

Abstract
Laminopathies are a group of genetic disorders caused by LMNA mutations; they include muscular dystrophies, lipodystrophies and progeroid syndromes. We identified a novel heterozygous LMNA mutation, L59R, in a patient with the general appearance of mandibuloacral dysplasia and progeroid features. Examination of the nuclei of dermal fibroblasts revealed the irregular morphology characteristic of LMNA mutant cells. The nuclear morphological abnormalities of LMNA mutant lymphoblastoid cell lines were less prominent compared to those of primary fibroblasts. Since it has been reported that progeroid features are associated with increased extracellular matrix in dermal tissues, we compared a subset of these components in fibroblast cultures from LMNA mutants with those of control fibroblasts. There was no evidence of intracellular accumulation or altered mobility of collagen chains, or altered conversion of procollagen to collagen, suggesting that skin fibroblast-mediated matrix production may not play a significant role in the pathogenesis of this particular laminopathy.
Keywords: Progeroid syndrome, Laminopathy, Collagen, Aging
Introduction
The LMNA gene encodes lamin A/C nuclear intermediate filaments [1] . Mutations at the LMNA locus are responsible for a number of disorders collectively referred to as laminopathies. Most laminopathies involve tissues of mesenchymal origins, resulting in such features as cardiac and/or skeletal muscular dystrophy, lipodystrophy and progeroid syndromes [2, 3]. A small subset of patients exhibit neuropathies known as Charcot-Marie-Tooth disease type 2B1 [4].
There are two broad classes of LMNA mutations – missense mutations and splicing variants. Amino acid substitutions comprise the majority of disease-causing LMNA mutations [2]. These are located across the coding region and can result in Emery-Dreifuss muscular dystrophy (EDMD2, autosomal dominant, and EDMD3, autosomal recessive) [5], dilated cardiomyopathy (DCM1A)[6] , limb-girdle muscular dystrophy type 1B (LGMD1B) [7], Dunnigan-type familial partial lipodystrophy (FPLD2) [8], mandibuloacral dysplasia (MAD) [9], and atypical Werner syndrome [10]. A small number of truncation mutations are also reported in DCM and muscular dystrophies [2]. Overlapping features of these laminopathies are not unusual [11, 12], and, in the case of DCM, include conduction defects, arrhythmias, and left ventricular dilations that can lead to heart failure [6, 13, 14]. The lipid disorders in patients with lipodystrophies are additional risk factors for cardiovascular disease.
Splicing mutations of exon 11 comprise the second type of LMNA mutations. This group of mutations results in the in-frame deletion and elimination of proteolytic cleavage sites at the C-teminus of lamin A. Cleavage by the ZMPSTE24 protein is required for the maturation of pre-lamin A to mature lamin A [15]. The most common mutation responsible for the Hutchinson-Gilford progeria syndrome (HGPS), G608G, is an exon 11 splice mutation that results in a fifty amino acid deletion [16, 17]. Milder forms of HGPS are due to a different 35 amino acid deletion [18]. The most severe laminopathy, restrictive dermopathy, results in neonatal death. This disease is due to a splicing mutation that skips 90 amino acids of exon 11 [19]. Interestingly, ZMPSTE24 mutations are also reported in MAD and restrictive dermopathy [15, 19]. The most striking cardiovascular manifestations of HGPS is a form of severe atherosclerosis. Patients usually die from myocardial infarction and/or congestive heart failure at a median age of 13.
The pathological changes in the arteries of individuals with HGPS have been described as being very similar to that of the atherosclerosis seen in the general population [20]. The arteries of HGPS patients, however, show particularly severe depletion of smooth muscle cells and derangement of basement membrane [21]. It might therefore be best described as an arterial smooth muscle dystrophy. Moreover, unlike common forms of atherosclerosis, type I collagen has been shown to be dramatically reduced in the arterial adventitia and media and in both external and internal basement membranes of HGPS patients. Additionally, type I collagen is overrepresented in the arterial intimas of these patients [22]. Type III collagen is distributed in all three layers of arterial wall, as seen in control arteries [22]. Microarray studies of HPGS fibroblasts have so far failed to elucidate the pathogenesis [23, 24].
