

Abstract
Array comparative genomic hybridization (CGH) is being widely used to screen for recurrent genomic copy number alterations in neoplasms, with imbalances typically detected through the application of gain and loss thresholds. Review of array CGH publications for the year 2005 showed that a wide range of thresholds are used. However, the effect of sample ploidy on the sensitivity of these thresholds for single copy alterations (SCAs) has not been evaluated. Here, we describe a method to evaluate the detection accuracy of thresholds for detecting SCAs in cell line array CGH data. By applying a hidden Markov model-based method, we segmented array CGH data from well-karyotyped cell lines and generated ploidy-specific sensitivity-specificity plots, from which we identified optimum thresholds relevant to sample ploidy. We demonstrate that commonly used nonploidy-specific thresholds are suboptimal in their ability to call SCAs, particularly when applied to hypertriploid or tetraploid cell lines. We conclude that the use of ploidy-specific thresholds improves the sensitivity of thres-hold-based array CGH for detecting SCAs in cell lines. Because polyploidy is a common feature of cancer cells, the application of ploidy-specific thresholds to cell lines (and potentially to clinical samples) may improve the detection sensitivity of SCAs of biological significance.
The acquisition of genomic DNA copy number alterations and corresponding changes in expression of genes involved in cellular growth and survival pathways are key events in the development and progression of human cancers. Array comparative genomic hybridization (CGH) represents an efficient approach to screening entire genomes for regions with DNA copy number alterations by providing global information on characteristics of the genome structure. There is considerable interest in applying the technique to identify copy number alterations in neoplasms, using cell lines and clinical samples. With the emergence of increasing array CGH data sets, there is a critical need for an approach that identifies copy number alterations with high sensitivity.
In a typical array CGH experiment, genomic DNA is isolated from test and reference samples, differentially labeled, and hybridized to DNA microarrays containing elements mapped to the genome sequence.1 The addition of Cot-1 DNA suppresses the hybridization of highly repetitive sequences. Relative differences in signal intensity ratios between test and reference DNA reflect copy number alterations in the test DNA. Before analysis, the data are usually normalized by setting the median of the intensity ratios from the entire genome to 1 on a linear scale.2 After normalization, the most commonly used method to identify regions of gain or loss is to set thresholds, either arbitrarily or at multiples of the SD (log2 ratio value) of the mean from normal-normal hybridizations.3
Table 1 summarizes our review of array CGH publications within the year 2005 and the thresholds used therein to define gains and losses. Although threshold-based analysis is in widespread use, justification for the choice of thresholds used is frequently neglected in array CGH publications. Verification of ratio profiles may be limited to the use of fluorescence in situ hybridization (FISH) on a few loci to show the absence of false-positives, whereas the potential presence of false-negative results is not addressed. Importantly, the accuracy (particularly the sensitivity) of commonly used thresholds at calling single copy gains and losses has not been evaluated adequately. Because the linear relationship between intensity ratio and copy number is dependent on the ploidy of the sample,4 we would expect the thresholds to be ploidy-dependent. As such, true single copy gains and losses may be missed by arbitrarily selected thresholds. This limitation in detection sensitivity is of critical importance in applying threshold-based array CGH to screening cells from neoplasms because polyploidy is a common feature of malignancies and premalignancies.
Table 1.
Thresholds Used to Call Gains and Losses in Array CGH Publications in the Year 2005
| Reference number | Thresholds used (log2 ratio values unless otherwise stated) | Reason for selection |
|---|---|---|
| 13 | ±0.25 | None identified |
| 14, 15, 16 | ±0.3 | |
| 17, 18 | ±0.4 | |
| 19 | ±0.5 | |
| 20, 21, 22 | ±0.2 | ±2 SDs from normal hybridizations |
| 23 | ±0.08 | ±3 SDs from normal hybridizations |
| 24 | ±0.13 | |
| 25 | ±0.3 | |
| 26 | ±0.42 | |
| 27, 28 | ±2 SDs of each sample profile | ±2 SDs of each sample profile |
| 29 | ±2.5 SDs of each sample profile | ±2.5 SDs of each sample profile |
| 30 | ±3 SDs of all clones | ±3 SDs of all clones |
| 31 | ±3 SDs of the normal regions of each sample | ±3 SDs of the normal regions of each sample |
| 32 | Gain 0.39; loss −0.5 | None identified |
| 33 | ± 0.4 | Gaussian modeling |
| 34, 35 | Loss by comparison with X chromosome controls; gains using arbitrary values | X chromosome controls for losses |
| 36 | Gain 1.2; loss 0.69 (absolute ratio values) | FISH validation of true positives |
| 37, 38, 39 | Gain 1.2; loss 0.8 (absolute ratio values) | None identified |
| 40 | Gain 1.5; loss 0.5 (absolute ratio values) | |
| 41 | Amplified 1.8; deleted 0.55 (absolute ratio values) |
The literature search was done via the National Library of Medicine (NCBI) search engine (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi), using the search term ′array CGH.′ This search resulted in 90 studies (up to the end of December, 2005). The studies were surveyed for their relevance by reading the abstracts, when available. Twenty-nine relevant studies were found, and these were assessed in more detail.
