

Abstract
Alveolar cells of the lung are injured and killed when exposed to elevated levels of inspired oxygen. Damaged tissue architecture and pulmonary function is restored during recovery in room air as endothelial and type II epithelial cells proliferate. Although excessive fibroblast proliferation and inflammation occur when abnormal remodeling occurs, genes that regulate repair remain unknown. Our recent observation that hyperoxia inhibits proliferation through induction of the cyclin-dependent kinase inhibitor p21Cip1/WAF1/Sdi1, which also facilitates DNA repair, suggested that p21 may participate in remodeling. This hypothesis was tested in p21-wild-type and -deficient mice exposed to 100% FiO2 and recovered in room air. p21 increased during hyperoxia, remained elevated after 1 day of recovery before returning to unexposed levels. Increased proliferation occurred when p21 expression decreased. In contrast, higher and sustained levels of proliferation, resulting in myofibroblast hyperplasia and monocytic inflammation, occurred in recovered p21-deficient lungs. Cells with DNA strand breaks and expressing p53 were observed in hyperplastic regions suggesting that DNA integrity had not been restored. Normal recovery of endothelial and type II epithelial cells, as assessed by expression of cell-type-specific genes was also delayed in p21-deficient lungs. These results reveal that p21 is required for remodeling the oxygen-injured lung and suggest that failure to limit replication of damaged DNA may lead to cell death, inflammation, and abnormal remodeling. This observation has important implications for therapeutic strategies designed to attenuate long-term chronic lung disease after oxidant injury.
Supplemental oxygen (hyperoxia) is used to reduce tissue hypoxia for patients suffering from respiratory distress. Unfortunately, oxygen levels greater than 90% FiO2 kill microvascular endothelial and type I epithelial cells, resulting in capillary leak into alveolar and interstitial spaces. Additional injury can occur by the oxidant burst released by inflammatory cells that are recruited as cells die. Ultrastructural studies in adult rats revealed that cells died by necrotic swelling of cytoplasm, nuclei, and mitochondria that culminated in disintegration of the plasma membrane. 1 Dead cells are not replaced during exposure because oxygen inhibits proliferation. 2 However, increased proliferation and tissue remodeling occur when injured lungs are returned to normoxic conditions. 3-5 Pulse-chase labeling using 3H thymidine in rodents showed that alveolar type II epithelial cells proliferate between days 2 and 4 of recovery and differentiate into type I cells. Type II cells may also participate in vascular remodeling because they express vascular endothelial growth factor (VEGF), a potent endothelial cell mitogen. 6 Although increased proliferation of fibroblasts and matrix deposition are important for normal healing, failure to terminate these processes can lead to progressive fibrosis. 7 In fact, abnormal proliferation of fibroblasts and epithelial cells is observed in bronchopulmonary dysplasia, a disease associated with oxygenation and ventilation of the premature lung. 8,9 All together, these findings suggest that carefully regulated cell proliferation is required for remodeling the oxygen-injured lung.
Cell proliferation is regulated by growth stimulatory and inhibitory signals that modify the activity of cyclin-dependent kinases responsible for phosphorylating proteins essential for cell cycle progression. 10 Cell cycle checkpoints are activated by environmental stress and are thought to prevent fixation of DNA mutations and allow additional time for repair to occur. The tumor-suppressor protein p53 is the major inhibitor of cell proliferation after environmental stress. It accumulates in cells with DNA strand breaks and limits replication of damaged DNA or induces apoptosis. 11 The cyclin-dependent kinase inhibitor p21Cip1/WAF1/Sdi1 (hereafter p21) is the predominant target of p53-mediated growth suppression. p21 has two domains that prevent cells from exiting G1 phase of the cell cycle. 12 The amino-terminus inhibits G1 cyclin-dependent kinases while the carboxy-terminus blocks association of DNA polymerases with proliferating cell nuclear antigen (PCNA). 13 p21 may also participate in long-patch base excision repair or nucleotide excision repair of damaged DNA through its interactions with PCNA. 14 Thus, p21 limits replication and promotes repair of damaged DNA.
Although the genotoxic effects of oxygen on cells have been known for nearly 10 years, 15 it is only recently appreciated that the growth suppressive activities of oxygen are mediated, in part, through induction of p21. Using SV40-transformed type II epithelial cells exposed to hyperoxia, Corroyer and colleagues 16 were the first to show that hyperoxia increased p21, which decreased G1 cyclin E-dependent kinase activity. Subsequent studies in a variety of nontransformed cell lines confirmed that hyperoxia inhibited proliferation in G1 through induction of p21. 17-19 Hyperoxia also increased p21 mRNA and protein in terminal bronchiole epithelium and alveolar endothelial and type I and II epithelial cells of adult and newborn mice. 20,21 We recently showed that 72 hours of hyperoxia inhibited cell proliferation in adult p21-wild-type, but not p21-deficient mice. 22 Because p21 mediates the growth-arresting activities of oxygen and can participate in DNA repair, it may be a key molecule required for remodeling the injured lung. The current study tests this hypothesis by exposing p21-wild-type and -deficient mice to hyperoxia and recovering in room air where cell proliferation and tissue remodeling occur.
Materials and Methods
Mice and Oxygen Exposures
Adult p21-wild-type and -deficient mice (10 to 12 weeks old) were kept in room air or exposed to filtered humidified 100% oxygen for 60 hours as previously described. 22 Mice were recovered in room air by removing their cages from hyperoxia. Two hours before sacrifice, mice were injected intraperitoneally with 5-bromo-2′-deoxyuridine (BrdU) as recommended by the manufacturer (Zymed, South San Francisco, CA). The lungs were exposed and the right lobe inflation fixed with 10% neutral buffered formalin and the left lobe removed for isolation of DNA, RNA, or protein. The University of Rochester’s Committee on Animal Resources approved all protocols involving the mice.
Analysis of Gene Expression
Total RNA was isolated from lungs, separated on 1.0% agarose-formaldehyde gels, and transferred to Nytran. Blots were hybridized with a 32P-labeled 454-bp cDNA containing the second exon of the mouse p21 gene, a 553-bp cRNA containing the murine histone H3.2 open-reading frame or a cDNA containing the murine L32 cDNA. 22 Radiolabeled cRNA probes were generated by in vitro transcription and cDNA probes were labeled by random prime labeling. Differences in gene expression were visualized using a PhosphorImage screen and normalized to expression of L32 RNA using ImageQuant analysis (Amersham Pharmacia Biotech, Piscataway, NJ).
Anti-sense and sense 33P-radiolabeled histone H3.2 riboprobes were synthesized using T3 and T7 RNA polymerases to a specific activity of 3 × 109 dpm/μg for in situ hybridizations. Sections were prehybridized for 3 hours and hybridized for 16 hours at 53°C as previously described. 22 After washes and digestion with RNase A, sections were washed stringently in 0.1× standard saline citrate for 30 minutes at 68°C, dipped in a 1:1 dilution of NTB-2 emulsion (Eastman Kodak, Rochester, NY), and exposed at 4°C before developing and counterstaining with hematoxylin and eosin. Slides were visualized with a Nikon E800 microscope (Nikon, Melville, NY) and images captured with a SPOT-RT digital camera (Diagnostic Instruments, Sterling Heights, MI).