We recently identified a novel LMNA mutation in a 16 year-old female originally referred to our International Registry of Werner Syndrome (University of Washington, Seattle, WA) because of progeroid features and a differential diagnosis that included a collagen disorder. No mutations could be detected in the coding sequence or the promoter of the WRN gene, and Western analysis revealed that the levels of WRN protein were normal Abnormal nuclear morphology of cultured somatic cells was consistent with a laminopathy disorder. Given the clinical suspicion of a collagen disorder and the importance of mesenchmal pathology in progeroid syndromes, we examined type I and III collagen expression in fibroblast cultures from this patient and controls. The results failed to document altered matrix expression in cells from our patient.
Materials And Methods
Patients and samples
The patient was referred to the International Registry of Werner Syndrome (University of Washington, Seattle, WA) (http:www.wernersyndrome.org). A lymphoblasotid cell line (LCL) was established by transformation of B lymphocytes from a peripheral blood sample via a preparation of Epstein-Barr virus. Primary fibroblast cultures were established from a punch biopsy of skin of the medial aspect of the mid upper arm [25]. The protocols were approved by the Human Subjects Review Committee of the University of Washington, Seattle, WA.
Nucleotide sequencing
Genomic DNA was isolated from the LCL cells. Each exon of the LMNA gene was amplified and sequenced with an ABI PRIZM 310 (Applied Biosystems, Foster City, CA) according to the manufacturer's instruction [10].
Cell culture and nuclear morphology
Primary fibroblasts were established via explants from the skin biopsy and maintained under standard tissue culture conditions (give these conditions, including media, additions to media, including amount and type of serum, CO2 and oxygen, pH, temp., humidity, conditions of passaging, including trypsin/versene, degree of confluence when passaged, etc., unless given in another publication) . Fractions of the dividing cells were determined by 3H-thymidine incorporation for 72hrs followed by autoradiography. The percentage of labeled nuclei (labeling index) was determined by counting a minimum of 200 cells [26]
For nuclear morphology, exponentially growing culture were plated on glass coverslips, fixed with 3% paraformaldhyde in PBS, pH 7.4, permealilized with 1% Triton-X in PBS, and mounted with DAPI (diaminophenylindole) on the coverglass. Nuclei were visualized with the Nikon Upright Microscope at the Keck Center for Imaging, University of Washington. Seattle, WA [10, 26]. For the quantitation of nuclear irregularity, nuclear contour ratio was determined in minimum of a hundred randomly chosen nuclei with the MetaMorph program. Statistical significance was determined by the student t-test and f-test [26].
Assay of collagen production
Production of collagens in the culture media were determined by proline incorporation as previously described [27, 28]. Primary fibroblasts were grown in the presence of ascorbic acid containing 1 μCi 14C proline/ml for 20hrs. Media and cell layers were harvested separately, lyophilized and dissolved in 0.5M acetic acid. Samples were separated by 5% SDS-PAGE, and visualized by autoradiography. For the conversions of procollagens to collagens, the samples were digested with 50 μg/ml pepsin for 4 hr at 15°C and stopped by 0.5 μg/ml pepstatin.
Results
Case Report
Our patient is a 17 year-old Caucasian female of North European origin. No significant health issues were reported at birth or during her first 5 years of life. Around age 5, she developed recurrent shoulder dislocations and joint contractures, most noticeably of her small fingers. Osteoporosis was diagnosed at age 8, along with abnormal skin findings including teleangiectasias, sclerodactyly, and poikilodermia. She had a history of poor wound healing. On examination, she was 151cm tall (−2SD) and weighed 39kg (−2.5SD). Her facial features were unusual; there were small ears, a narrow nose and a very small chin. In addition to the above skin findings, soft tissue calcification on the left elbow was noted. She had a Levine II/VI pan systolic murmur. An echocardiogram revealed mild-to-moderate mitral regurgitation. She was later diagnosed with cardiomyopathy. Her vision was normal with no evidence of cataracts.
The patient was referred to the International Registry of Werner Syndrome with a possible diagnosis of Werner syndrome. Sequencing analysis of the WRN gene showed no mutation, and the WRN protein was of the expected size and expressed at a level comparable to those of controls. Studies were initiated to rule out atypical Werner syndrome and associated mutations in the LMNA gene.