In this report, we describe a method to evaluate the detection accuracy of thresholds for single copy alterations (SCAs) in array CGH data from carcinoma cell lines. We identify optimum thresholds relevant to sample ploidy and evaluate the accuracy of these in comparison to standard thresholds (arbitrarily set at ±3 SDs of the mean log2 ratio value from normal-normal hybridizations) at calling SCAs known to exist in well-karyotyped cell lines. We verify that these thresholds are more accurate than standard thresholds at calling SCAs in cell lines. Moreover, our preliminary observations in tumor tissue indicate that these ploidy-specific thresholds may be applicable to clinical samples of known ploidies.
Materials and Methods
Cervical Cell Lines and Clinical Sample
We used six cervical keratinocyte cell lines derived from squamous cell carcinomas (SCCs) of the uterine cervix (Table 2). All were obtained from the American Type Culture Collection (Manassas, VA) and cultured as described by the American Type Culture Collection. We also used snap-frozen and formalin-fixed, paraffin-embedded tissue from a cervical SCC that had been shown previously by interphase FISH using centromeric probes to be hypertriploid or tetraploid. The tissue was obtained from the Department of Histopathology, Addenbrooke’s Hospital, Cambridge, UK, with local research ethics committee approval. In the frozen sample of the tumor, at least 80% of the cells were malignant.
Table 2.
Details of Cervical Squamous Cell Carcinoma Cell Lines Used
| Cell line | ATCC number | Modal chromosome number |
|---|---|---|
| C4I | CRL-1594 | 45 |
| C4II | CRL-1595 | 46 |
| ME180 | HTB-33 | 62 |
| SiHa | HTB-35 | 69 |
| SW756 | CRL-10302 | 80 |
| CaSki | CRL-1550 | 80 |
The table shows the cell line name and modal chromosome number determined from cytogenetic analysis (N. Foster, manuscript in preparation). ATCC, American Type Culture Collection.
DNA Isolation
Genomic DNA (gDNA) was isolated by conventional phenol/chloroform extraction. gDNA from peripheral blood lymphocytes of a healthy male was used as the reference for normal gene copy numbers. DNA concentrations and quality were determined using the Nanodrop UV spectrophotometer (Nanodrop Technologies, Wilmington, DE).
Array CGH Hybridization and Image Capture
The arrays used were kindly provided by Professor Barbara Weber, University of Pennsylvania (Philadelphia, PA), and contained 4134 bacterial artificial chromosome (BAC) clones that covered the human genome at 1 MB resolution. DNA labeling and hybridization were performed as described previously.5 One μg each of test DNA from cervical cell lines or tissue sample and reference DNA from normal male peripheral blood lymphocytes were labeled with Cy3-dCTP or Cy5-dCTP (with dye-swapping) using random-prime labeling (BioPrime Plus array CGH labeling module; Invitrogen Ltd., Paisley, UK). Hybridization was performed at 37°C in a shaking water bath for 72 hours. Hybridized arrays were washed in 2× standard saline citrate, 50% formamide, pH 7.0, at 45°C for 15 minutes and 2× standard saline citrate, 0.1% sodium dodecyl sulfate at 45°C for 30 minutes before a final wash in 0.2× standard saline citrate at room temperature for 15 minutes. The arrays were dried in a slide centrifuge before being scanned using an Axon 4000B scanner (Axon Instruments, Burlingame, CA). The acquired Cy3 and Cy5 images were preprocessed with GenePix Pro 4.1 imaging software (Axon Instruments, Foster City, CA). Differences in overall signal intensity between the Cy3 and Cy5 channels were adjusted by normalizing all signal intensities to a 1:1 ratio. For each spot, the median pixel intensities minus the median local backgrounds for both dyes were used to obtain the log2 value of test to reference copy number ratio. Fluorescence ratios of the clones were calculated as the average of paired dye-swapped arrays.
Defining Copy Number Segments
Cells lines of various ploidies were selected for the investigation. In accordance with commonly used procedures,6 we defined the ploidy of each cell line as the copy number of the majority of the genome; that is, present in at least 70% of 50 metaphases examined (N. Foster, I. Roberts, M. R. Pett, N. Coleman, manuscript in preparation). On this basis, we determined the cell lines to be diploid (C4I, C4II), triploid (ME180, SiHa), and hypertriploid (SW756, CaSki), in keeping with published American Type Culture Collection findings. The cytogenetic data from the diploid and triploid cell lines and SW756 were examined to identify regions showing SCAs in more than 70% of metaphases. Whole chromosomes or chromosome arms with 1-copy loss, 1-copy gain, and normal copies relative to the base ploidy of each cell line were selected for further investigation. Only chromosomes with unambiguous karyotype data were used. For more accurate definition of the boundaries of SCAs in the selected chromosome arms, segmentation was performed on the array CGH ratios from the selected chromosomes using the aCGH package for the R statistical language from Bioconductor (http://www.bioconductor.org/). This package contains a hidden Markov model7-based method that assigns clones to states with constant copy number. Clones lying within the segments defined as showing SCAs were selected for further analysis. Because of the limitations of segmentation, some clones within the defined segments remained stateless. To ensure accuracy of the selection, any clone showing focal (ie, isolated) aberrations after segmentation, together with those lying within three positions of the end of each defined SCA segment, were therefore excluded from the analysis.