The expression of surfactant protein (SP)-C and endothelial cell-specific genes was assessed by S1 or RNase protection analyses, respectively. S1 nuclease protection probes for exon 6 of murine SP-C and L32 were synthesized by PCR from plasmid templates and radiolabeled using polynucleotide kinase for 30 minutes at 37°C as described. 23,24 Probes were added to 5 μg of total RNA, denatured by boiling, and hybridized overnight at 50.5°C. S1 digestion mixture was immediately added and incubated for 60 minutes at room temperature. Radiolabeled RNA probes encoding Flt1, Flt4, Tie1, Tie2, CD31, and VEGF were synthesized at room temperature according to the RiboQuant Multi-Probe RNase Protection Assay System (PharMingen, San Diego, CA), hybridized to 5 μg of total RNA, and processed according to the kit. 25 Protected RNAs obtained after S1 or RNase digestion were ethanol precipitated and separated on a 6% polyacrylamide gel. The gel was dried and exposed to a PhosphorImage screen for visualization. Band intensities were normalized to L32 and quantified using ImageQuant analysis.
Movat Staining
Movat’s stain was modified by staining deparaffinized and hydrated slides for 30 minutes in Bouin’s solution preheated to 56°C. 26 Slides were washed in running tap water for 5 to 10 minutes, rinsed in distilled water, and stained in Alcian Blue solution. Sections were rewashed in water and stained for 15 minutes in Verhoeff solution. 27 Slides were washed with water and dipped in 5% ferric chloride before rinsing in water. The following stains were used according to Elbadawi’s protocol: Biebrich scarlet-woodstain scarlet-phosphotungstic acid, phosphotungstic-phosphomolybdic acid, naphthol green B, and alcoholic safran. Slides were rinsed in two changes of absolute ethanol, cleared in xylene, and visualized.
Immunohistochemistry
Slides for BrdU/α-smooth muscle actin immunostaining were deparaffinized, rehydrated, treated with hydrogen peroxide, and rinsed in water. Antigen retrieval was performed by incubating the slides in citrate buffer, pH 6.0, and heating in an autoclave to 121°C at a pressure to 15 PSI for 10 minutes. The slides were incubated an additional 10 minutes in the warm solution before washing in Tris-buffered saline and 0.1% Triton X-100 (TBST) and quenching endogenous peroxide with 3% hydrogen peroxide. Mouse monoclonal anti-BrdU from the Animal Research Kit (DAKO, Carpinteria, CA) was added and the slides incubated for 15 minutes at room temperature before washing and incubating with streptavidin-horseradish peroxidase for 30 minutes. After washing and reacting with diaminobenzidine, the slides were incubated in anti-smooth muscle actin antibody (DAKO) at 1:50 dilution for 30 minutes. The slides were washed and incubated with biotinylated secondary antibody (1:250) for 30 minutes. They were then washed again, incubated in streptavidin-alkaline phosphatase (Vector Laboratories, Burlingame, CA) at 1:500 for 15 minutes at room temperature before extensive washings. After rinsing, the slides were incubated with biotinylated goat anti-rabbit IgG diluted 1:200 in TBST and 2% normal goat serum (NGS) for 45 minutes, rinsed, and treated with ABC-alkaline phosphatase (Vector Laboratories) for 30 minutes. 5-Bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium stain was used to visualize cells expressing α-smooth muscle actin.
Terminal dUTP nick-end labeling (TUNEL) staining was performed using an ApopTag kit (Serologicals Corp., Norcross, GA) as previously described. 23 Slides for p53 staining were deparaffinized and hydrated before performing antigen retrieval. They were quenched with 1% hydrogen peroxide, blocked with 3% bovine serum albumin, and incubated with 1:250 dilution of phosphoserine 15-p53 primary antibody (Cell Signaling, Beverly, MA). After rinsing, biotinylated goat anti-rabbit secondary was applied for 30 minutes, washed, and then treated with ABC-alkaline phosphatase. The stain was visualized using diaminobenzidine (Sigma, St. Louis, MO).
Quantification of TUNEL and p53-Positive Cells
Random, noncontiguous fields of parenchymal airspaces were acquired using the ×40 objective of a Nikon E-800 fluorescent microscope fitted with a Spot-RT camera. Two fields were obtained from three separate animals and analyzed. Fields containing a large airway or blood vessel were rejected. Quantification measurements were performed using MetaMorph (Molecular Devices, Corp., Sunnyvale, CA). Brown and blue nuclei were counted and the ratio of brown nuclei to brown plus blue nuclei obtained.
DNA Laddering
Lungs were frozen in liquid nitrogen, ground into a powder using a mortar and pestle, and incubated overnight at 50°C in sample buffer from the TACS Apoptotic DNA Laddering kit (Trevigen Inc., Gaithersburg, MD). DNA was isolated, separated on a 1.2% agarose gel, and visualized with ethidium bromide (Bio-Rad Laboratories, Hercules, CA).
Bronchoalveolar Lavage
Experimental mice were euthanized with 400 mg/kg body weight of sodium pentobarbital administered intraperitoneally. The trachea was exposed and surgically cannulated with a blunted 18-gauge needle. A three-way stopcock was attached to the cannula, and the lungs were sequentially lavaged with eight, 1-μml vol of sterile saline. The collected lavage fluid was centrifuged at 250 × g for 10 minutes at 4°C to pellet cells. The recovered cells were washed once with ice-cold phosphate-buffered saline (PBS), pelleted again, and then resuspended in 1 ml of ice-cold PBS. Aliquots of the lavage cell suspension were diluted 1:2 with Turk’s solution, and cells quantified using a hemacytometer.
Hydroxyproline Assay
The amount of hydroxyproline in lung homogenates was used as an indicator of total collagen as previously described. 28 Lung tissue (0.5 to 0.8 g) was hydrolyzed in 20 ml of 6 N HCl for 16 hours at 116°C in an autoclave. The hydrolysate (200 μl) was normalized to pH 7.0 using NaOH resulting in a final volume of 2 ml. Chloramine T (1 ml) was added, incubated for 20 minutes, followed by 1 ml of perchloric acid for 5 minutes. P-dimethylamino benzaldehyde (1 ml) was reacted for 20 minutes at 60°C and the absorbance was read at 557 nm. A standard curve was prepared using trans-4-hydroxy-6-proline. All reagents were obtained from Sigma. The lower limit of detection was 1 μg/sample.
Statistical Analysis
Values are expressed as means ± SD. Group means were compared by analysis of variance with Fisher’s procedure post hoc analysis using StatView software (SAS, Institute Inc., Cary, NC) for Macintosh with P < 0.05 considered significant.