A Novel LMNA mutation associated with the abnormal nuclear morphology
Sequencing of the LMNA gene revealed a heterozygous T-to-C alteration at c.176 (Fig. 1). This alteration was confirmed by sequencing in the reverse direction. This change is expected to result in the substitution of arginine for leucine at amino acid 59 (L59R) within the LMNA gene. Neither of the parents carried this mutation, indicating that this was a de novo mutation. This change was not seen in any of 116 controls from a population-based sample of elderly US residents, most of whom were of northern European origin (DNA bank, National Long-Term Care Survey (Department of Pathology, University of Washington, Seattle, WA, USA) [10], thus making it unlikely that this was a polymorphism.
Figure 1.
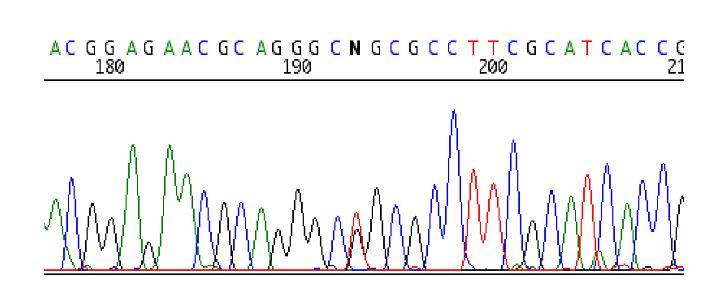
Detection of the heterozygous LMNA mutation. The nucleotide N indicates the presence of heterozygous alteration resulting in L59R (CTG->CCG) mutation.
One of the hallmarks of LMNA mutations is abnormal nuclear morphology. We therefore examined the nuclear morphology in the primary fibroblasts derived from this patient. Figure 2 shows typical confocal microscopic fields after DAPI staining. The labeling index of the control line, 82-6, was 88% and the index of our patient's (NEWFL1010) fibroblasts was 73%, indicating that both lines were at a relatively early passage. The nuclei of the LMNA mutant fibroblasts showed denting, blebbing and irregular margins compared to the control lines (Fig. 2).
Figure 2.
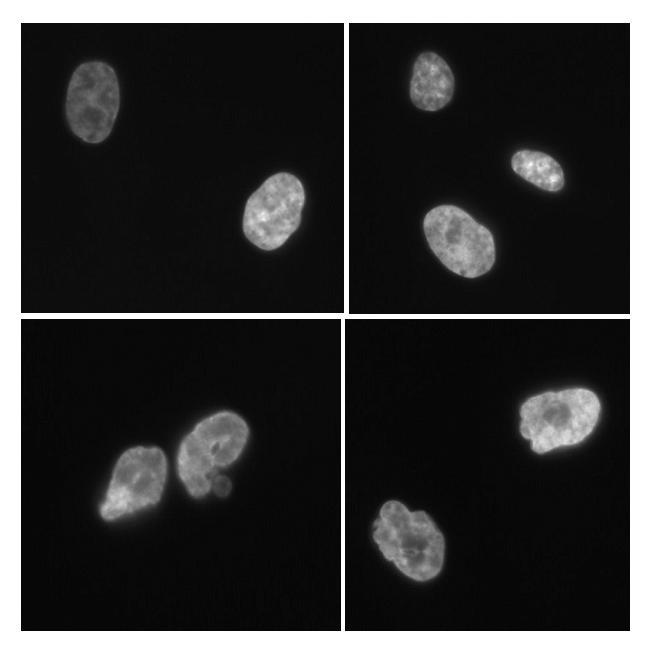
Nuclear morphology of the LMNA mutant fibroblasts. Top two panels show the DAPI staining of the control fibroblasts, 82-6. Bottom two panels show that of NEWFL1010 fibroblasts.
The degree of the abnormality of nuclear morphology was calculated by the nuclear contour ratio, (2πr/perimeter)2 , which would be 1 if the nucleus is completely round and less than 1 if it is not. These ratios were 0.724 (± 0.121) for NEWFL1010 compared to 0.842 (± 0.065) for 82-6, indicating that morphology of LMNA mutant nuclei were more irregular and varied compared to the control (t-test P < 0.001).