Identification and Evaluation of Thresholds
The ability of threshold-based array CGH to detect SCAs at different ploidies was evaluated by comparing receiver-operating characteristic (ROC) curves. For each ploidy, ROC curves were generated for single copy gains and losses by entering the appropriate array fluorescent intensity ratios into the statistical software SPSS 11.5 for Windows (SPSS Inc., Chicago, IL). Sensitivity and specificity at a range of gain and loss thresholds were calculated and plotted to identify optimum thresholds. We evaluated the accuracy of standard thresholds and compared this to the performance of optimum thresholds, when applied to cell lines of different ploidy. Standard thresholds were arbitrarily set at ±3 SDs (log2 ratio value) of the mean of normal-normal hybridizations. These normal hybridizations were of gDNA from normal cervical squamous epithelium, or normal female placenta, versus gDNA from normal male peripheral blood lymphocytes.
Application of Optimum Thresholds to Cell Lines
To evaluate the accuracy of the optimum thresholds for detecting true single copy gains and losses in cell lines, additional array CGH data from the hypertriploid cell lines SW756 and CaSki were analyzed. For the cell line SW756, the chromosomes selected for validation were different from those used in identifying optimum thresholds, whereas CaSki had not been used to identify optimum thresholds. SCAs were called from raw unsegmented data, without exclusion of any clone. A random sampling of clones that were identified as gained or lost by the optimum thresholds, but not the standard thresholds, were selected for verification by BAC-FISH. Metaphase spreads of the cell lines were prepared using standard procedures and FISH was performed as described by Hoglund and colleagues.8 BAC clones were obtained from BACPAC Resources (http://bacpac.chori.org/home.htm) and DNA was extracted as described previously.9 BAC DNA was labeled with biotin 16-dUTP (Roche, East Sussex, UK) or digoxigenin 11-dUTP (Roche) using nick translation (Vysis, Downers Grove, IL) according to the manufacturer’s instructions. After hybridization, probes labeled with biotin were detected with avidin-Cy5 (Amersham Biosciences, Uppsala, Sweden) (1:400) and biotin anti-avidin (1:300), whereas probes labeled with digoxigenin were detected with anti-digoxigenin rhodamine (Roche) (1:200). FISH images were captured with a fluorescence microscope equipped with a charge-coupled device camera, controlled by a Macintosh computer running the SmartCapture (Vysis) software.
Application to Primary Tumor Data
We performed a preliminary experiment to examine whether ploidy-specific thresholds would improve the sensitivity for detecting SCAs in clinical samples as well as cell lines. Array CGH data for gDNA from a frozen primary cervical SCC was analyzed with optimized thresholds selected using the approach described above and the results were compared with those obtained using the standard thresholds. The sample was known to be hypertriploid or tetraploid, as determined by FISH using centromeric probes on chromosomes 9 and 10, in which more than 70% of the cells examined had three or four centromeric signals (data not shown). Randomly selected clones identified as gained using both thresholds or using the optimum threshold only were used in interphase FISH on formalin-fixed paraffin-embedded sections from the same SCC. FISH on interphase nuclei was performed as described previously.9 Test and control probes were labeled with Spectrum Orange (Vysis) and biotin 16-dUTP, respectively, using nick translation (Vysis). Avidin-Cy5 (1:400) (Amersham) and biotin anti-avidin (1:300) were used to detect biotin-labeled probes. FISH images were captured as before.
Results
Selection of Copy Number Segments
The karyograms of the diploid (C4I and C4II), triploid (ME180 and SiHa), and hypertriploid (SW756) cell lines were examined, and chromosome segments with SCAs in more than 70% of metaphases were selected. The array CGH ratio profiles of the selected chromosomes were partitioned into copy number states using a hidden Markov model-based method implemented in the R statistical language. The segmentation allowed more precise definitions of the ends of the selected chromosome segments and verified the changes identified by cytogenetic analysis (Figure 1). BAC clones from two or three whole chromosomes or segments at each state (1-copy loss, 1-copy gain, normal) were selected from each cell line. Clones at the same state, from different cell lines of the same ploidy (ie, C4I and C4II; ME180 and SiHa), were pooled in downstream analysis. Pooling was done after Student’s t-tests showed no significant difference in the intensity ratio values of clones at the same state from cell lines of the same ploidy (Table 3).
Figure 1.

