

Abstract
Nuclear factor (NF)-κB regulates several genes implicated in the inflammatory response and represents an interesting therapeutic target. We examined the effects of gliotoxin (a fungal metabolite) and parthenolide (a plant extract), which possess anti-inflammatory activities in vitro, on the progression of experimental glomerulonephritis. In the anti-Thy 1.1 rat model, gliotoxin (75 μg/rat/day, 10 days, n = 18 rats) markedly reduced proteinuria, glomerular lesions, and monocyte infiltration. In anti-mesangial cell nephritis in mice, parthenolide (70 μg/mouse/day, 7 days, n = 17 mice) significantly decreased proteinuria, hematuria, and glomerular proliferation. NF-κB activity, localized in glomerular and tubular cells, was attenuated by either gliotoxin or parthenolide, in association with diminished renal expression of monocyte chemoattractant protein-1 and inducible nitric oxide synthase. In cultured mesangial cells and monocytes, gliotoxin and parthenolide inhibited NF-κB activation and expression of inflammatory genes induced by lipopolysaccharide and cytokines, by blocking the phosphorylation/degradation of the IκBα subunit. In summary, gliotoxin and parthenolide prevent proteinuria and renal lesions by inhibiting NF-κB activation and expression of regulated genes. This may represent a novel approach for the treatment of immune and inflammatory renal diseases.
Chronic glomerulonephritis is because of the release of many inflammatory mediators that can be produced by resident glomerular cells or infiltrating leukocytes. 1 The mechanisms for gene expression of these inflammatory proteins have been extensively studied, with special attention to transcription factors, such as nuclear factor (NF)-κB. NF-κB can be activated in several cell types by numerous physiological and pathological stimuli, such as cytokines, mitogens, virus, mechanical and oxidative stress, and chemical agents. 2,3 Studies in experimental and human nephritis have provided evidence supporting the involvement of NF-κB in the pathogenesis of glomerulonephritis. 4-6 In addition, expression of NF-κB-dependent genes, such as chemokines, adhesion molecules, inducible nitric oxide synthase (iNOS), and cyclooxygenase has been demonstrated in cultured glomerular cells, including mesangial cells (MCs). 7-9
The family of NF-κB proteins shares a conserved central region, the Rel domain, involved in the dimerization and interaction with the inhibitory subunit IκB, nuclear translocation, and DNA binding. The five members of this family (p50/p105, p65/RelA, c-Rel, RelB, and p52/p100) associate in different dimeric forms. The classic heterodimer is composed of p50 and p65. 10 In resting cells, NF-κB dimers remain in the cytoplasm as an inactive form bound to the inhibitory subunit IκB. The IκB family comprises IκBα (bound to the p50-p65 dimer and related with transient activation of NF-κB), IκBβ (implicated in a more stable activation), and IκBε. 10,11 Upon stimulation, IκB is phosphorylated by the IκB kinase (IKK) complex, ubiquitinated, and ultimately degraded by the proteasome system. Then, free NF-κB dimers expose the nuclear localization sequence and translocate into the nucleus, where they activate the transcription of target genes. 4,10,11
The pharmacological modulation of the inflammatory responses is the main objective in the design of anti-inflammatory drugs. Various drugs, such as steroids and aspirin, are effective in inhibiting NF-κB-inducible gene expression. 3,5,12,13 In general, the strategies regulating NF-κB can be classified in relation with the step in the NF-κB pathway: antioxidants, 14 protease inhibitors, 15 nuclear translocation inhibitors, 16 and DNA binding blockers. 17 Antioxidants are the most frequently used and inhibit chemokine expression in cultured renal cells, 8,18 and prevent proteinuria and cytokine expression in experimental nephritis. 19
Recently, several natural products, such as gliotoxin and parthenolide, have been emerged as possible anti-inflammatory agents. 20,21 Gliotoxin, a metabolite from the fungus Aspergillus fumigatus, inhibits the activation and phagocytosis in leukocytes and the catalytic activity of 20S proteasome, and can also induce apoptosis. 20,22-25 Parthenolide, a sesquiterpene lactone derived from Mexican-Indian medicinal plants, inhibits the gene expression of interleukin-8, iNOS, and cyclooxygenase in several cell lines. 26-28 Previous studies reported that low doses of gliotoxin and parthenolide prevent the activation of NF-κB by a variety of stimuli in cultured cells, 29-31 but the mechanisms of their potential anti-inflammatory effects, and the possible use in inflammatory and immune diseases are not well examined.
Because many forms of primary glomerulonephritis do not consistently respond to current therapy, in this study we attempted novel therapeutic approaches for these diseases, focusing on NF-κB as a possible target. Our purpose is to modify in vivo the expression of proinflammatory genes by using novel NF-κB inhibitors. First, we evaluated the effects of gliotoxin and parthenolide on NF-κB activation and expression of regulated genes (MCP-1 and iNOS) in cultured MCs and monocytes stimulated with inflammatory stimuli. Second, we analyzed whether these inhibitors modulate (or prevent) the initiation and progression of glomerular injury in two experimental models of mesangial proliferative glomerulonephritis. The administration of these agents could represent a novel therapeutic approach to progressive glomerulonephritis.
Materials and Methods
Reagents and Chemicals
Gliotoxin, parthenolide, and lipopolysaccharide (LPS) were purchased from Sigma Chemical Co. (St. Louis, MO). The cytokines [human tumor necrosis factor-α (TNF-α), human interferon-γ (IFN-γ) and human interleukin-1β (IL-1β)] were purchased from Immunogenex Corp. (Los Angeles, CA). The specific antibodies (Abs) used were as follows: pIκBα, IκBα, p65, and c-Rel (Santa Cruz Biotechnology, Santa Cruz, CA), ED1 (Serotec, Oxford, UK), BrdU (Calbiochem, La Jolla, CA), iNOS and p50 (Chemicon International, Temecula, CA), fluorescein isothiocyanate-labeled IgG (The Binding Site, Birmingham, UK), peroxidase-labeled IgG (Amersham, Buckinghamshire, UK), and alkaline phosphatase-labeled IgG (Roche, Barcelona, Spain).
Cell Cultures
MCs from human and rat kidneys 32 were cultured in RPMI 1640 with 25 mmol/L HEPES, pH 7.4, 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mmol/L glutamine (Life Technologies, Paisley, Scotland) and characterized by phase contrast microscopy, positive staining for desmin and vimentin, and negative staining for factor VIII and cytokeratin. The human monocytic cell line THP-1 [American Type Culture Collection (ATCC), Rockville, MD] was cultured in RPMI with 10% FCS. Cells were made quiescent by 24 hours of incubation in medium with 0.5% FCS, then preincubated for 90 minutes with the inhibitors, and washed before stimulation with LPS and cytokines. Cell viability was assessed by trypan blue exclusion.