Results
p21-Deficient Lungs Have Enhanced Proliferation during Recovery
p21 mRNA expression was assessed in mice exposed to room air; 60 hours of hyperoxia; or after 24, 72, or 240 hours of recovery in room air. p21 mRNA increased nearly ninefold in p21-wild-type mice exposed to hyperoxia, remained elevated after 24 hours of recovery before returning to unexposed levels at 72 and 240 hours (Figure 1) ▶ . As we previously reported, 21 hyperoxia increased p21 mRNA and protein in terminal bronchiolar epithelial cells and uniformly throughout the parenchyma. p21 mRNA or protein was not detected in tissue homogenates or in situ after 72 hours of recovery, nor was it ever detected in p21-deficient mice.
Figure 1.

Hyperoxia increases p21 mRNA expression. RNA was isolated from p21-wild-type (+/+) and p21-deficient (−/−) lungs exposed to room air (0 hours); hyperoxia for 60 hours; or recovered in room air for 24, 72, or 240 hours. A: Representative Northern blot probed for p21 or the ribosomal RNA L32. B: The expression of p21 was normalized to L32 and graphed relative to room air-exposed values. Values represent mean ±SD (n = 3). p21 mRNA was significantly increased in lungs exposed to hyperoxia or recovered in room air for 24 hours. *, P < 0.01.
The expression of the replication-dependent histone H3.2 gene was used to assess whether the absence of p21 altered cell proliferation during recovery. Expression of H3.2 is restricted to the S phase of the cell cycle and is not expressed by cells repairing damaged DNA. 29,30 H3.2 was faintly detected in p21-wild-type mice exposed to room air or hyperoxia (Figure 2) ▶ . It remained low after 24 hours of recovery and increased fourfold by 72 hours before returning to control levels after 240 hours. Similarly, low levels of H3.2 were expressed in p21-deficient lungs exposed to room air or hyperoxia. It increased 10-fold after 72 hours and remained elevated after 240 hours compared to unexposed mice. Although H3.2 expression was comparable between p21-wild-type and -deficient mice during exposure, it was significantly higher in p21-deficient mice at 72 and 240 hours of recovery compared to wild-type mice.
Figure 2.

Cell proliferation during and after hyperoxia. A: Northern blot analysis of histone H3.2 and L32 in p21-wild-type (+/+) and p21-deficient (−/−) lungs exposed and recovered from hyperoxia. B: Graphic representation of H3.2 expression compared to room air-exposed samples. Values represent mean ± SD (n = 4 to 6). H3.2 was significantly increased in recovered p21-wild-type (*, P < 0.0001) and p21-deficient (†, P < 0.008) lungs compared to unexposed controls. It was also significantly higher in p21-deficient lungs compared to wild-type lungs after 72 and 240 hours (§, P < 0.02).
In situ hybridization was used to investigate where proliferation was occurring. A randomly distributed proliferating cell was detected in the parenchyma of p21-wild-type and -deficient lungs exposed to room air or hyperoxia (Figure 3; A to D) ▶ . Proliferation remained low in p21-wild-type mice recovered for 24 hours, increased throughout the parenchyma after 72 hours, and returned to low levels by 240 hours (Figure 3; E, G, and I) ▶ . In contrast, proliferation increased modestly in p21-deficient lungs recovered for 24 hours, markedly increased at 72 hours, and remained elevated at 240 hours (Figure 3; F, H, and J) ▶ . Closer examination of lungs recovered for 240 hours revealed that cell proliferation was prevalent in hyperplastic regions of the parenchyma. Minimal proliferation of terminal bronchiole epithelial cells was observed in p21-wild-type or -deficient lungs at any time during recovery. Adjacent sections hybridized with sense H3.2 probe had minimal signal (data not shown). 22 Assessing PCNA expression by immunohistochemistry and Western blot analysis confirmed that proliferation was elevated in p21-deficient lungs (data not shown).
Figure 3.
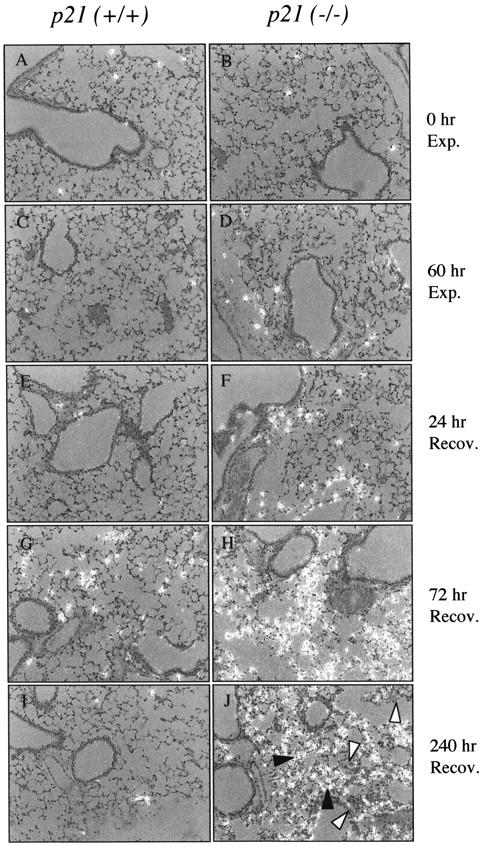
Localization of H3.2 mRNA by in situ hybridization. p21-wild-type (A, C, E, G, I) and p21-deficient (B, D, F, H, J) mice were exposed to room air (A, B); 60 hours of hyperoxia (C, D); or recovered in room air for 24 (E, F), 72 (G, H), and 240 (I, J) hours. Sections were hybridized with anti-sense radiolabeled H3.2 cRNA probe. The filled arrowhead identifies proliferating cells and the open arrowhead identifies nonproliferating cells (J).
p21-Deficient Lungs Have Inflammation and Myofibroblasts
Because fibroblasts were observed in the hyperplastic areas, a modified Movat stain was performed that distinguishes collagen, proteoglycans, elastic fibers, fibroblasts, and muscle. 26,27 Lungs of recovered p21-wild-type mice had normal alveolarization with minimal inflammation and thickening of the airway or alveolar septae (Figure 4A) ▶ . In contrast, normal and abnormal architecture was observed in lungs of recovered p21-deficient mice (Figure 4B) ▶ . Increased numbers of inflammatory cells were observed in alveolarized regions that had normal architecture and minimal septal thickening (Figure 4C) ▶ . This histological evidence of increased inflammatory cells was confirmed by counting inflammatory cells in bronchoalveolar lavage fluid. p21-deficient mice had nearly threefold more inflammatory cells compared to wild-type mice, of which 98% were macrophages (Table 1) ▶ . Although the percentage of macrophages, neutrophils, or lymphocytes were comparable between the mice, a small increase in eosinophils was detected in p21-deficient but not in p21-wild-type lungs. Hyperplasia was also observed in recovered p21-deficient lungs (Figure 4D) ▶ . Intense α-smooth muscle actin and BrdU immunostaining was observed in the hyperplastic regions, suggesting the presence of proliferating myofibroblasts (Figure 4, E and F) ▶ . α-Smooth muscle actin staining was not observed in the alveolarized regions of p21-deficient mice. It was also never observed in the parenchyma of recovered p21-wild-type mice. Although faint collagen (light green stain) and proteoglycan (light blue stain) were observed in the hyperplastic regions, hydroxyproline was not significantly different between wild-type and deficient mice. Recovered p21-wild-type lungs (n = 5) contained 1.34 ± 0.17 mg of hydroxyproline/g of lung tissue whereas recovered p21-deficient lungs (n = 8) contained 1.26 ± 0.15 mg of hydroxyproline/g of lung tissue (P = 0.332). In summary, normal architecture and minimal inflammation were observed in recovered p21-wild-type mice. Increased inflammation and myofibroblast hyperplasia, without deposition of mature collagen, were observed in p21-deficient lungs.