Selection and segmentation of chromosomal regions showing SCAs. A: Selection of chromosome segments with SCAs in cell line SiHa. Karyogram of chromosomes 1 and 11 (normal copies), 4 and 13 (1-copy losses), and 9 and 15 (1-copy gains), as seen in the majority of metaphases in the triploid cell line SiHa. B: Ratio profiles before and after segmentation, performed using a hidden Markov model-based method and implemented in the R statistical language. Profiles for chromosomes 11, 13, and 15 are shown. The arrowed distal segment of chromosome 15 indicates the region from which clones with 1-copy gains were selected. The orange dots indicate focal aberrations representing true narrow copy changes, mismapped clones, or natural copy number polymorphisms. These aberrations need to be investigated further using alternative techniques and were therefore excluded from analysis. Red dashed line, centromere; green and blue lines, start and end of state transitions, respectively.
Table 3.
Results of Student’s t-tests Performed on Fluorescence Intensity Ratios of BAC Clones at the Same State (1-Copy Loss, 1-Copy Gain, Normal) from Cell Lines of the Same Ploidy
| Ploidy | Copy number state | Cell line | Mean ratio value (n) | P value |
|---|---|---|---|---|
| Diploid | 1-Copy loss | C4I | 0.8164 (85) | 0.15 |
| C4II | 0.7086 (90) | |||
| Normal copies | C4I | 1.0156 (410) | 0.21 | |
| C4II | 1.0067 (478) | |||
| 1-Copy gain | C4I | 1.3370 (23) | 0.99 | |
| C4II | 1.3366 (116) | |||
| Triploid | 1-Copy loss | ME180 | 0.7847 (207) | 0.16 |
| SiHa | 0.7777 (281) | |||
| Normal copies | ME180 | 1.0279 (189) | 0.067 | |
| SiHa | 1.0081 (349) | |||
| 1-Copy gain | ME180 | 1.2762 (75) | 0.070 | |
| SiHa | 1.2403 (77) |
No significant difference between ratio values was detected (P > 0.05). Ratio values of clones at the same state within each ploidy were therefore pooled in downstream analyses.
Comparison of ROC Curves
ROC curves were used to evaluate the power of array CGH to detect SCAs at various sample ploidies. Figure 2 shows the plot of sensitivity versus intervals of “1 minus specificity”, corresponding to different thresholds for the detection of single copy loss. The performance of array CGH to detect single copy loss is best for the diploid cell line, as evidenced by the highest sensitivity for any given specificity. This is followed by the triploid cell line, with discrimination of single copy loss being the poorest in the hypertriploid cell line. The same performance trend is observed for single copy gains (data not shown). Overall performance is reflected in the area under the ROC curve (Table 4).
Figure 2.

ROC curves illustrating the power of array CGH to detect single copy losses for samples of different ploidy.
Table 4.
Power of Array CGH to Detect Single Copy Alterations for Samples of Different Ploidy, as Determined by the Area Under the ROC Curves
| Ploidy | Copy number alteration | Area under ROC curve |
|---|---|---|
| Diploid | 1-Copy loss | 0.997 |
| 1-Copy gain | 0.954 | |
| Triploid | 1-Copy loss | 0.970 |
| 1-Copy gain | 0.930 | |
| Hypertriploid | 1-Copy loss | 0.825 |
| 1-Copy gain | 0.802 |
A larger area indicates better performance.
Setting of Optimum Thresholds
From plots of sensitivity and specificity throughout the range of gain/loss thresholds for each ploidy, optimum thresholds were selected at the crossing points of these graphs, where the probability of error is minimized. Figure 3 illustrates the plots obtained from the triploid cell lines and the selection of optimum thresholds. Table 5 lists the optimum ploidy-specific thresholds for detection of SCAs, together with corresponding sensitivities, specificities, and error probability values. This demonstrates that for the optimum detection of single copy changes, the stringency of the thresholds must necessarily decrease with increasing ploidy.
Figure 3.

Plots of array CGH sensitivity and specificity versus the range of gain and loss thresholds for triploid cell lines. The optimum threshold is obtained from the crossing point of the sensitivity and specificity graphs, where the probability of error is minimized. Dotted lines show the locations of optimized gain (A) and loss (B) thresholds for triploid cell lines.
Table 5.
Optimum Thresholds For Cells of Different Ploidies, Obtained from the Crossing Points of Plots of Sensitivity and Specificity throughout the Range of Threshold Values Tested
| Ploidy | Gain threshold (sensitivity/specificity) (error probability) | Loss threshold (sensitivity/specificity) (error probability) |
|---|---|---|
| Diploid | 1.14 | 0.83 |
| (0.91) | (0.97) | |
| (0.18) | (0.060) | |
| Triploid | 1.13 | 0.86 |
| (0.85) | (0.92) | |
| (0.30) | (0.16) | |
| Hypertriploid | 1.04 | 0.92 |
| (0.71) | (0.74) | |
| (0.58) | (0.52) |
The table also lists sensitivity, specificity, and error probability values at each optimum threshold. By definition, the sensitivity and specificity values are the same at each crossing point.