Electrophoretic Mobility Shift Assay (EMSA)
Cell nuclear proteins (10 μg) were incubated in buffer containing 50 μg/ml of poly(dI-dC) (Amersham), and 0.035 pmol of [32P]NF-κB or [32P]AP-1 consensus oligonucleotides, and the reactions were analyzed on a 4% nondenaturing polyacrylamide gel and autoradiographed. Specificity of the binding reaction was confirmed using a 100-fold excess of unlabeled probe. NF-κB subunits were identified by incubation with Abs against p50, p65, and c-Rel. 8
Western Blot Analysis
Cytosolic proteins (20 μg) were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Immobilon P; Millipore, Bedford, MA). Membranes were blocked with 5% nonfat milk, and then incubated with 10 μg/ml of goat anti-IκBα or 1 μg/ml of monoclonal anti-pIκBα, followed by peroxidase-conjugated secondary Ab and chemiluminescent detection system.
Cell Proliferation Assay
Human MCs were pretreated for 90 minutes with serial dilutions of inhibitors, then washed and incubated for an additional 24 hours in medium containing 10% FCS. Cell proliferation was determined in 96-well plates by colorimetric assay of methylene blue incorporation. 33 Alternatively, cell proliferation was studied after 24 hours of incubation in medium with 10% FCS and inhibitors. Data were expressed as percentage of proliferating cells in relation to control conditions.
Analysis of mRNA Expression
Total RNA from cells and tissues was obtained by the acid guanidine-thiocyanate-phenol-chloroform method. In cultured cells, MCP-1 mRNA expression was analyzed by Northern blot using cDNA probes for human MCP-1 and 28S ribosomal RNA (JE/pGEM-hJE34 and HHCD07; ATCC). Membranes containing RNA were hybridized, autoradiographed, and densitometered. Relative amounts of MCP-1 were established in relation to 28S. In renal tissues the mRNA expression was analyzed by reverse transcriptase-polymerase chain reaction (RT-PCR) using the following primers: rat MCP-1 sense, 5′-TTCTGGGCCTGTTGTTCACA-3′; anti-sense, 5′-GGTCACTTCTACAGAAGTCC-3′; mouse MCP-1 sense, 5′-AGCACCAGCCAACTCTCACT-3′; anti-sense, 5′-TCTGGACCCATTCCTTCTTG-3′; rat iNOS sense, 5′-CTTCAACCCCAAGGTTGTCTGCAT-3′; anti-sense, 5′-ATGTCATGAGCAAAGGGGCAGAAC-3′. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control.
Determination of Nitrites
NO released from cells was determined by the accumulation of nitrites and measured by fluorescent reaction with 1 mmol/L of 2,3-diaminonaphthalene. 34 The absorbance at 548 nm was measured and compared with a standard of NaNO2, and data were expressed as nmol NO2−/mg protein.
Transient Transfection and Luciferase Assay
Growth-arrested cells (1 × 105) were co-transfected with piNOS-Luc (a gift from Dr. Santiago Lamas; CIB, Madrid, Spain) 9 and a normalizing reporter vector (pRL-TK with the Renilla luciferase gene; Promega, Madison WI) by using the FuGENE reagent (Roche) and then stimulated for 24 hours. The luciferase activity in cleared lysate was assayed using a luminometer. Luciferase activity was normalized by total protein concentration and variations in transfection efficiency.
Isolation of Anti-MC Abs
Monoclonal Ab (IgG1) against rat mesangial antigen Thy 1.1 was isolated from ascitic BALB/c mice injected with hybridome cells OX-7 (European Collection of Cell Cultures, Salisbury, UK) by affinity chromatography on protein G-Sepharose. Anti-murine MC serum was obtained by sheep immunization with protein extract form SV40-transformed mouse cells MES13 (Yo Y, Mobaraki H, Lu H, Braun M, Owens J, King C, Kopp JB, manuscript submitted). Control serum was obtained from sheep before immunization. For all experiments, sera were decomplemented by heating at 56° for 30 minutes.
Experimental Models of Mesangial Proliferative Glomerulonephritis
Anti-Thy 1.1 nephritis was induced in male Sprague-Dawley rats (200 g) at day 0 by injection of anti-Thy1.1 IgG (5 mg/kg in 350 μl of phosphate-buffered saline) into a tail vein. Mesangial glomerulonephritis was induced in female C57/BL6 mice (18 to 20 g) at day 0 by injection of sheep anti-murine MC antiserum (200 μl/20 g body weight). Groups of study: I, rats (n = 18) and mice (n = 17) treated daily with gliotoxin (75 μg/200 g body weight, i.p.) and parthenolide (70 μg/20 g body weight, i.p.), respectively; II, rats (n = 20) and mice (n = 10) with spontaneous development of nephritis with injection of vehicle (saline); III and IV, healthy control rats (n = 17) and mice (n = 8) receiving the respective amount of inhibitors and saline. Treatment with gliotoxin or parthenolide was started the day before and followed for 10 or 7 days, respectively. Urine samples were taken at regular intervals starting on day 0. Proteinuria was measured by the sulfosalicylic method (rats) or the Knight’s method (mice). 35 Urinary hemoglobin was measured by Aution sticks (Arkray Factory, Shiga, Japan). All studies were performed in accordance to the European Union normative. Animals were sacrificed at days 2, 4, 7, and 10 (rat model) and days 5 and 7 (mouse model).
Kidney Histology and Immunostaining
Light microscopic analysis of renal cortex was performed on paraffin sections (5-μm thick) stained with hematoxylin and eosin, and Masson’s trichrome. The examination of infiltrating cells (ED1+, rat macrophage) and iNOS production was performed by indirect immunoperoxidase. For determining proliferating cells, mice were intraperitoneally injected with 2 mg of BrdU for 16, 8, and 4 hours before sacrifice, and then immunoperoxidase staining with anti-BrdU was performed on paraffin-embedded samples.