Variation of the nuclear contour ratio among LMNA mutant cell lines
In order to assess the significance of the alterations in nuclear shape, we examined other cell lines with LMNA mutations (Table 1). Since there is considerable variation of mean nuclear contour ratios among control fibroblasts lines [26], a comparison of the variances using f-tests is more suitable than a comparison of the averages using t-tests. Using f-tests, the nuclear contour ratio of primary fibroblasts from cells with the R133L mutation (PORTU8020) was 0.800 ± 0.152 whereas fibroblasts obtained from a wild type female sibling control, PORTU8010, was 0.815 ± 0.073 (P<0.0001). On the other hand, the nuclear contour ratio of another LMNA mutant cell line, ATLAN1010, an A57P mutation, was 0.841 ± 0.080, which was not statistically different from either of two control lines (P = 0.220, 0.347). These data indicate that the abnormal nuclear morphology, though characteristic of somatic cells from patients with mutations in LMNA and LMNA-related genes, is not always prominent in primary skin fibroblasts.
Table 1.
Nuclear contour ratios of the LMNA mutant fibroblasts and lymphoblastoid cell lines (LCL).
| Registry number | LMNA Mutation |
Labeling Index (%) |
Nuclear Contour Ratio |
P (f-test) | |
|---|---|---|---|---|---|
| Primary fibroblasts | |||||
| NEWLF1010 | L59R | 73 | 0.724 ± 0.121 | <0.002, | |
| <0.001 * | |||||
| PORTU8010 | R133L | 70 | 0.800 ± 0.152 | <0.0001, | |
| <0.0001 * | |||||
| ATLAN1010 | A57P | 65 | 0.841 ± 0.080 | 0.220, 0.347 * | |
| PORTU8020 | wt | 81 | 0.815 ± 0.073 | 0.502 ** | |
| 82-6 | wt | 88 | 0.842 ± 0.065 | NA | |
| LCL | |||||
| PORTU8010 | R133L | NA | 0.765 ± 0.203 | <0.0002 | |
| PORTU8020 | wt | NA | 0.768 ± 0.159 | ||
| ATLAN1010 | A57P | NA | 0.799 ± 0.224 | 0.504 | |
| ATLAN1020 | wt | NA | 0.722 ± 0.229 |
vs. 82-6 and vs. PORTU8020
vs. 82-6
Examination of the nuclear morphology of LCLs revealed a similar trend. Comparison of the variances within the PORTU pedigree again revealed a significant difference between the patient and the sibling (0.765 ± 0.203 vs 0.768 ± 0.159, <0.0002). The variance was not significantly different between the ATLAN patient and sibling (0.799 ± 0.224 vs 0.722 ± 0.229, P = 0.504). The differences in the variances of patient and controls for the LCL pairs was less that those of primary fibroblasts for both the PORTU and ATLAN pedigrees.
Extracellular matrix expression in LMNA mutant fibroblasts
SDS-PAGE analysis of the medium showed that the levels of proline incorporation of the bands corresponding to the procollagen chains, α1(III), α1(I) and α2(I), in LMNA mutant fibroblasts were similar to those of the control fibroblasts (Fig. 3A). Following pepsin treatment, the patterns of collagens, α1(III), α1(I) and α2(I) showed no difference between LMNA mutant and control fibroblasts (Fig. 3B). Similarly, assays of cell lysates showed no evidence of intracellular accumulation, altered mobility of chains, or altered conversion of procollagen to collagen (data not shown). These results indicate that the LMNA mutations we investigated do not quantitatively alter the synthesis of these extracellular matrix components at least in the primary skin fibroblasts.
Figure 3.
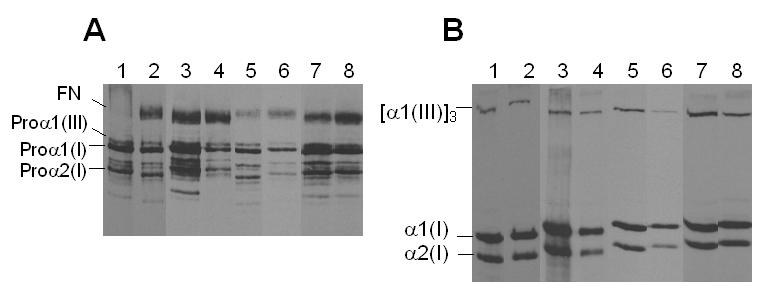
Collagen production of the LMNA mutant fibroblasts. SDS-PAGE of radiolabeled procollagens (A) and collagens (B) from the culture media . Lane 1, 3, 5 and 7, control fibroblasts, A8; lane 2, ATLAN1010; lane 4, NEWFL1010; lane 6, PORTU8010; and lane 8 PORTU8020. All test sample data are paired with the control data used within the experiments.