Evaluation of Standard Thresholds
Using the sensitivity/specificity and error probability plots, we measured the detection accuracy of standard thresholds for SCAs (Table 6). Except for a 17% increase in error rate using the standard gain threshold for the diploid cell line, the standard thresholds had comparable performance to the optimized thresholds when applied to cell lines with diploid and triploid genomes. For the triploid cell line, the application of standard thresholds resulted in an increase in false-negative rates (ie, reduced sensitivity), which was balanced by a decrease in false-positive rates (ie, improved specificity), resulting in a similar error rate overall to that of the optimized thresholds. In contrast, the standard thresholds performed poorly with the hypertriploid cell line. The application of a standard gain threshold resulted in a 70% increase in false-negative rate and an overall 43% increase in error probability, while a standard loss threshold gave a 49% increase in false-negative rate and a 29% increase in error probability.
Table 6.
Effect of Applying Standard Thresholds (Compared to Optimized Thresholds) to Diploid, Triploid, and Hypertriploid Samples
| Ploidy | Diploid | Triploid | Hypertriploid |
|---|---|---|---|
| Gain threshold applied | 1.18 | ||
| [log2 ratio value] | [0.24] | ||
| Sensitivity | 0.85 | 0.74 | 0.21 |
| (% change) | (−6.6) | (−13) | (−70) |
| Specificity | 0.94 | 0.96 | 0.96 |
| (% change) | (+3.3) | (+13) | (+35) |
| Probability of error | 0.21 | 0.30 | 0.83 |
| (% change) | (+17) | (0) | (+43) |
| Loss threshold applied | 0.85 | ||
| [log2 ratio value] | [−0.24] | ||
| Sensitivity | 0.99 | 0.90 | 0.38 |
| (% change) | (+2.1) | (−2.2) | (−49) |
| Specificity | 0.95 | 0.93 | 0.95 |
| (% change) | (−2.1) | (+1.1) | (+28) |
| Probability of error | 0.060 | 0.17 | 0.67 |
| (% change) | (0) | (+6.3) | (+29) |
Standard thresholds are at ±3 SDs of the mean of normal-normal hybridizations. Numbers in brackets indicate percent change from the optimized rates obtained using ploidy-specific thresholds.
Evaluation of Accuracy of Optimum Thresholds in Cell Lines
We analyzed additional array CGH data obtained from two hypertriploid cell lines (CaSki and SW756) and performed BAC-FISH with 16 randomly selected BAC clones, to verify the improved performance of the optimum thresholds over standard thresholds. Of eight randomly selected clones that were identified by the optimum threshold but not the standard threshold as showing gain, six were correctly classified by the optimum threshold, as confirmed by BAC-FISH analysis. All eight randomly selected clones identified by the optimum threshold but not the standard threshold as showing loss were correctly classified by the optimum threshold (Figure 4).
Figure 4.

Validation of optimized thresholds on additional array CGH data derived from the hypertriploid cervical SCC cell lines CaSki and SW756. Standard thresholds miss true single copy gains and losses detected by optimized thresholds and confirmed by FISH. Optimum thresholds (blue lines) call single copy changes of additional clones (red dots) classified as normal by the standard thresholds (green lines). Arrows in black or blue indicate the locations of clones selected for FISH validation that were correctly or wrongly classified by the optimum thresholds respectively. A: Log2 ratio profile of chromosome 2 in CaSki. The optimum loss threshold (blue line) but not the standard loss threshold (green line) detects a single copy loss of clone RP11-114I18 located on 2q. FISH on a metaphase spread of CaSki confirms 1-copy loss (three copies) of clone RP11-114I18 (red) and four copies of the control clone RP11-526G13 (green). Arrows indicate the locations of clones (black dots) for which FISH confirmed that classification of loss using the optimum threshold was correct. B: Log2 ratio profile of chromosome 7 in CaSki. The optimum gain threshold (blue line) but not the standard gain threshold (green line) detects a single copy gain of clone RP11-82A15. FISH on a metaphase spread of CaSki confirms 1-copy gain (five copies) of clone RP11-82A15 (green) and four copies of the control clone RP11-31N8 (red). Arrows indicate the locations of clones (cyan dots) selected for FISH validation. C: Log2 ratio profile of chromosome 11 in SW756. The optimum loss threshold (blue line) but not the standard loss threshold (green line) detects a single copy loss of clone RP11-307I15. FISH on a metaphase spread of SW756 confirms 1-copy loss (three copies) of clone RP11-307I15 (red) and four copies of control clone RP11-509D23 (green). Arrows indicate the locations of clones (black dots) selected for FISH validation. D: Log2 ratio profile of chromosome 7 in SW756. The optimum gain threshold (blue line) but not the standard gain threshold (green line) detects a single copy gain of clone RP11-008H7. FISH on a metaphase spread of SW756 confirms 1-copy gain (five copies) of clone RP11-008H7 (red) and four copies of the control clone RP11-81B20 (green). Arrows indicate the locations of clones (cyan dots) selected for FISH validation.