In Situ Detection of Activated Transcription Factors
Paraffin-embedded tissue sections fixed in 0.5% paraformaldehyde were incubated with 50 pmol of digoxigenin-labeled NF-κB and AP-1 probes in buffer containing 0.25% bovine serum albumin and 1 μg/ml of poly(dI-dC), followed by alkaline phosphatase-conjugated anti-digoxigenin IgG and colorimetric detection. A 200-fold excess of unlabeled probe was used to test the specificity of the technique. 36
Evaluation of Tissue Staining and Statistical Analysis
Semiquantification of morphological changes in the animal models was made in regard to glomerular lesions (number of glomeruli with hypercellularity, mesangiolysis, or immune deposits/30 glomeruli), tubular lesions (number of tubules with atrophy or degeneration/20 fields at ×40 magnification), and interstitial infiltration (number of focus/20 fields at ×40 magnification). Samples from each animal were examined in a blinded manner, and semiquantitatively graded on a scale from 0 to 3. Quantification of iNOS staining and infiltrating/proliferating cells was made by determining the total number of positive-labeled cells in 20 randomly chosen areas. The results are given as mean ± SD or representative experiments, when indicated. Values were analyzed by ANOVA and Tukey-Kramer tests using Instat (Graphpad Software, San Diego, CA). A P value <0.05 was considered significant.
Results
Gliotoxin and Parthenolide Inhibit the Activation of NF-κB in MCs and Monocytes
Human MCs were preincubated for 90 minutes with parthenolide or gliotoxin (1 to 3 μg/ml), then washed and incubated for 1 hour with the combination of LPS (10 μg/ml) and cytokines (TNF-α, 100 ng/ml; IFN-γ, 100 U/ml; and IL-1β, 100 U/ml). NF-κB activity in nuclear extracts was analyzed by EMSA. As indicated in Figure 1A ▶ , the strong DNA-binding activity induced by LPS/cytokines (8-fold increase versus control) was dose dependently attenuated by gliotoxin and parthenolide (75 ± 6% and 69 ± 9% inhibition at 3 μg/ml, respectively). Gliotoxin and parthenolide did not inhibit the activation of other transcription factors, such as AP-1 (Figure 1A) ▶ . Similar results were observed in nuclear extracts from rat MCs pretreated with gliotoxin or parthenolide (1 μg/ml) and then stimulated with 1 μg/ml of LPS (69 ± 8% and 72 ± 8% inhibition, respectively, n = 3). In the monocytic cell line THP-1, NF-κB activation induced by LPS/cytokines was also prevented by treatment with 1 μg/ml of gliotoxin and 2.5 μg/ml of parthenolide (79 ± 4% and 73 ± 12% inhibition, n = 3). In some cases, extracts from stimulated cells were incubated with gliotoxin or parthenolide before EMSA. Both inhibitors, even at higher doses, did not influence the interaction of NF-κB complex with the consensus oligonucleotide in vitro (not shown). These data exclude the possibility that both agents directly affect the DNA-binding ability of activated NF-κB.
Figure 1.

Gliotoxin and parthenolide specifically attenuate the NF-κB activation in human MCs. A: Cells were preincubated for 90 minutes with two doses (1 and 3 μg/ml) of gliotoxin and parthenolide, then washed and stimulated for 60 minutes with the combination of LPS and inflammatory cytokines (TNF-α, IFN-γ, and IL-1β). Binding activities of NF-κB and AP-1 in nuclear extracts were analyzed by gel shift assay as described. Specific bands were identified by competition with a 100-fold excess of unlabeled NF-κB and AP-1 oligonucleotides. B: The NF-κB activation was also measured as phosphorylation and subsequent degradation of IκBα. Cells were preincubated with 1 μg/ml of inhibitors before LPS/cytokine stimulation, and then cytosolic proteins were immunoblotted with specific Abs against complete IκBα protein or the phosphorylated form (pIκBα). Gels are representative of two to three experiments.
NF-κB activation was also evaluated by measuring phosphorylation and degradation of the inhibitory subunit IκBα. Western blot analysis of cytosolic proteins from LPS/cytokine-stimulated human MCs revealed an increase in phosphorylated IκBα and a subsequent degradation of IκBα (Figure 1B) ▶ . Preincubation with parthenolide prevented the phosphorylation and degradation of IκBα, whereas gliotoxin did not affect phosphorylation but protected IκBα from proteolysis (Figure 1B) ▶ . The effects of both agents on monocytic NF-κB activation were very similar (not shown). These results suggested that IκBα is the common step in the signaling cascade leading to NF-κB activation that is targeted by gliotoxin and parthenolide.
Effects of Gliotoxin and Parthenolide on Cell Viability and Proliferation
By trypan blue exclusion we observed that more than 95% of cells (MC and THP-1) were viable after 90 minutes of treatment with 1 μg/ml of gliotoxin. Viability remained high (>75%) at 3 μg/ml, but a toxic effect was noted using concentrations >5 μg/ml (viability around 30%). No toxic effect was observed with parthenolide even at doses up to 6 μg/ml.
In proliferation assays (methylene blue incorporation) we observed that preincubation for 90 minutes with serial dilutions of gliotoxin and parthenolide (from 0.75 to 12 μg/ml) did not diminish the number of proliferating MCs (maximal percentages of inhibition: 23 ± 4% and 16 ± 3%, respectively). MC proliferation was only inhibited by using gliotoxin at higher doses and longer incubation periods (24 μg/ml at 24 hours, 66 ± 8% inhibition, n = 4).
Gliotoxin and Parthenolide Inhibit the Expression of NF-κB-Regulated Genes
In these experiments we checked the effects of these agents on the expression of genes transcriptionally regulated by NF-κB, such as MCP-1 and iNOS. 7-9 In human MCs, preincubation with gliotoxin or parthenolide (1 μg/ml) prevented the MCP-1 mRNA expression induced by LPS/cytokines (72% and 58% inhibition; Figure 2A ▶ ). In a reported gene assay using human MCs transfected with piNOS-Luc plasmid, both agents inhibited the activation of the iNOS promoter induced by LPS/cytokines (75% inhibition, Figure 2B ▶ ). According to this data, the mesangial production of nitrites induced by LPS/cytokines after 24 hours was also decreased by treatment with gliotoxin and parthenolide (79% and 74% inhibition, Figure 2C ▶ ). In THP-1 cells, gliotoxin down-regulated the MCP-1 mRNA expression (69 ± 7% inhibition n = 3), and parthenolide decreased the iNOS transcription (69 ± 3% inhibition at 24 hours, n = 3), and the NO generation induced by LPS/cytokines (183 ± 11 versus 52 ± 7 nmol/mg protein at 24 hours, n = 4, P < 0.05).
Figure 2.