Figure 4.
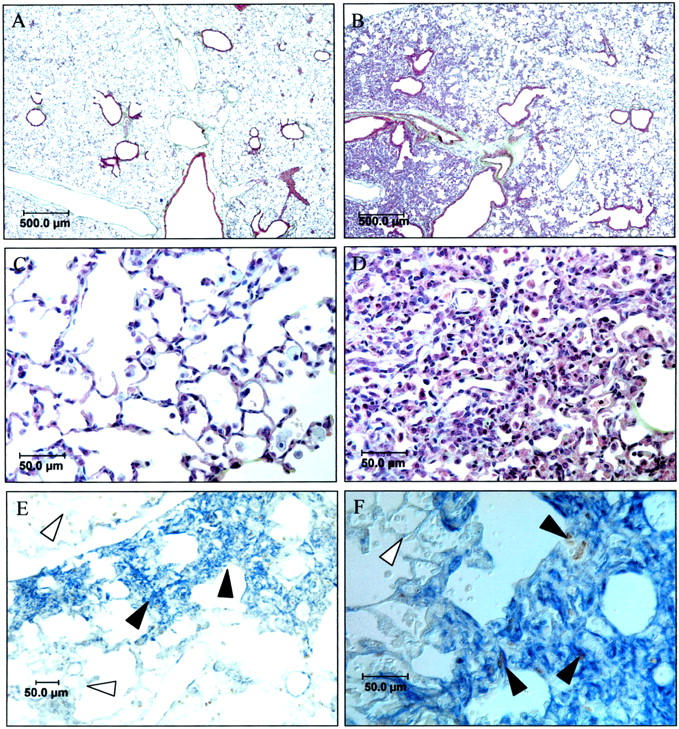
Histopathology of mice recovered for 10 days. A modified Movat stain was performed on p21-wild-type (A) and deficient lungs (B–D). C and D represent higher power views of alveolarized (C) and hyperplastic regions (D) of p21-deficient lungs. Note minimal collagen (light green) or proteoglycan (light blue) staining in A–D. p21-deficient (E, F) lungs were immunostained for α-smooth muscle actin and BrdU. α-Smooth muscle actin stained blue and BrdU stained brown. Filled arrow denotes cells expressing α-smooth muscle actin and open arrow denotes cells lacking expression.
Table 1.
Inflammatory Cells in Bronchoalveolar Lavage Fluid
| Total cells × 105/ml | Percent macrophages | Percent PMNs | Percent lymphocytes | Percent eosinophils | |
|---|---|---|---|---|---|
| p21 (+/+) | 5.3 ± 0.3 | 99.5 ± 0.5 | 0.32 ± 0.3 | 0.19 ± 0.4 | 0 |
| p21 (−/−) | 13.9 ± 1.6* | 98.2 ± 1.1 | 0.64 ± 0.9 | 0.27 ± 0.4 | 0.75 ± 0.4* |
Total number and percentage of inflammatory cells obtained from bronchoalveolar lavage fluid of mice exposed to hyperoxia for 60 hours and recovered for 8 days. Values represent mean ± SD (n = 4). Total number of cells and percent of eosinophils were significantly increased in p21-deficient mice (*, P < 0.01).
DNA Strand Breaks Are Present in Recovered p21-Deficient Lungs
Previous studies showed that hyperoxia induces DNA strand breaks that may be visualized as a smear by electrophoretic size fractionation of genomic DNA (DNA laddering) or TUNEL. 23,25,31 Because p21 participates in DNA repair, DNA laddering was performed on recovered lungs to assess whether DNA integrity had been restored during recovery. As shown in Figure 5 ▶ , intact DNA was isolated from recovered p21-wild-type lungs indicating that DNA integrity was restored. In contrast, a marked smear was obtained from p21-deficient lungs. TUNEL staining was used to visualize cells with fragmented DNA (Figure 6) ▶ . Approximately 2% of p21-wild-type parenchymal cells had TUNEL-positive staining. This low percentage reveals that DNA integrity was restored because ∼60% of parenchymal cells become TUNEL-positive during exposure. 32,33 In contrast, TUNEL-positive cells were readily detected in both the alveolarized and hyperplastic regions of p21-deficient lungs. Closer examination revealed that ∼12% of cells within alveolarized areas and 40% of cells in hyperplastic regions were TUNEL-positive. TUNEL staining was detected in pyknotic and morphologically intact nuclei.
Figure 5.

DNA laddering in recovered lungs. Total genomic DNA (2 μg) isolated from p21-wild-type and -deficient lungs recovered for 8 days were separated by size on a 1.2% agarose gel and stained with ethidium bromide. Lane M contains size marker DNA and the remaining lanes contain DNA obtained from individual mice.
Figure 6.