Application to Primary Tumor Data
In a preliminary investigation to examine the applicability of our findings to clinical samples, we examined array CGH data generated from a frozen primary cervical SCC sample that was previously shown by interphase FISH to be hypertriploid or tetraploid. We assessed the performance of thresholds optimized for hypertriploid cell lines using our approach, together with standard thresholds determined from normal-normal hybridizations, in calling gains and losses. To test the accuracy of gains detected using optimized thresholds, we performed interphase FISH analysis on formalin-fixed paraffin-embedded tissue from the same SCC using two randomly selected BAC clones from chromosome 18 that showed gain (Figure 5). One clone (RP11-439A16) was gained at high amplitude and was called using both optimum and standard thresholds, whereas the other (RP11-45M11) was gained at lower amplitude and called using the optimum threshold only. FISH indicated that the additional call made using the optimum threshold was correct, enabling detection of a region of gain that would have been missed using the standard threshold.
Figure 5.

Comparison of optimized thresholds with standard thresholds when applied to array CGH data generated from a frozen primary cervical SCC sample known to be hypertriploid or tetraploid. The gain threshold optimized to hypertriploid samples was 1.04 whereas the standard +3 SD gain threshold was 1.18. The ratio profile of chromosome 18, together with interphase FISH on cells of the same SCC sample, are illustrated. The optimum gain threshold detects gain of both clones RP11-45M11 and RP11-439A16, whereas the standard gain threshold fails to detect gain of RP11-45M11. FISH on interphase nuclei confirms gain of RP11-45M11 (red) as well as RP11-439A16 (green).
Discussion
Array CGH is a powerful screening tool for the detection of copy number alterations in cell lines and tissue samples. Our survey of array CGH articles published in the year 2005 confirmed that threshold-based analysis is in most common use and showed that gain and loss thresholds were either set arbitrarily or selected based on ratio profiles obtained from normal-normal hybridizations. Despite being widely used, the accuracy of such standard thresholds in detecting SCAs in cell lines of different ploidy has not previously been evaluated. Fluorescence intensity ratio profiles of cells with nondiploid genomes typically have closely spaced ratio levels, and the application of fixed standard thresholds to these samples may not be ideal. Our investigation into the detection power of threshold-based array CGH for SCAs in cell lines of various ploidies demonstrates weaker performance at higher ploidies. Because inappropriate thresholds will compromise the sensitivity of detecting SCAs, this observation highlights the importance of accurate selection of thresholds that are relevant to the sample ploidy in array CGH screening studies.
Mohapatra and colleagues4 describe a simple model of the relationship between CGH ratios and copy number data determined by FISH measurements. Ideally, this relationship is given by:
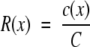 |
1 |
where R(x) is the CGH ratio at location x in the genome, c(x) is the FISH copy number at that location, and C is the average copy number for the genome or the ploidy of the sample (Figure 6). As copy number alterations will therefore give rise to CGH ratios that depend on sample ploidy, gain and loss thresholds that are chosen should take ploidy into account.
Figure 6.

Ideal relationship between CGH ratios and actual copy numbers determined by FISH for samples of different ploidies.
Before copy number assignment, several data-processing approaches have been proposed to segment the raw intensity ratio values obtained from an array CGH experiment into sets with the same copy number. Willenbrock and colleagues10 compared segmentation-based and threshold-based approaches for identifying copy number alterations (although effects of sample ploidy on the latter were not addressed). Of the segmentation-based approaches, a nonparametric change-point method (DNAcopy)11 was found to have the best operational characteristics in terms of sensitivity. Although there was a substantial difference in the proportions of clones declared to be altered between segmentation-based and threshold-based approaches (33 versus 5%, respectively), our current data suggests that the use of nonploidy-specific thresholds on paraffin-embedded samples with high heterogeneity and experimental noise may have impaired the performance of the threshold-based method used by Willenbrock and colleagues.10
Segmentation methods of array CGH data analysis generally assume that copy number changes involve chromosome segments, such that the ratios of contiguous loci should be identical, except at transitions to another level. Although the assumption facilitates breakpoint identification and noise reduction, it necessarily limits the abilities of these methods to detect single clone aberrations that may be highly informative.12 In contrast to threshold-based approaches (where every clone is considered), a change that affects a single clone cannot reliably be evaluated with segmentation algorithms, and an aberrant clone is either ignored in the state assignments or assigned to the same state as its neighboring clones. This may lead to loss of data when true single clone changes are smoothed to the level of the surrounding clones. This limitation is likely to be of greatest significance in lower-density arrays (eg, 1 MB arrays) in which each clone represents a substantial genomic region, rather than higher density (eg, tiling path) arrays in which each genomic region is represented by a large number of clones and changes are likely to be supported by more than a single clone.
In the present study, we therefore investigated the optimal thresholds relevant to sample ploidy for use in array CGH. Such threshold-based analysis has no prior need for data segmentation. In our study, we used segmentation only to define accurately genomic segments showing SCAs for subsequent identification of optimized thresholds. We had not compared the performance of threshold-based approaches with segmentation-based approaches. A further motivation for seeking to improve the accuracy of threshold-based methods is that the implementation of complex segmentation algorithms to large array CGH data sets incurs high computational overheads and typically requires dedicated software that may not be readily available and/or easy for biologists to use. Significantly, threshold-based approaches are in widespread use in biological studies (Table 1) and are worthy of further methodological investigations.