Gliotoxin and parthenolide inhibit the mRNA expression of MCP-1 and iNOS in human MCs. A: Human MCs were pretreated with either gliotoxin or parthenolide (1 μg/ml), then washed and incubated with LPS/cytokines. The MCP-1 mRNA expression at 6 hours was analyzed by Northern blot. Relative amounts of mRNA were established in relation to 28S expression as fold increases versus control cells. B: The iNOS promoter activity was analyzed in human MCs transfected with piNOS-Luc and stimulated for 24 hours in medium alone (▪) or containing LPS/cytokines (□). Data of luciferase activity were normalized to Renilla activity and protein amounts, and are expressed as fold increases, compared with control. C: The NO production at 24 hours in medium alone (▪) or containing LPS/cytokines (□) was analyzed by measuring the absorbance of a fluorescent reaction. Data of nitrite production (nmol/mg protein) are mean ± SD of two to five experiments made in duplicate (*, P < 0.05 versus control without inhibitor).
Gliotoxin Treatment in Experimental Model of Mesangial Proliferative Glomerulonephritis
Because both gliotoxin and parthenolide can inhibit NF-κB activation and regulated-genes in cultured cells, we designed studies to examine their in vivo effect in experimental models of mesangial proliferative glomerulonephritis.
The evolution of urinary protein excretion in rats with anti-Thy 1.1 nephritis is shown in Figure 3A ▶ . Proteinuria slightly increased until day 4 (39 ± 6 mg/24 hours), and thereafter decreased to control levels. Daily treatment with gliotoxin (75 μg/day) prevented the development of proteinuria, with nonsignificant differences between gliotoxin-treatment and healthy control groups. Previous studies have shown that the injection of anti-Thy 1.1 Ab in rats is associated with an acute complement-dependent mesangiolysis, followed by a MC proliferation phase that begins at day 2, peaks between days 3 and 5, and is resolved by day 14. 37,38 This pattern was observed in vehicle-treated rats (Figure 3B) ▶ , with a marked increase in the glomerular cell number, interstitial cell infiltration, and tubular atrophy compared with normal rats. The glomerular damage quantification after Masson’s staining is summarized in Table 1 ▶ . Gliotoxin treatment drastically reduced the excessive mesangial proliferative response (65% inhibition at day 7), the collapse of capillaries and the tubular atrophy (Figure 3C) ▶ . Gliotoxin also significantly reduced the early glomerular influx of monocytes/macrophages (Table 1) ▶ . Normal animals receiving daily injection of gliotoxin (ranging from 75 to 200 μg/rat) for 10 days did not show any toxic symptoms or renal morphological changes when compared with normal rats injected with saline, used as control (Table 1) ▶ .
Figure 3.

Gliotoxin improves proteinuria and renal damage in mesangial proliferative glomerulonephritis. A: Proteinuria in rats with anti-Thy 1.1 nephritis injected with saline (•) or gliotoxin (▴) and in healthy control rats receiving saline (○) was analyzed daily and expressed in mg/24 hours (*, P < 0.05 versus untreated nephritic rats). B and C: Representative micrographs show morphological changes at day 7 in untreated nephritic (B) and gliotoxin-treated rat (C). Original magnifications, ×200.
Table 1.
Gliotoxin Treatment Reduces Pathological Changes in Anti-Thy 1.1 Glomerulonephritis
| Day | Glomerular lesions | ED1+ cells | ||||||
|---|---|---|---|---|---|---|---|---|
| Control | Nephritis | Control | Nephritis | |||||
| Saline | Gliotoxin | Saline | Gliotoxin | Saline | Gliotoxin | Saline | Gliotoxin | |
| 2 | 0.5 ± 0.1 | ND | 1.3 ± 0.3† | 0.5 ± 0.3*‡ | 0.9 ± 0.1 | ND | 6.0 ± 1.4† | 3.0 ± 0.7*‡ |
| 4 | 0.8 ± 0.1 | 0.3 ± 0.2* | 2.2 ± 0.5† | 1.0 ± 0.2*‡ | 0.9 ± 0.1 | 1.1 ± 0.6* | 7.9 ± 1.8† | 2.5 ± 0.7*‡ |
| 7 | 0.5 ± 0.1 | ND | 2.9 ± 0.2† | 1.0 ± 0.1*‡ | 0.9 ± 0.1 | ND | 9.5 ± 2.0† | 2.0 ± 0.1*‡ |
| 10 | 0.3 ± 0.2 | 0.4 ± 0.2* | 2.0 ± 0.5† | 1.0 ± 0.1†‡ | 1.1 ± 0.3 | 0.9 ± 0.1* | 5.6 ± 1.7† | 0.8 ± 0.6*‡ |
Glomerular damage was scored from 0 to 3. Infiltrating monocytes/macrophages were expressed as number of ED1+ cells/glomerular cross section (gcs). Values are mean ± SD.
*†, Not significant and P < 0.05 compared to control rats receiving saline, respectively.
‡P < 0.05 versus untreated nephritis rats (injected with saline). ND, not determined.
The effect of gliotoxin on NF-κB activation in vivo was analyzed by EMSA. Extracts from nephritic rats showed a marked increase in the intensity of the NF-κB bands at day 2, that remained elevated until 10 days after injection of the Abs. Gliotoxin administration attenuated this activation (Figure 4A) ▶ . The composition of activated complexes (p50/p65 heterodimer, without c-Rel subunit) was not different between untreated nephritic and gliotoxin-treated rats, as determined by supershift assay (not shown).
Figure 4.

Gliotoxin attenuates NF-κB activation in experimental glomerulonephritis. A: Nuclear proteins from untreated nephritic, gliotoxin-treated, and control rats were analyzed by EMSA. Competition (c) with an excess of unlabeled probe was used as negative control. Specific NF-κB bands were densitometered, and data are expressed as increase versus control rats (▪, untreated nephritis; □, gliotoxin-treated; *, P < 0.05 versus nephritis). B–E: In situ localization of activated NF-κB in renal tissues. Control rats receiving gliotoxin (B) presented a weak staining. Nephritic animals presented a strong NF-κB activation in glomerular and tubular cells at day 4 (C), that was attenuated by gliotoxin treatment (D). Specificity of DNA-protein interaction in nephritic rats (same sample as C) was determined by competition with a 200-fold excess of unlabeled oligonucleotide (E). Original magnifications, ×200.
The localization of activated NF-κB in the kidneys was analyzed in situ by Southwestern histochemistry. Very weak nuclear staining was observed in kidneys from healthy rats receiving daily injection of gliotoxin (Figure 4B) ▶ or its vehicle (not shown), as control. In nephritic rats, NF-κB activation increased (Figure 4C) ▶ and was widely distributed in glomerular (mesangial and infiltrating cells), tubular (proximal and distal), and interstitial infiltrating cells. In gliotoxin-treated rats, the number and intensity of positive cells were decreased (Figure 4D) ▶ . Specificity was verified in samples from nephritic animals by competition with unlabeled probe (Figure 4E) ▶ .