Discussion
LMNA mutations have been identified in an increasing numbers of genetic conditions, including muscular dystrophies, lipodystrophies and progeroid syndromes. The international Registry of Werner Syndrome (University of Washington, Seattle, WA) has been receiving a spectrum of Werner-like patients for diagnosis. As a result, we have identified LMNA mutations in four patients with atypical Werner syndrome [10]. We report a novel LMNA mutation, L59R, in a fifth patient presenting with what we have operationally defined as an atypical Werner syndrome. Given the molecular findings, however, this subject might be more appropriately referred to atypical or novel laminopathy.
Identification of this LMNA mutation in association with the clinical phenotype of our patient suggests the diagnosis of a laminopathy, most likely an atypical form of MAD. The diagnosis of laminopathy is further supported by the demonstration of abnormal nuclear morphology. The variance, rather than average, of the nuclear contour ratio appears to be more suitable for the assessment of the degree of abnormality. However, such abnormalities are not always seen in LMNA mutant cells. Filesi et al [29] reported a MAD case with homozygous R527H mutations that showed minimal degree of nuclear blebbings accompanied by low level of pre-lamin A accumulation.
Because the causal relationship between LMNA mutations and a range of progeroid syndromes has only recently been established, LMNA mutants have not been extensively studied by biogerontologists. Reduced production of type I and III collagens has been described in primary fibroblasts derived from the aged skin [30, 31]. These changes are associated with an increased expression of matrix metalloproteinase and a reduced expression of tissue inhibitor of metalloproteinases in skin samples from aged people [32]. Such findings were also reported in dermal fibroblasts that underwent senescence in culture [33]. An additional report describes how collagen production determined by H3-labeled proline incorporation is increased in fibroblasts obtained from LMNA null mice compared to mutant mice complemented with lamin A and C cDNA [34]. It is therefore if importance to investigate the dynamics of extracellular matrix in a variety of human laminopathies. The present study makes a beginning effort in that direction. The results show that primary skin fibroblasts with LMNA mutations produce collagen type I and III at levels similar to that of control fibroblasts.
The manifestations of aging are thought to be particularly pronounced in the extracelular matrix, the primary component of which is collagen. The characteristic change of aged extracellular matrix is the accumulation of collagen glycoxidation generated by the so-called Maillard reaction. These advanced glycation endproducts (AGEs) are seen in aged skin from a wide variety of mammalian species [35], and has been implicated in the apoptosis of dermal fibroblasts [36]. Our initial investigation of collagen protein production therefore needs to be expanded to include the examination of these biomarkers of aging. To this end, all biological materials described in this report will be made available to qualified researchers via the International Registry of Werner Syndrome (http://www.wernersyndrome.org).