In this study, we describe a method to evaluate the accuracy of thresholds for detecting SCAs in array CGH data from cell lines and use it to identify optimal thresholds relevant to sample ploidy. Using array CGH data from cell lines of known ploidy, we compared the performance of the optimized thresholds with that of standard thresholds at calling single copy gains and losses. For diploid and triploid cell lines, the use of standard thresholds, instead of the optimized ones, generally resulted in a decrease in sensitivity and increase in specificity, with minimal change to the overall error rate. However, because array CGH frequently serves as a first-round screening tool, used to highlight recurrent genomic changes in large sample sets for further confirmatory testing, the choice of the more sensitive optimized thresholds would generally be preferable.
The standard thresholds had the poorest detection accuracy with the hypertriploid cell line. The application of standard gain and loss thresholds resulted in large error rates of 83 and 67%, respectively, which were principally attributable to the high false-negative calls. The use of the stringent standard thresholds resulted in the failure to detect a large proportion of single copy changes. This is in agreement with Equation 1, which implies that thresholds applied to samples of higher ploidy must necessarily be less stringent to detect the smaller magnitude ratio change produced by SCAs.
To validate the improved performance of thresholds optimized for hypertriploid cell lines over standard thresholds, we applied the optimized thresholds to additional array CGH data from two hypertriploid cell lines. Six of eight randomly selected clones identified as gained by the optimum threshold but not the standard threshold were verified by FISH to be correctly classified by the optimized threshold. All of eight randomly selected clones classified as lost by the optimum threshold but not the standard threshold were confirmed by FISH to be lost.
Moreover, in a preliminary investigation, we used interphase FISH analysis of a cervical SCC that was previously shown to be hypertriploid or tetraploid to demonstrate that ploidy-specific thresholds also provide increased sensitivity in analyzing array CGH data from a clinical sample. We therefore suggest that the optimized thresholds derived from cell lines may have direct relevance to studies using threshold-based array CGH to screen for copy number imbalances in clinical samples. Nevertheless, the application to clinical samples requires further validation, because in addition to sample ploidy, other factors such as contamination from normal cells and tissue chimerism, are likely to impact threshold optimization.
In summary, as many neoplasms show abnormal ploidy levels, analyses of array CGH data using standard thresholds are likely to fail to detect many copy number imbalances, including those likely to be of biological significance. Our data suggest that initial analysis of the genomic content of cell lines (and possibly clinical samples) under investigation in a threshold-based array CGH study would enable selection of the most appropriate thresholds and increase sensitivity of detection of imbalances, including SCAs.
Acknowledgments
We thank Professor Barbara Weber for providing the BAC arrays used in this study.
Footnotes
Supported by the Medical Research Council; Cancer Research UK, and the Agency for Science, Technology, and Research, Singapore (National Science Scholarships to G.N. and J.H.).
References
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Fridlyand J, Pejavar S, Olshen AB, Korkola JE, DeVries S, Carroll P, Kuo WL, Pinkel D, Albertson D, Cordon-Cardo C, Jain AN, Waldman FM. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–2880. [PubMed] [Google Scholar]
- Mohapatra G, Moore DH, Kim DH, Grewal L, Hyun WC, Waldman FM, Pinkel D, Feuerstein BG. Analyses of brain tumor cell lines confirm a simple model of relationships among fluorescence in situ hybridization, DNA index, and comparative genomic hybridization. Genes Chromosom Cancer. 1997;20:311–319. doi: 10.1002/(sici)1098-2264(199712)20:4<311::aid-gcc1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EH, van Rijk A, Smeets D, van Ravenswaaij-Arts CM, Knoers NV, van der Burgt I, de Jong PJ, Brunner HG, van Kessel AG, Schoenmakers EF, Veltman JA. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CP, Lu XY, Narayan G, Singh B, Murty VV, Rao PH. Comprehensive molecular cytogenetic characterization of cervical cancer cell lines. Genes Chromosom Cancer. 2003;36:233–241. doi: 10.1002/gcc.10158. [DOI] [PubMed] [Google Scholar]
- Fridlyand J, Snijders AM, Pinkel D, Albertson DG, Jain AN. Hidden Markov models approach to the analysis of array CGH data. J Multivar Anal. 2004;90:132–153. [Google Scholar]