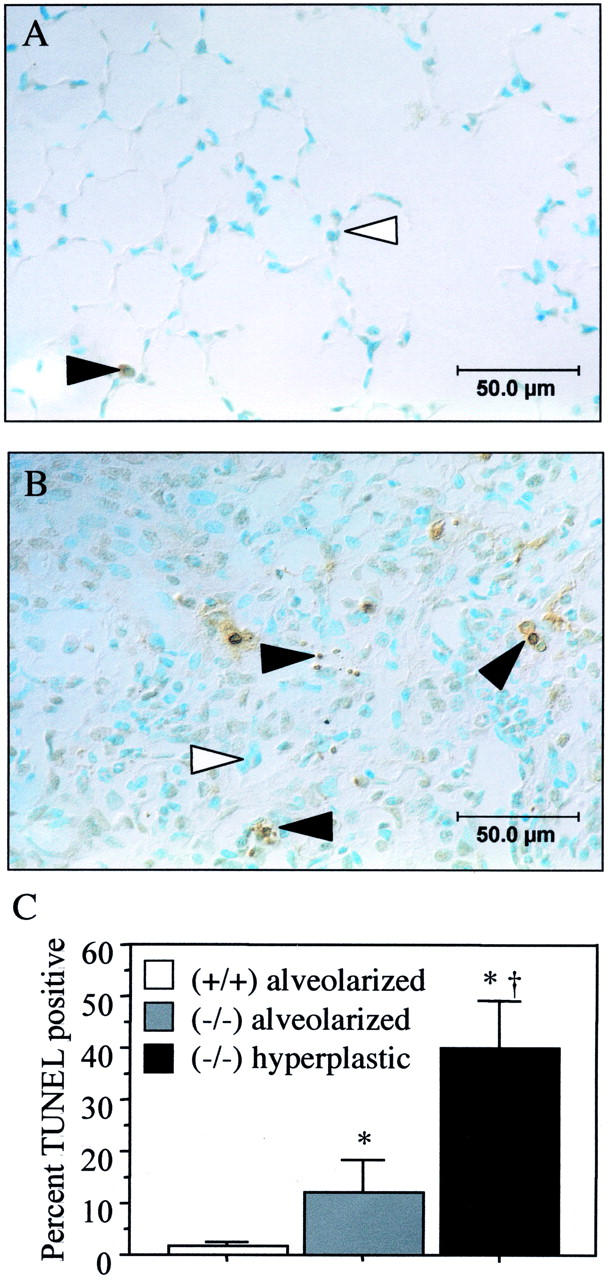
TUNEL staining in recovered lungs. TUNEL staining of p21-wild-type (A) and hyperplastic regions of p21-deficient (B) lungs recovered for 10 days. Filled arrowhead denotes TUNEL-positive nuclei and open arrowhead denotes negative nuclei that stain blue because of methyl green counterstain. C: The percentage of TUNEL-positive cells was increased in p21-deficient mice (*, P < 0.01) and was significantly higher in hyperplastic regions compared to alveolarized areas of their lungs (†, P < 0.0001).
The expression of p53, which increases during hyperoxia, was used to confirm that DNA damage signaling was still prevalent in recovered p21-deficient lungs. p53 was detected in ∼2% of parenchymal p21-wild-type cells, consistent with the low percentage of TUNEL-positive cells (Figure 7) ▶ . Similarly, ∼2% of parenchymal cells in alveolarized regions of p21-deficient lungs were TUNEL-positive. In contrast, p53 was detected in 10% of cells within hyperplastic regions, consistent with a greater percentage of TUNEL-positive cells. Thus, DNA integrity was restored in recovered p21-wild-type but not deficient lungs.
Figure 7.
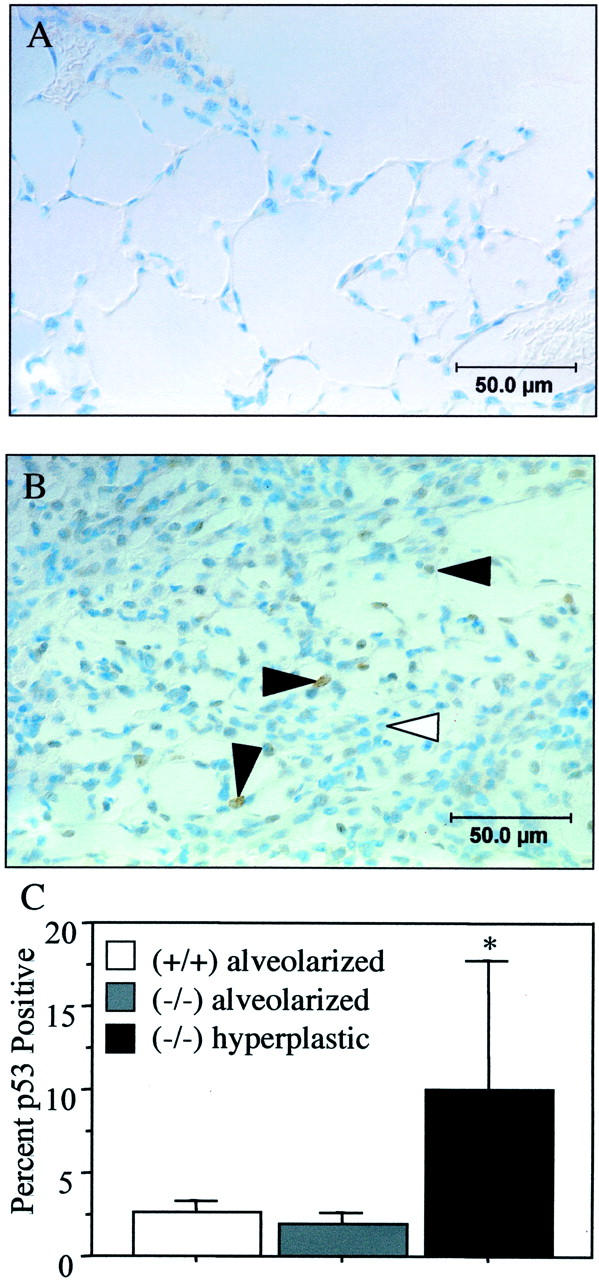
p53 expression in recovered lungs. p53 staining of p21-wild-type (A) and hyperplastic regions of p21-deficient (B) lungs recovered for 10 days. Filled arrowhead denotes cells expressing p53. C: The percentage of p53-positive cells was increased in hyperplastic regions of p21-deficient mice compared to their alveolarized regions and compared to parenchyma of p21-wild-type mice (*, P < 0.01).
Expression of Epithelial and Endothelial Cell Genes Are Delayed in p21-Deficient Mice
Because normal remodeling is dependent on proliferation of epithelial type II and endothelial cells, the expression of cell-type-specific genes was used to determine whether these cells were affected by the absence of p21. Hyperoxia decreased expression of the type II cell-specific gene SP-C. It remained low after 24 hours of recovery, modestly increased after 72 hours, and returned to unexposed levels by 240 hours (Figure 8) ▶ . Hyperoxia also decreased SP-C mRNA in p21-deficient lungs. It remained low after 24 hours and 72 hours of recovery before returning to control levels by 240 hours. Although SP-C expression was restored in p21-deficient mice, it was significantly delayed compared to wild-type mice. In contrast, SP-B expression, which is also expressed by bronchiolar epithelial cells, was not different between the mice at any time during or after hyperoxia (data not shown). These findings suggest that type II cells may recover more slowly in the absence of p21.
Figure 8.

Expression of SP-C is delayed in p21-deficient lungs. p21-wild-type and -deficient mice were exposed to room air (0) or recovered from 60 hours of hyperoxia for 24, 72, and 240 hours. The expression of SP-C and L32 mRNA was assessed by S1 protection analysis and graphed as mean ± SD (n = 3) after normalizing to L32. SP-C expression was significantly reduced in p21-deficient mice compared to wild-type mice after 72 hours of recovery (*, P < 0.04).