We analyzed the effect of gliotoxin on expression of NF-κB-dependent genes. Our results revealed increased MCP-1 mRNA levels in renal cortex of nephritic rats, maximal at day 4 (Figure 5) ▶ , in temporal association with the ED1+ cell infiltration (Table 1) ▶ , that was reduced by gliotoxin treatment (76% inhibition, Figure 5 ▶ ). Similarly, the iNOS mRNA expression increased with the severity of the disease and was attenuated in the gliotoxin-treated group (Figure 5) ▶ . By immunohistochemistry we observed an increase in iNOS protein in the glomeruli of nephritic animals (mesangial and infiltrating cells), that was diminished by gliotoxin treatment (glomerular score, day 4: control, 0.7 ± 0.2; untreated nephritis, 2.8 ± 0.2, P < 0.001 versus control; gliotoxin treatment, 1.3 ± 0.2, P < 0.01 versus nephritis).
Figure 5.

Renal mRNA expression of MCP-1 and iNOS is reduced by gliotoxin treatment. Top: The RNA from renal cortex of healthy control, untreated nephritic, and gliotoxin-treated rats was analyzed by RT-PCR (33 cycles) with specific primers for rat MCP-1, iNOS, and GAPDH as a control. Blot representative of four. After densitometry of MCP-1 and iNOS bands and correction by GAPDH, data are expressed as fold increases in relation to control. Each bar (▪, untreated nephritis; □, gliotoxin-treated) represents the mean ± SD of the total number of animals from each group (*, P < 0.05 versus untreated nephritis).
Parthenolide Reduces Glomerular Injury in Murine Experimental Glomerulonephritis
Injection of sheep anti-mouse MC serum to normal mice was associated with mesangial deposition of heterologous sheep Ab leading to acute glomerulopathy characterized by severe proteinuria and hematuria. This model, similar to the anti-Thy 1.1 nephritis in rats, is characterized by a phase of reduced glomerular cell number followed by a phase of restorative cell proliferation (Yo Y, Mobaraki H, Lu H, Braun M, Owens J, King C, Kopp JB, submitted). Pathological proteinuria was even detected on day 1 (Figure 6A) ▶ , and histological changes were maximal on day 5 (Figure 6, B and C) ▶ . No pathological reactions (proteinuria and glomerular lesions) were observed with control nonimmune serum (not shown).
Figure 6.

Beneficial effects of parthenolide on proteinuria and renal injury in murine glomerulonephritis. A: Evolution of proteinuria along the period of study in mice receiving a single dose of anti-MC Ab and a daily injection of saline (•) or parthenolide (▴), and in control normal mice injected with saline (○). B–D: Renal samples of each group were analyzed by Masson’s staining and glomerular and tubulointerstitial lesions were semiquantitatively scored (B) and expressed as mean ± SD (▪, untreated nephritis; □, parthenolide-treated; *, P < 0.05 versus nephritis). Representative glomerulus at day 5 from untreated nephritic (C) and parthenolide-treated mice (D). Original magnifications, ×200.
In a pilot experiment we analyzed the effects of gliotoxin in this mouse model. Although this treatment partially decreased proteinuria levels, the peritoneal administration (70 to 100 μg/day) caused peritonitis in ∼30% of mice. Thus, these findings, together with the in vitro data revealing nontoxic effects of parthenolide on cultured MCs, led us to investigate the effects of parthenolide in the mouse model. Mice injected with anti-murine MCs and receiving daily parthenolide treatment (70 μg/day) developed less proteinuria than the untreated mice did (Figure 6A) ▶ . Hematuria was also decreased by the treatment (1.2 ± 0.6 versus 3.4 ± 0.4 score at day 5; 0.2 ± 0.1 versus 3 ± 0.8 at day 7; P < 0.05). Likewise, the morphological lesions including glomerular (hypercellularity, mesangial expansion, matrix accumulation, and deposits), tubular (atrophy, epithelial degeneration, cast and cell infiltration around the damaged tubules) and interstitial areas (fibrosis and infiltrates) were significantly reduced by the treatment (Figure 6; B, C, D) ▶ . No adverse reactions were observed in either control or nephritic mice receiving this dose of parthenolide. Glomerular cell proliferation (BrdU+ cells) was increased in mice with glomerulonephritis at days 5 and 7 when compared with controls, whereas parthenolide treatment was associated with a marked reduction in the number of glomerular proliferating cells (Figure 7) ▶ .
Figure 7.

Prevention of excessive glomerular cell proliferation by parthenolide. Cellular proliferation in glomeruli was evaluated by in vivo BrdU incorporation. Representative glomeruli from untreated nephritic (A) and parthenolide-treated mice (B) at day 5 of the study are shown. C: Data of glomerular quantification are expressed as cells/glomerular cross-section (gcs) and are mean ± SD (▪, untreated nephritis; □, parthenolide-treated;*, P < 0.05 versus untreated nephritis). Original magnifications, ×200.
In this model, increased activation of NF-κB was observed at days 5 and 7 as assessed by in vitro and in situ oligonucleotide hybridization, with a wide distribution in glomerular, tubular, and infiltrating cells (Figure 8, A and B) ▶ . Treatment with parthenolide prevented the activation of this transcription factor, detecting levels near to control animals in all cell types (61% inhibition at day 7; Figure 8, A and C ▶ ). Parthenolide administration also reduced the increased mRNA expression of MCP-1 detected in this model of glomerulonephritis, as determined by RT-PCR analysis (increases versus control: untreated nephritis, 2.6-fold; parthenolide treatment, 1.1-fold; n = 4). In this experimental model, the activation of other transcription factors, such as AP-1, was also investigated. Although induction of the nephritis caused activation of AP-1 (Figure 8D) ▶ , no decrease in the intensity and number of AP-1-positive cells were observed after parthenolide treatment (Figure 8E) ▶ , confirming the specificity of this compound for NF-κB pathway.
Figure 8.