Acknowledgements
The studies on collagen biosynthesis were performed in the Collagen Diagnostic Laboratory, Department of Pathology, University of Washington. This work was supported by the grant CA78088 from NIH/NIA and NCI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–16326. [PubMed] [Google Scholar]
- 2.Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Mol Genet Metab. 2006;87:289–302. doi: 10.1016/j.ymgme.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Smith ED, Kudlow BA, Frock RL, Kennedy BK. A-type nuclear lamins, progerias and other degenerative disorders. Mech Ageing Dev. 2005;126:447–460. doi: 10.1016/j.mad.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 4.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–736. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 6.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr., Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 7.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 8.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 9.Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 11.Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112:549–555. doi: 10.1016/s0002-9343(02)01070-7. [DOI] [PubMed] [Google Scholar]
- 12.Hegele R. LMNA mutation position predicts organ system involvement in laminopathies. Clin Genet. 2005;68:31–34. doi: 10.1111/j.1399-0004.2005.00447.x. [DOI] [PubMed] [Google Scholar]
- 13.Sylvius N, Tesson F. Lamin A/C and cardiac diseases. Curr Opin Cardiol. 2006;21:159–165. doi: 10.1097/01.hco.0000221575.33501.58. [DOI] [PubMed] [Google Scholar]
- 14.Sebillon P, Bouchier C, Bidot LD, Bonne G, Ahamed K, Charron P, DrouinGarraud V, Millaire A, Desrumeaux G, Benaiche A, Charniot JC, Schwartz K, Villard E, Komajda M. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet. 2003;40:560–567. doi: 10.1136/jmg.40.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 16.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuchi K, Katsuya T, Sugimoto K, Kuremura M, Kim HD, Li L, Ogihara T. LMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:e67. doi: 10.1136/jmg.2003.014688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, Faivre L, Verloes A, Van Essen T, Flori E, Hennekam R, Beemer FA, Laurent N, Le Merrer M, Cau P, Levy N. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 20.Ackerman J, Gilbert-Barness E. Hutchinson-Gilford progeria syndrome: a pathologic study. Pediatr Pathol Mol Med. 2002;21:1–13. doi: 10.1080/pdp.21.1.1.13. [DOI] [PubMed] [Google Scholar]
- 21.Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999;8:29–39. doi: 10.1016/s1054-8807(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 22.Stehbens WE, Delahunt B, Shozawa T, Gilbert-Barness E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol. 2001;10:133–136. doi: 10.1016/s1054-8807(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 23.Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging. Science. 2000;287:2486–2492. doi: 10.1126/science.287.5462.2486. [DOI] [PubMed] [Google Scholar]
- 24.Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 25.Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, LucciCordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27:558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang S, Chen L, Libina N, Janes J, Martin GM, Campisi J, Oshima J. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum Genet. 2005;118:444–450. doi: 10.1007/s00439-005-0051-7. [DOI] [PubMed] [Google Scholar]
- 27.Korkko J, Ala-Kokko L, De Paepe A, Nuytinck L, Earley J, Prockop DJ. Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfecta type I: identification of common sequences of null-allele mutations. Am J Hum Genet. 1998;62:98–110. doi: 10.1086/301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarze U, Schievink WI, Petty E, Jaff MR, Babovic-Vuksanovic D, Cherry KJ, Pepin M, Byers PH. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989–1001. doi: 10.1086/324123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filesi I, Gullotta F, Lattanzi G, D'Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, Maraldi NM, Biocca S, Novelli G. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23:150–158. doi: 10.1152/physiolgenomics.00060.2005. [DOI] [PubMed] [Google Scholar]
- 30.Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861–1868. doi: 10.2353/ajpath.2006.051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenkins G. Molecular mechanisms of skin ageing. Mech Ageing Dev. 2002;123:801–810. doi: 10.1016/s0047-6374(01)00425-0. [DOI] [PubMed] [Google Scholar]
- 32.Varani J, Warner RL, Gharaee-Kermani M, Phan SH, Kang S, Chung JH, Wang ZQ, Datta SC, Fisher GJ, Voorhees JJ. Vitamin A antagonizes decreased cell growth and elevated collagen-degrading matrix metalloproteinases and stimulates collagen accumulation in naturally aged human skin. J Invest Dermatol. 2000;114:480–486. doi: 10.1046/j.1523-1747.2000.00902.x. [DOI] [PubMed] [Google Scholar]
- 33.West MD. The cellular and molecular biology of skin aging. Arch Dermatol. 1994;130:87–95. [PubMed] [Google Scholar]
- 34.Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, van Leeuwen RE, Crijns HJ, Ramaekers FC, Hutchison CJ, Pinto YM. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet. 2005;14:2839–2849. doi: 10.1093/hmg/ddi316. [DOI] [PubMed] [Google Scholar]
- 35.Sell DR, Lane MA, Johnson WA, Masoro EJ, Mock OB, Reiser KM, Fogarty JF, Cutler RG, Ingram DK, Roth GS, Monnier VM. Longevity and the genetic determination of collagen glycoxidation kinetics in mammalian senescence. Proc Natl Acad Sci U S A. 1996;93:485–490. doi: 10.1073/pnas.93.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem. 2005;280:12087–12095. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]