The expression of genes involved in endothelial cell function was also examined. Hyperoxia decreased expression of Flt1, Flt4, Tie1, Tie2, CD31, and VEGF (Figure 9) ▶ . Although the expression of all genes was restored by 240 hours of recovery in p21-wild-type mice, they showed a delayed response in p21-deficient mice. The expression of CD31 was lower in p21-deficient mice at both 24 and 72 hours of recovery (P < 0.003), while the expression of all other genes was lower at 72 hours (P < 0.01). Tie1 and Tie2 receptor mRNAs were most significantly reduced compared to the other genes (P < 0.001). Thus, reduced expression of endothelial cell-specific genes suggests that vascularization may be delayed in p21-deficient mice.
Figure 9.

Expression of endothelial genes is delayed in p21-deficient lungs. p21-wild-type and -deficient mice were exposed to room air; 60 hours of hyperoxia; or recovered in room air for 24, 72, and 240 hours. The expression of Flt1, Flt4, Tie1, Tie2, CD31, and VEGF was assessed by RNase protection and graphed as mean ± SD (n = 5 to 6) after normalizing to L32. Gene expression was significantly reduced in p21-deficient lungs compared to p21-wild type (*, P < 0.01).
Discussion
Previous studies showed that hyperoxia inhibits parenchymal cell proliferation, damages DNA, and kills microvascular endothelial and type I epithelial cells. 1 Increased proliferation of endothelial and type II epithelial cells occurs during recovery in room air as tissue remodeling occurs. 3,5 Using p21-deficient mice, we recently showed that hyperoxia inhibits proliferation through induction of p21. 22 The present study extends these findings by demonstrating that p21 also participates in normal repair of the injured lung. Specifically, increased and sustained proliferation of parenchymal cells was observed in p21-deficient lungs that progressed into hyperplastic regions enriched in myofibroblasts. Enhanced proliferation was associated with DNA fragmentation, inflammation, and delayed recovery of genes expressed by endothelial and type II epithelial cells. Because p21 inhibits DNA replication and facilitates DNA repair, it is likely that one or both processes are required for normal remodeling.
The p21 protein was independently identified as a cdk2-interacting protein (Cip1), 34 a p53-inducible transcript that inhibited proliferation (WAF1), 35 and as a gene expressed by senescent cells (Sdi1). 36 It contains two functional domains that can independently inhibit DNA synthesis. 13 The amino-terminal domain binds and inhibits cyclin E/cdk2 kinase activity, which is required for entry into S phase. The carboxy-terminus inhibits replication-dependent association of PCNA with DNA polymerases δ and ε. We previously demonstrated in adult mice exposed to hyperoxia that p21 mRNA and protein increased in terminal bronchiolar epithelium and throughout the parenchyma. It decreased after 2 days of recovery in room air when cell proliferation occurs. 21 Because proliferation resumes when p21 decreased, 3,5 it seemed likely that DNA replication cannot occur until p21 levels diminish. This hypothesis was not fully supported by the current findings. Although there was a trend toward increased proliferation in p21-deficient mice recovered for 24 hours compared to wild-type mice, it was not statistically significant. Cell proliferation continued to increase throughout time resulting in marked hyperplasia by 10 days. Based on BrdU labeling, the predominant proliferating cell was the myofibroblast. BrdU-labeled cells were also observed to a lesser extent in alveolarized regions. Although the identity of these cells remains to be determined, a rare type II epithelial cell was observed to stain for BrdU. Continued cell proliferation is not likely to be caused by an inability to terminate proliferation, because p21 is not expressed in wild-type mice when proliferation normally ceases. 21 In fact, molecules that terminate proliferation when remodeling is concluded remain unknown.
Studies with cell lines may explain why proliferation did not occur immediately when p21-deficient mice were removed from hyperoxia. Cells that express p21 growth arrest in G1 phase of the cell cycle when exposed to hyperoxia. In contrast, p21-deficient HCT116 colon carcinoma or Mv1Lu epithelial cells, which fail to express p21, growth arrest in S and G2 phases. 17,18 Thus, proliferation may not occur immediately in recovered p21-deficient mice because other growth inhibitory pathways might be active. On the other hand, DNA replication may conclude faster in cells that arrest in S phase compared to those that arrest in G1. Abnormal tissue repair could be a consequence of disorganized proliferation of epithelial, endothelial, or fibroblast cells that progress from different phases of the cell cycle. Supportive evidence comes from studies showing that slower proliferation of oxygen-injured epithelium leads to fibrosis. 7 Because type II epithelial cells inhibit fibroblast proliferation, maintaining the proper balance between these cell populations may be critical for suppressing fibrosis. 37 Fibrosis also occurred when fibroblast proliferation was inhibited with the proline analog L-azetidine carboxylic acid. 38 This suggests that reciprocal interactions between fibroblasts and epithelial cells are critical for normal repair. Thus, abnormal repair in p21-deficient lungs may simply be because of dysregulated proliferation of different cell types required for normal remodeling.
Abnormal tissue repair might also occur when cells die as damaged DNA is replicated. Studies that examined the effects of ionizing radiation on HCT116 cells support this concept. 39 Ionizing radiation induced p21 in the parental p21-wild-type cells resulting in growth arrest in G1. In contrast, p21-deficient HCT116 cells failed to arrest in G1. They instead progressed through S phase and accumulated in G2 where they continued to replicate DNA without cytokinesis and died by apoptosis. We showed that the same p21-deficient HCT116 cells arrested in S phase during hyperoxia and had decreased ability to resume proliferation on recovery compared to wild-type cells. 18 Continuous damage to S phase cells inhibits DNA polymerase resulting in unreplicated DNA in the daughter strand. 40 Although unproven, p21-deficient cells recovering from hyperoxia may be less viable because they fail to successfully complete DNA replication during recovery. p21 may simply protect cells from injury, and therefore enhance tissue repair, by preventing replication of damaged DNA. Epithelial or endothelial cells that attempt to replicate damaged DNA may die thereby compromising reciprocal interactions required for tissue remodeling. Inflammatory cells that are recruited to remove dead cells may promote additional injury and death to resident cells of the lung. Although macrophages were the principle inflammatory cell in p21-deficient lungs, recent studies indicate that neutrophils promote DNA damage and contribute to abnormal development of the oxygen-injured lung. 41 Thus, oxygen-induced cell injury coupled with inflammation-induced injury might compromise repair in p21-deficient lungs.
In addition to its role in DNA replication, p21 may also participate in long-patch base excision repair or nucleotide excision repair of damaged DNA through its interactions with PCNA, a component of both processes. Evidence that p21 participates in nucleotide excision repair comes from studies showing that p21-deficient HCT116 colon carcinoma cells are hypersensitive to nitrogen mustard, cis-platin, and UV-induced damage. 42,43 p21 also enhanced survival and repair of DLD1 colorectal carcinoma cells exposed to UV. 44 p21 may also participate in base excision repair because p21-deficient HCT116 cells are sensitive to hyperoxia. 18 TUNEL staining, indicative of DNA strand breaks, and p53 expression were observed in hyperplastic regions of p21-deficient lungs. TUNEL identifies DNA with a free 3′hydroxyl group on the ribose sugar. This would indicate that DNA is simply damaged, undergoing repair, or fragmenting because of apoptosis or necrosis. Based on the smear observed by DNA laddering, it is most likely that cells are dying, in part, because of catastrophic loss of DNA integrity. It is unclear whether cells are dying by apoptosis or necrosis because smeared DNA could form when laddered DNA is severely damaged by reactive oxygen species. The presence of pyknotic nuclei however suggests that some cells are dying by apoptosis. Because DNA damage induces p53 expression that can stimulate apoptosis, future studies will investigate whether p53-dependent apoptosis is responsible for cell death in p21-deficient mice.