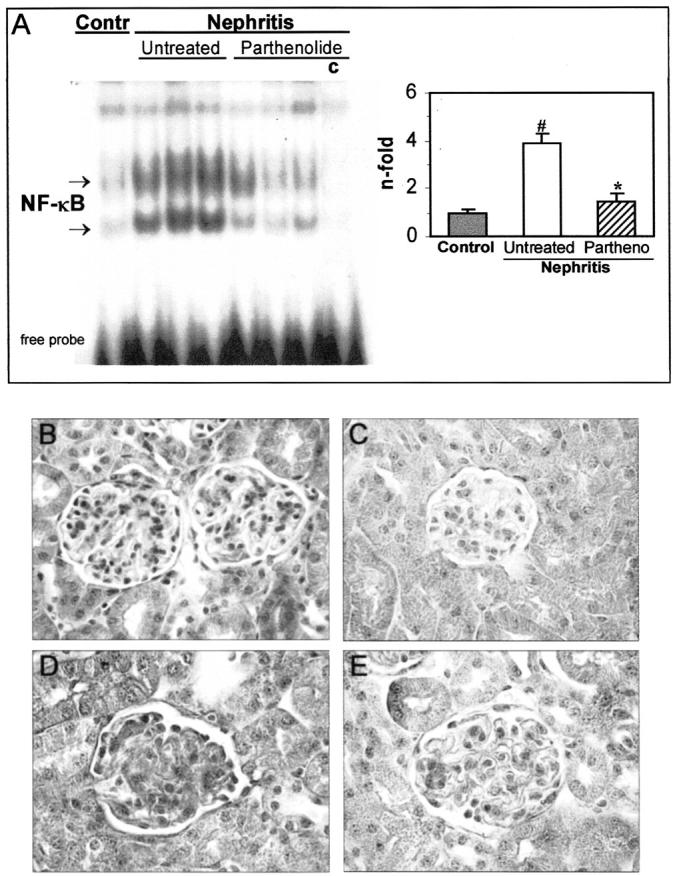
NF-κB activation in murine glomerulonephritis is attenuated by parthenolide treatment. A: Gel shift assay from control, untreated nephritis, or parthenolide-treated mice at day 7. Competition (c) with an excess of unlabeled NF-κB probe was used as a negative control. Densitometric analysis of specific bands revealed a decrease in NF-κB activation by parthenolide treatment (*, P < 0.05 versus nephritis; #, P < 0.05 versus control). The in situ localization of activated NF-κB (B and C) and AP-1 (D and E) was made by Southwestern histochemistry. At day 7, untreated nephritic mice showed strong nuclear staining for NF-κB (B) and AP-1 (D) in glomerular and tubular cells. Parthenolide treatment inhibited the activation of NF-κB (C), but not AP-1 (E). Original magnifications, ×200.
Discussion
In this work we firstly demonstrate that the injection of anti-MC Abs causes NF-κB activation and expression of NF-κB-dependent genes, both in renal resident and infiltrating cells, thus confirming the participation of this transcription factor in the pathogenesis of mesangial proliferative glomerulonephritis, as previously reported in other human and experimental nephritis. 4,6,13,19,39,40
The wide spectrum of immunologically relevant genes regulated by NF-κB and the studies of NF-κB-deficient mice had emphasized the importance of this transcription factor as a mediator of immune and inflammatory responses. 10 In this regard, several drugs have emerged as potential NF-κB modulators, although their mechanisms are not well described. 3-5,13-15 New promising strategies include cell-permeable peptides containing the nuclear localization domain and decoy double-stranded oligonucleotides, with the ability to inhibit NF-κB nuclear translocation and gene transactivation, respectively. 16,17,41 However, it is important to identify new compounds with specificity for this activation pathway without affecting other routes of cell signaling.
The NF-κB pathway is regulated by diverse transduction cascades. One important step is IκB phosphorylation mediated by the IKK complex (kinase subunits α and β, and regulatory subunit γ), leading to ubiquitination and subsequent degradation by the 26S proteasome. Then free NF-κB complexes translocate into the nucleus, bind to target DNA elements, and regulate gene transcription. 10,11 Based on that, we hypothesized that the inhibition of IκB proteins could be a possible target for specific therapies. In this sense, the use of gliotoxin and parthenolide could be more reasonable, because their inhibitory action is due to, at least partially, the blockade of the IκB phosphorylation/degradation pathway. In this work we demonstrate in cultured renal cells that both inhibitors, at nontoxic doses, modulate the NF-κB activation induced by inflammatory stimuli (LPS and cytokines) and the expression of NF-κB-regulated genes (iNOS and MCP-1). The inhibitory effects of both agents seem to be specific for NF-κB, because they did not influence the activity of other transcription factors, as previously reported. 29,30 This inhibition of DNA binding was paralleled by the prevention of IκBα degradation. In addition, determination of phosphorylated IκBα levels revealed a loss of induced phosphorylation in the presence of parthenolide, but not gliotoxin. Both inhibitors act at short periods and relatively low concentrations, suggesting a rapid cell absorption and persistent effect.
The in vivo effects of gliotoxin and parthenolide were examined in experimental models of mesangial proliferative glomerulonephritis. The inflammatory response of rats and mice receiving daily injections of these agents was lower than those of untreated animals, as demonstrated by reduced proteinuria and glomerular lesions. The treatment was also effective in the prevention of glomerular recruitment of mononuclear cells and excessive mesangial proliferation. Indeed, the activation of NF-κB in glomeruli and tubules of nephritic animals, as well as the renal expression of iNOS and MCP-1, were importantly attenuated by the treatment. To our knowledge, this is the first study to show the anti-inflammatory properties of gliotoxin and parthenolide administration in renal diseases. Other authors reported that, in dextran sodium sulfate-induced colitis in rodents, gliotoxin reduced the disease activity and colonic levels of inflammatory cytokines. 42,43 Gliotoxin also presented immunomodulating activity for the prevention of autoimmune diabetes mellitus in rats. 44 To date, there are no reports describing the in vivo effects of parthenolide.
The molecular mechanisms of gliotoxin and parthenolide are not well established. Parthenolide could interfere with some enzymes of the IκB phosphorylation complex, probably with the regulating subunit IKKγ or other not yet identified proteins. 31 A recent report speculates that sesquiterpene lactones directly alkylate the p65 subunit of NF-κB, thereby inhibiting DNA binding. 45 In the future, binding assays using radiolabeled parthenolide could help to identify the structures that this inhibitor may inactivate. Moreover, gliotoxin decreases the expression of NF-κB-dependent genes, such as chemokines, cytokines, and adhesion molecules in several cell types 29,42,43,46,47 and inhibits specifically the activation of NF-κB by blocking the degradation of IκB. 29 However, gliotoxin may also cause the inactivation of other enzymes in different signaling cascades. This fact probably explains its narrow therapeutic range 43 and potential side effects, such as induction of apoptosis. 20 In this sense, recent reports indicate that gliotoxin induces apoptosis in murine macrophages in a dose-dependent and cell cycle-independent manner 23 and inhibits proliferation of human umbilical vein endothelial cells and macrophages. 24,48
In summary, our data show that gliotoxin and parthenolide reduce inflammatory responses in experimental glomerulonephritis, such as mononuclear cell infiltration and MC proliferation. These agents may exert their effects through the blockade of NF-κB activation and subsequent expression of inflammatory genes. Our findings support the idea that drugs modulating NF-κB may represent a novel therapeutic approach in progressive renal diseases and other inflammatory conditions. Moreover, the potential selective effect of these new compounds could represent an additional advantage, allowing to approach separately the different intracellular signaling cascades.
