
- 56.Ni W, Egashira K, Kitamoto S, Kataoka C, Koyanagi M, Inoue S, Imaizumi K, Akiyama C, Nishida Ki K, Takeshita A: New anti-monocyte chemoattractant protein-1 gene therapy attenuates atherosclerosis in apolipoprotein E-knockout mice. Circulation 2001, 103:2096-2101 [DOI] [PubMed] [Google Scholar]
- 57.Davies MJ: A macro and micro view of coronary vascular insult in ischemic heart disease. Circulation 1990, 82:II38-II46 [PubMed] [Google Scholar]
- 58.Ross R: Atherosclerosis: an inflammatory disease. N Engl J Med 1999, 340:115-126 [DOI] [PubMed] [Google Scholar]
- 59.Libby P: What have we learned about the biology of atherosclerosis? The role of inflammation Am J Cardiol 2001, 88:3-6 [DOI] [PubMed] [Google Scholar]
- 60.Crosby JR, Tappan KA, Seifert RA, Bowen-Pope DF: Chimera analysis reveals that fibroblasts and endothelial cells require platelet-derived growth factor receptorβ expression for participation in reactive connective tissue formation in adults but not during development. Am J Pathol 1999, 154:1315-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cochran BH, Reffel AC, Stiles CD: Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell 1983, 33:939-947 [DOI] [PubMed] [Google Scholar]
- 62.Inaba T, Shimano H, Gotoda T, Harada K, Shimada M, Ohsuga J, Watanabe Y, Kawamura M, Yazaki Y, Yamada N: Expression of platelet-derived growth factor β receptor on human monocyte-derived macrophages and effects of platelet-derived growth factor BB dimer on the cellular function. J Biol Chem 1993, 268:24353-24360 [PubMed] [Google Scholar]
- 63.Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F: Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998, 97:242-244 [DOI] [PubMed] [Google Scholar]
- 64.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS: Interferon-γ elicits arteriosclerosis in the absence of leukocytes. Nature 2000, 403:207-211 [DOI] [PubMed] [Google Scholar]
- 65.Whitman SC, Ravisankar P, Elam H, Daugherty A: Exogenous interferon-γ enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol 2000, 157:1819-1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wuttge DM, Eriksson P, Sirsjo A, Hansson GK, Stemme S: Expression of interleukin-15 in mouse and human atherosclerotic lesions. Am J Pathol 2001, 159:417-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rollins BJ, Morrison ED, Stiles CD: Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc Natl Acad Sci USA 1988, 85:3738-3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rollins BJ, Walz A, Baggiolini M: Recombinant human MCP-1/JE induces chemotaxis, calcium flux, and the respiratory burst in human monocytes. Blood 1991, 78:1112-1116 [PubMed] [Google Scholar]
- 69.Liao F, Berliner JA, Mehrabian M, Navab M, Demer LL, Lusis AJ, Fogelman AM: Minimally modified low density lipoprotein is biologically active in vivo in mice. J Clin Invest 1991, 87:2253-2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zang YC, Samanta AK, Halder JB, Hong J, Tejada-Simon MV, Rivera VM, Zhang JZ: Aberrant T cell migration toward RANTES and MIP-1 α in patients with multiple sclerosis: overexpression of chemokine receptor CCR5. Brain 2000, 123:1874-1882 [DOI] [PubMed] [Google Scholar]