TUNEL-positive cells may be endothelial or type I epithelial cells, which are rapidly damaged during hyperoxia. They may also be type II epithelial cells that fail to replicate damaged DNA properly. Injury to type II cells could alter endothelial cell proliferation because type II cells express VEGF. The delayed expression of SP-C and VEGF, which are expressed by type II epithelial cells, is consistent with this concept. Delayed expression of genes expressed by endothelial cells suggests that these cells may also be affected by the absence of p21. Two receptor tyrosine kinase families control endothelial cell proliferation, differentiation, and formation of capillaries. The VEGF receptors Flt1 and Flt2 are members of one family. Based on the phenotype of knockout mice, Flt1 is thought to signal capillary formation and permeability. 45 Cytokines that promote the fibroblastic phenotype could leak into the alveolar space of p21-deficient lungs that lack adequate Flt1 expression. Tie1 and Tie2 are a second class of receptors that bind angiopoietin 1 or 2. Studies with knockout mice reveal that Tie1 regulates endothelial cell proliferation and blood vessel integrity. 46 Tie2 is important for sprouting and branching from pre-existing vessels. Failure to restore Tie2 levels in p21-deficient lungs could result in insufficient numbers of vascular branches required for normal alveolarization and appropriate gas exchange. The reduced expression of CD31 (PECAM) at 24 and 72 hours of recovery implies that p21-deficient lungs have fewer endothelial cells. Although additional studies are required to establish whether reduced expression of genes expressed by endothelial or epithelial cells signifies absence of these cells or failure to express differentiated genes, the fact that gene expression was altered indicates that cell phenotype was affected.
Fibrosis occurred in lung explants isolated from oxygen-exposed rodents. 7 The extent of fibrosis was dependent on the degree of epithelial cell injury during exposure. Fibroblast proliferation and collagen deposition were not observed when only endothelial cell damage occurred. Because explants did not contain inflammatory cells, Adamson and colleagues 7 concluded that interactions between type II epithelial cells and fibroblasts are critical for normal repair. Even though fibroblast proliferation was observed in the current study, hydroxyproline levels were not different between p21-wild-type and -deficient mice. Movat and Trichrome (not shown) staining indicated that mature collagen was not deposited. This suggests that additional factors required for the fibrotic phenotype fail to become established. Because normal lung architecture is restored in p21-deficient mice recovered for 2 months (data not shown), it will be important to understand how fibroblast hyperplasia is resolved.
In summary, the present study demonstrates that p21 promotes normal repair of the oxygen-injured lung. The absence of p21 results in premature and extended proliferation of parenchymal cells resulting in hyperplastic regions enriched in proliferating fibroblasts. Although other cell types may also be proliferating, our examination of epithelial and endothelial cell genes suggests that future studies should focus on proliferation, differentiation, and death of these cells earlier in the recovery period. In addition, these mice will be useful for understanding how inflammatory cells participate in lung remodeling. Because p21 is induced in lungs exposed to radiation or bleomycin as well as in patients with idiopathic fibrosis, 47,48 our findings may also have important implications for understanding remodeling after other forms of injuries.
Acknowledgments
We thank Susan Reynolds for assistance in the S1 studies, Charles Churukian for advice on Movat staining, Patricia Sime for discussions about myofibroblasts, and the Environmental Health Sciences Center at Rochester for supplying the core facilities for animal exposures.
Footnotes
Address reprint requests to Michael A. O’Reilly, Ph.D., Department of Pediatrics (Neonatology), Box 850, School of Medicine and Dentistry, University of Rochester, 601 Elmwood Ave., Rochester, NY 14642. E-mail: michael_oreilly@urmc.rochester.edu.
Supported in part by National Institutes of Health grants HL58774 (to M. A. O.) and HL63400 (to W. M. M.) and grant ES01247 (to the Environmental Health Sciences Center at Rochester).
References
- 1.Crapo JD, Barry BE, Foscue HA, Shelburne J: Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis 1980, 122:123-143 [DOI] [PubMed] [Google Scholar]
- 2.Evans MJ, Hackney JD, Bils RF: Effects of a high concentration of oxygen on cell renewal in the pulmonary alveoli. Aerospace Med 1969, 40:1365-1368 [Google Scholar]
- 3.Tryka AF, Witschi H, Gosslee DG, McArthur AH, Clapp NK: Patterns of cell proliferation during recovery from oxygen injury. Species differences. Am Rev Respir Dis 1986, 133:1055-1059 [DOI] [PubMed] [Google Scholar]
- 4.Evans MJ, Hackney JD: Cell proliferation in lungs of mice exposed to elevated concentrations of oxygen. Aerospace Med 1972, 43:620-622 [PubMed] [Google Scholar]
- 5.Adamson IY, Bowden DH: The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab Invest 1974, 30:35-42 [PubMed] [Google Scholar]
- 6.Maniscalco WM, Watkins RH, Finkelstein JN, Campbell MH: Vascular endothelial growth factor mRNA increases in alveolar epithelial cells during recovery from oxygen injury. Am J Respir Cell Mol Biol 1995, 13:377-386 [DOI] [PubMed] [Google Scholar]
- 7.Adamson IY, Young L, Bowden DH: Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 1988, 130:377-383 [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brodovich HM, Mellins RB: Bronchopulmonary dysplasia. Unresolved neonatal acute lung injury. Am Rev Respir Dis 1985, 132:694-709 [DOI] [PubMed] [Google Scholar]
- 9.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM: Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 2001, 164:1971-1980 [DOI] [PubMed] [Google Scholar]
- 10.Elledge SJ: Cell cycle checkpoints: preventing an identity crisis. Science 1996, 274:1664-1672 [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Ko LJ, Jayaraman L, Prives C: p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 1996, 10:2438-2451 [DOI] [PubMed] [Google Scholar]
- 12.Chen IT, Akamatsu M, Smith ML, Lung FD, Duba D, Roller PP, Fornace AJ, Jr, O’Connor PM: Characterization of p21 Cip1/Waf1 peptide domains required for cyclin E/Cdk2 and PCNA interaction. Oncogene 1996, 12:595-607 [PubMed] [Google Scholar]