Footnotes
Address reprint requests to Carmen Gómez-Guerrero, Ph.D., Renal and Vascular Research Lab, Fundación Jiménez Díaz Avda, Reyes Católicos, 2 28040 Madrid, Spain. E-mail: cgomez@fjd.es.
Supported by the Fondo de Investigaciones Sanitarias (grants FIS 99/0425, 00/0111), the Comunidad de Madrid (grants 08.3/0002/1998, 08.4/0019.1/2000), the Spanish Society of Nephrology, and the Japan Health Science Foundation and Alumni Association of Juntendo University (to Y. S.).
References
- 1.Clynes R, Dumitru C, Ravetch JV: Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 1998, 279:1052-1054 [DOI] [PubMed] [Google Scholar]
- 2.Chen F, Castranova V, Shi X, Demers LM: New insights into the role of nuclear factor-κB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem 1999, 45:7-17 [PubMed] [Google Scholar]
- 3.Lee JI, Burckart GJ: Nuclear factor-κB: important transcription factor and therapeutic target. J Clin Pharmacol 1998, 38:981-993 [DOI] [PubMed] [Google Scholar]
- 4.Guijarro C, Egido J: Transcription factor-κB (NF-κB) and renal disease. Kidney Int 2001, 59:415-424 [DOI] [PubMed] [Google Scholar]
- 5.Sakurai H, Shigemori N, Hisada Y, Ishizuka T, Kawashima K, Sugita T: Suppression of NF-κB and AP-1 activation by glucocorticoids in experimental glomerulonephritis in rats. Biochim Biophys Acta 1997, 1362:252-262 [DOI] [PubMed] [Google Scholar]
- 6.Gómez-Guerrero C, Duque N, Casado MT, Pastor C, Blanco J, Mampaso F, Vivanco F, Egido J: Administration of IgG Fc fragments prevents glomerular injury in experimental immune complex nephritis. J Immunol 2000, 164:2092-2101 [DOI] [PubMed] [Google Scholar]
- 7.Rovin BH, Dikerson JA, Tan LC, Hebert CA: Activation of nuclear factor-κB correlates with MCP-1 expression by human mesangial cells. Kidney Int 1995, 48:1263-1271 [DOI] [PubMed] [Google Scholar]
- 8.Duque N, Gómez-Guerrero C, Egido J: Interaction of IgA with Fcα receptors of human mesangial cells activates transcription factor nuclear factor-κB and induces expression and synthesis of monocyte chemoattractant protein-1, IL-8 and IFN-inducible protein 10. J Immunol 1997, 159:3474-3482 [PubMed] [Google Scholar]
- 9.Saura M, Zaragoza C, Diaz-Cazorla M, Hernandez-Perera O, Eng E, Lowenstein CJ, Perez-Sala D, Lamas S: Involvement of transcriptional mechanisms in the inhibition of NOS2 expression by dexamethasone in rat mesangial cells. Kidney Int 1998, 53:38-49 [DOI] [PubMed] [Google Scholar]
- 10.Tak PP, Firestein GS: NF-κB: a key role in inflammatory diseases. J Clin Invest 2001, 107:7-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Gaynor RB: Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest 2001, 107:135-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin MJ, Yamamoto Y, Gaynor RB: The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature 1998, 396:77-80 [DOI] [PubMed] [Google Scholar]
- 13.Seto M, Kim S, Yoshifusa H, Nakamura Y, Masuda T, Hamaguchi A, Yamanaka S, Iwao H: Effects of prednisolone on glomerular signal transduction cascades in experimental glomerulonephritis. J Am Soc Nephrol 1998, 9:1367-1376 [DOI] [PubMed] [Google Scholar]
- 14.Zieger-Heitbrock HW, Sternsdorf T, Liese J, Belohradsky B, Weber C, Wedel A, Schreck R, Bauerle P, Strobel M: Pyrrolidine dithiocarbamate inhibits NF-κB mobilization and TNF production in human monocytes. J Immunol 1993, 151:6986-6993 [PubMed] [Google Scholar]
- 15.Fiedler MA, Wernke-Dollries K, Stark JM: Inhibition of TNF-α-induced NF-κB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol 1998, 19:259-268 [DOI] [PubMed] [Google Scholar]
- 16.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawigen J: Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 1995, 270:14255-14258 [DOI] [PubMed] [Google Scholar]
- 17.Yokoseki O, Suzuki J, Kitabayashi H, Watanabe N, Wada Y, Aoki M, Morishita R, Kaneda Y, Ogihara T, Futamatsu H, Kobayashi Y, Isobe M: Cis element decoy against nuclear factor-κB attenuates development of experimental autoimmune myocarditis in rats. Circ Res 2001, 89:899-906 [DOI] [PubMed] [Google Scholar]
- 18.Satriano J, Schlondorff D: Activation and attenuation of transcription factor NF-κB in mouse glomerular mesangial cells in response to tumor necrosis factor-α, immunoglobulin G, and adenosine 3′:5′-cyclic monophosphate. J Clin Invest 1994, 94:1629-1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakurai H, Hisada Y, Ueno M, Sugiura M, Kawashima K, Sugira T: Activation of transcription factor NF-κB in experimental glomerulonephritis in rats. Biochem Biophys Acta 1996, 1316:132-138 [DOI] [PubMed] [Google Scholar]
- 20.Sutton P, Newcombe NR, Waring P, Mullbacher A: In vivo immunosuppressive activity of gliotoxin, a metabolite produced by human pathogenic fungi. Infect Immun 1994, 62:1192-1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinrich M, Ankli A, Frei B, Weimann C, Sticher O: Medicinal plants in Mexico: healers’ consensus and cultural importance. Soc Sci Med 1998, 47:1859-1871 [DOI] [PubMed] [Google Scholar]
- 22.Elsharkawy AM, Wright MC, Hay RT, Arthur MJ, Hughes T, Bahr MJ, Degitz K, Mann DA: Persistent activation of nuclear factor-κB in cultured rat hepatic stellate cells involves the induction of potentially novel Rel-like factors and prolonged changes in the expression of IκB family proteins. Hepatology 1999, 30:761-769 [DOI] [PubMed] [Google Scholar]
- 23.Lakics V, Medvedev AE, Okada S, Vogel SN: Inhibition of LPS-induced cytokines by Bcl-xL in a murine macrophage cell line. J Immunol 2000, 165:2729-2737 [DOI] [PubMed] [Google Scholar]
- 24.Muller K, Dulku S, Hardwick SJ, Skepper JN, Mitchinson MJ: Changes in vimentin in human macrophages during apoptosis induced by oxidized low density lipoprotein. Atherosclerosis 2001, 156:133-144 [DOI] [PubMed] [Google Scholar]
- 25.Kroll M, Arenzana-Seisdedos F, Bachelerie F, Thomas D, Friguet B, Conconi M: The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem Biol 1999, 6:689-698 [DOI] [PubMed] [Google Scholar]
- 26.Mazor RL, Menendez IY, Ryan MA, Fiedler MA, Wong HR: Sesquiterpene lactones are potent inhibitors of interleukin 8 gene expression in cultured respiratory epithelium. Cytokine 2000, 12:239-245 [DOI] [PubMed] [Google Scholar]
- 27.Hwang D, Fischer NH, Jang BC, Tak H, Kim JK, Lee W: Inhibition of the expression of inducible cyclooxygenase and proinflammatory cytokines by sesquiterpene lactones in macrophages correlates with the inhibition of MAP kinases. Biochem Biophys Res Commun 1996, 226:810-818 [DOI] [PubMed] [Google Scholar]
- 28.Wong HR, Menendez IY: Sesquiterpene lactones inhibit inducible nitric oxide synthase gene expression in cultured rat aortic smooth muscle cells. Biochem Biophys Res Commun 1999, 262:375-380 [DOI] [PubMed] [Google Scholar]
- 29.Pahl HL, Krauss B, Schulze-Osthoff K, Decker T, Traenckner EB, Vogt M, Myers C, Parks T, Warring P, Muhlbacher A, Czernilofsky AP, Baeuerle PA: The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-κB. J Exp Med 1996, 183:1829-1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Droge W, Schmitz ML: Sesquiterpene lactones specifically inhibit activation of NF-κB by preventing the degradation of IκB-α and IκB-β. J Biol Chem 1998, 273:1288-1297 [DOI] [PubMed] [Google Scholar]
- 31.Hehner SP, Hofmann TG, Droge W, Schmitz ML: The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-κB by targeting the IκB kinase complex. J Immunol 1999, 163:5617-5623 [PubMed] [Google Scholar]
- 32.Gómez-Guerrero C, Duque N, Egido J: Stimulation of FcαR induces tyrosine phosphorylation of phospholipase C-γ1, phosphatidylinositol phosphate hydrolysis, and Ca2+ mobilization in rat and human mesangial cells. J Immunol 1996, 156:4369-4376 [PubMed] [Google Scholar]
- 33.Gómez-Guerrero C, López-Armada MJ, González E, Egido J: Soluble IgA and IgG aggregates are catabolized by cultured rat mesangial cells and induce production of TNF-α and IL-6, and proliferation. J Immunol 1994, 153:5247-5255 [PubMed] [Google Scholar]
- 34.Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG: A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem 1993, 214:11-16 [DOI] [PubMed] [Google Scholar]
- 35.Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C: Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int 1998, 54:1166-1174 [DOI] [PubMed] [Google Scholar]
- 36.Hernández-Presa MA, Gomez-Guerrero C, Egido J: In situ non-radioactive detection of nuclear factors in paraffin sections by Southwestern histochemistry. Kidney Int 1999, 55:209-214 [DOI] [PubMed] [Google Scholar]
- 37.Bagchus WM, Hoedemaeker PJ, Rozing J, Bakker WW: Glomerulonephritis induced by monoclonal anti-Thy 1.1 antibodies. A sequential histological and ultrastructural study in the rat. Lab Invest 1986, 55:680-687 [PubMed] [Google Scholar]
- 38.Johnson RJ, Raines EW, Floege J, Yoshimura A, Pritzl P, Alpers CE, Ross R: Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J Exp Med 1992, 175:1413-1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mezzano SA, Barria M, Droguett MA, Burgos ME, Ardiles LG, Flores C, Egido J: Tubular NF-κB and AP-1 activation in human proteinuric renal disease. Kidney Int 2001, 60:1366-1377 [DOI] [PubMed] [Google Scholar]
- 40.Rangan GK, Wang Y, Tay YC, Harris DC: Inhibition of nuclear factor-κB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int 1999, 56:118-134 [DOI] [PubMed] [Google Scholar]
- 41.D’Aquisto F, Ialenti A, Ianaro A, Di Vaio R, Carnuccio R: Local administration of transcription factor decoy oligonucleotides to nuclear factor-κB prevents carrageenan-induced inflammation in rat hind paw. Gene Therapy 2000, 7:1731-1737 [DOI] [PubMed] [Google Scholar]
- 42.Herfarth H, Brand K, Rath HC, Rogler G, Scholmerich J, Falk W: Nuclear factor-κB activity and intestinal inflammation in dextran sulphate sodium (DSS)-induced colitis in mice is suppressed by gliotoxin. Clin Exp Immunol 2000, 120:59-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzpatrick LR, Wang J, Le T: In vitro and in vivo effects of gliotoxin, a fungal metabolite: efficacy against dextran sodium-induced colitis in rats. Dig Dis Sci 2000, 45:2327-2336 [DOI] [PubMed] [Google Scholar]
- 44.Liu H, Jackman S, Driscoll H, Larsen B: Immunological effects of gliotoxin in rats: mechanism for prevention of autoimmune diabetes. Ann Clin Lab Sci 2000, 30:366-378 [PubMed] [Google Scholar]
- 45.Garcia-Piñeres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, Merfort I: Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem 2001, 276:39713-39720 [DOI] [PubMed] [Google Scholar]
- 46.Li QQ, Bever CT: Glatiramer acetate blocks interleukin-2-dependent nuclear factor-κB activation and RANTES expression in human U-251 MG astroglial cells. Brain Res Mol Brain Res 2001, 87:48-60 [DOI] [PubMed] [Google Scholar]
- 47.Friedland JS, Constantin D, Shaw TC, Stylianu E: Regulation of interleukin-8 gene expression after phagocytosis of zymosan by human monocytic cells. J Leukoc Biol 2001, 70:447-454 [PubMed] [Google Scholar]
- 48.Lee HJ, Lee JH, Hwang BY, Kim HS, Lee JJ: Anti-angiogenic activities of gliotoxin and its methylthioderivative, fungal metabolites. Arch Pharm Res 2001, 24:397-401 [DOI] [PubMed] [Google Scholar]