- 13.Luo Y, Hurwitz J, Massague J: Cell-cycle inhibition by independent CDK and PCNA binding domains in p21 Cip1. Nature 1995, 375:159-161 [DOI] [PubMed] [Google Scholar]
- 14.Li R, Waga S, Hannon GJ, Beach D, Stillman B: Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature 1994, 371:534-537 [DOI] [PubMed] [Google Scholar]
- 15.Cacciuttolo MA, Trinh L, Lumpkin JA, Rao G: Hyperoxia induces DNA damage in mammalian cells. Free Radic Biol Med 1993, 14:267-276 [DOI] [PubMed] [Google Scholar]
- 16.Corroyer S, Maitre B, Cazals V, Clement A: Altered regulation of G1 cyclins in oxidant-induced growth arrest of lung alveolar epithelial cells. Accumulation of inactive cyclin E-DCK2 complexes. J Biol Chem 1996, 271:25117-25125 [DOI] [PubMed] [Google Scholar]
- 17.Rancourt RC, Keng PC, Helt CE, O’Reilly MA: The role of p21 Cip1/WAF1 in growth of epithelial cells exposed to hyperoxia. Am J Physiol 2001, 280:L617-L626 [DOI] [PubMed] [Google Scholar]
- 18.Helt CE, Rancourt RC, Staversky RJ, O’Reilly MA: p53-dependent induction of p21 Cip1/Waf1/Sdi1 protects against oxygen-induced toxicity. Toxicol Sci 2001, 63:214-222 [DOI] [PubMed] [Google Scholar]
- 19.Shenberger JS, Dixon PS: Oxygen induces S-phase growth arrest and increases p53 and p21(WAF1/CIP1) expression in human bronchial smooth-muscle cells. Am J Respir Cell Mol Biol 1999, 21:395-402 [DOI] [PubMed] [Google Scholar]
- 20.McGrath SA: Induction of p21 WAF/CIP1 during hyperoxia. Am J Respir Cell Mol Biol 1998, 18:179-187 [DOI] [PubMed] [Google Scholar]
- 21.O’Reilly MA, Staversky RJ, Watkins RH, Maniscalco WM: Accumulation of p21(Cip1/WAF1) during hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 1998, 19:777-785 [DOI] [PubMed] [Google Scholar]
- 22.O’Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, Keng PC: The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol 2001, 24:703-710 [DOI] [PubMed] [Google Scholar]
- 23.O’Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN: Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol 1998, 18:43-50 [DOI] [PubMed] [Google Scholar]
- 24.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC: Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994, 264:713-716 [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly MA, Staversky RJ, Huyck HL, Watkins RH, LoMonaco MB, D’Angio CT, Baggs RB, Maniscalco WM, Pryhuber GS: Bcl-2 family gene expression during severe hyperoxia induced lung injury. Lab Invest 2000, 80:1845-1854 [DOI] [PubMed] [Google Scholar]
- 26.Elbadawi A: Hexachrome modification of Movat’s stain. Stain Technology 1976, 51:249-253 [DOI] [PubMed] [Google Scholar]
- 27.Russell HK, Jr: A modification of Movat’s pentachrome stain. Arch Pathol 1972, 94:187-191 [PubMed] [Google Scholar]
- 28.Woessner JF, Jr: The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys 1961, 93:440-447 [DOI] [PubMed] [Google Scholar]
- 29.Konishi H, Steinbach G, Terry NH, Lee JJ, Dubin JA, Glober GA, Fujita K, Spaulding D, Cass L, Hittelman WN: Histone H3 messenger RNA in situ hybridization correlates with in vivo bromodeoxyuridine labeling of S-phase cells in rat colonic epithelium. Cancer Res 1996, 56:434-437 [PubMed] [Google Scholar]
- 30.Alterman RB, Ganguly S, Schulze DH, Marzluff WF, Schildkraut CL, Skoultchi AI: Cell cycle regulation of mouse H3 histone mRNA metabolism. Mol Cell Biol 1984, 4:123-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF: Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol 1998, 19:573-581 [DOI] [PubMed] [Google Scholar]
- 32.O’Reilly MA, Staversky RJ, Watkins RH, Maniscalco WM, Keng PC: p53-independent induction of GADD45 and GADD153 in mouse lungs exposed to hyperoxia. Am J Physiol 2000, 278:L552-L559 [DOI] [PubMed] [Google Scholar]
- 33.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA: Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest 1998, 101:1970-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75:805-816 [DOI] [PubMed] [Google Scholar]
- 35.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B: WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75:817-825 [DOI] [PubMed] [Google Scholar]
- 36.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR: Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 1994, 211:90-98 [DOI] [PubMed] [Google Scholar]
- 37.Pan T, Mason RJ, Westcott JY, Shannon JM: Rat alveolar type II cells inhibit lung fibroblast proliferation in vitro. Am J Respir Cell Mol Biol 2001, 25:353-361 [DOI] [PubMed] [Google Scholar]
- 38.Bowden DH, Young L, Adamson IY: Fibroblast inhibition does not promote normal lung repair after hyperoxia. Exp Lung Res 1994, 20:251-262 [DOI] [PubMed] [Google Scholar]
- 39.Waldman T, Lengauer C, Kinzler KW, Vogelstein B: Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996, 381:713-716 [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann JS, Johnson NP, Villani G: Conversion of monofunctional DNA adducts of cis-diamminedichloroplatinum (II) to bifunctional lesions. Effect on the in vitro replication of single-stranded DNA by Escherichia coli DNA polymerase I and eukaryotic DNA polymerases alpha. J Biol Chem 1989, 264:15130-15135 [PubMed] [Google Scholar]
- 41.Auten RL, Jr, Mason SN, Tanaka DT, Welty-Wolf K, Whorton MH: Anti-neutrophil chemokine preserves alveolar development in hyperoxia-exposed newborn rats. Am J Physiol 2001, 281:L336-L344 [DOI] [PubMed] [Google Scholar]
- 42.McDonald ER, III, Wu GS, Waldman T, El-Deiry WS: Repair defect in p21 WAF1/CIP1 −/− human cancer cells. Cancer Res 1996, 56:2250-2255 [PubMed] [Google Scholar]
- 43.Fan S, Chang JK, Smith ML, Duba D, Fornace AJ, Jr, O’Connor PM: Cells lacking CIP1/WAF1 genes exhibit preferential sensitivity to cisplatin and nitrogen mustard. Oncogene 1997, 14:2127-2136 [DOI] [PubMed] [Google Scholar]
- 44.Sheikh MS, Chen YQ, Smith ML, Fornace AJ, Jr: Role of p21 Waf1/Cip1/Sdi1 in cell death and DNA repair as studied using a tetracycline-inducible system in p53-deficient cells. Oncogene 1997, 14:1875-1882 [DOI] [PubMed] [Google Scholar]
- 45.Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 46.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y: Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376:70-74 [DOI] [PubMed] [Google Scholar]
- 47.Mishra A, Doyle NA, Martin WJC: Bleomycin-mediated pulmonary toxicity: evidence for a p53-mediated response. Am J Respir Cell Mol Biol 2000, 22:543-549 [DOI] [PubMed] [Google Scholar]
- 48.Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N: P21 Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1996, 154:477-483 [DOI] [PubMed] [Google Scholar]