- Hoglund M, Johansson B, Pedersen-Bjergaard J, Marynen P, Mitelman F. Molecular characterization of 12p abnormalities in hematologic malignancies: deletion of KIP1, rearrangement of TEL, and amplification of CCND2. Blood. 1996;87:324–330. [PubMed] [Google Scholar]
- Shing DC, McMullan DJ, Roberts P, Smith K, Chin SF, Nicholson J, Tillman RM, Ramani P, Cullinane C, Coleman N. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 2003;63:4568–4576. [PubMed] [Google Scholar]
- Willenbrock H, Fridlyand J. A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics. 2004;21:4084–4091. doi: 10.1093/bioinformatics/bti677. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RC, Albertson DG. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene. 2005;24:4232–4242. doi: 10.1038/sj.onc.1208601. [DOI] [PubMed] [Google Scholar]
- Knijnenburg J, van der Burg M, Nilsson P, Ploos van Amstel HK, Tanke H, Szuhai K. Rapid detection of genomic imbalances using micro-arrays consisting of pooled BACs covering all human chromosome arms. Nucleic Acids Res. 2005;33:e159. doi: 10.1093/nar/gni161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani I, Ramona RF, Roversi G, Beghini A, Pfundt R, Schoenmakers EF, Larizza L. Identification of oligodendroglioma specific chromosomal copy number changes in the glioblastoma MI-4 cell line by array-CGH and FISH analyses. Cancer Genet Cytogenet. 2005;161:140–145. doi: 10.1016/j.cancergencyto.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Veltman I, Veltman J, Janssen I, Hulsbergen-van de Kaa C, Oosterhuis W, Schneider D, Stoop H, Gillis A, Zahn S, Looijenga L, Gobel U, van Kessel AG. Identification of recurrent chromosomal aberrations in germ cell tumors of neonates and infants using genomewide array-based comparative genomic hybridization. Genes Chromosom Cancer. 2005;43:367–376. doi: 10.1002/gcc.20208. [DOI] [PubMed] [Google Scholar]
- Rosenberg C, Knijnenburg J, Chauffaille Mde L, Brunoni D, Catelani AL, Sloos W, Szuhai K, Tanke HJ. Array CGH detection of a cryptic deletion in a complex chromosome rearrangement. Hum Genet. 2005;116:390–394. doi: 10.1007/s00439-004-1248-x. [DOI] [PubMed] [Google Scholar]
- Izumi H, Inoue J, Yokoi S, Hosoda H, Shibata T, Sunamori M, Hirohashi S, Inazawa J, Imoto I. Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non-small cell lung cancers. Hum Mol Genet. 2005;14:997–1007. doi: 10.1093/hmg/ddi092. [DOI] [PubMed] [Google Scholar]
- Takada H, Imoto I, Tsuda H, Sonoda I, Ichikura T, Mochizuki H, Okanoue T, Inazawa J. Screening of DNA copy-number aberrations in gastric cancer cell lines by array-based comparative genomic hybridization. Cancer Sci. 2005;96:100–110. doi: 10.1111/j.1349-7006.2005.00016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin S, Rickert P, Kastury K, McMahon M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosom Cancer. 2005;44:37–51. doi: 10.1002/gcc.20216. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24:1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Suguro M, Tsuzuki S, Matsuo K, Karnan S, Ohshima K, Okamoto M, Morishima Y, Nakamura S, Seto M. Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood. 2005;106:1770–1777. doi: 10.1182/blood-2005-02-0542. [DOI] [PubMed] [Google Scholar]
- Nakashima Y, Tagawa H, Suzuki R, Karnan S, Karube K, Ohshima K, Muta K, Nawata H, Morishima Y, Nakamura S, Seto M. Genome-wide array-based comparative hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK-cell leukemia and extranodal NK/T-cell lymphoma, nasal type. Genes Chromosom Cancer. 2005;44:247–255. doi: 10.1002/gcc.20245. [DOI] [PubMed] [Google Scholar]
- Reis-Filho JS, Simpson PT, Jones C, Steele D, Mackay A, Iravani M, Fenwick K, Valgeirsson H, Lambros M, Ashworth A, Palacios J, Schmitt F, Lakhani SR. Pleomorphic lobular carcinoma of the breast: role of comprehensive molecular pathology in characterization of an entity. J Pathol. 2005;207:1–13. doi: 10.1002/path.1806. [DOI] [PubMed] [Google Scholar]
- Coe BP, Henderson LJ, Garnis C, Tsao MS, Gazdar AF, Minna J, Lam S, Macaulay C, Lam WL. High-resolution chromosome arm 5p array CGH analysis of small cell lung carcinoma cell lines. Genes Chromosom Cancer. 2005;42:308–313. doi: 10.1002/gcc.20137. [DOI] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, Kashork CD, Shaffer LG. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- van Duin M, van Marion R, Watson JE, Paris PL, Lapuk A, Brown N, Oseroff VV, Albertson DG, Pinkel D, de Jong P, Nacheva EP, Dinjens W, van Dekken H, Collins C. Construction and application of a full-coverage, high-resolution, human chromosome 8q genomic microarray for comparative genomic hybridization. Cytometry A. 2005;63:10–19. doi: 10.1002/cyto.a.20102. [DOI] [PubMed] [Google Scholar]
- Hughes S, Damato BE, Giddings I, Hiscott PS, Humphreys J, Houlston RS. Microarray comparative genomic hybridisation analysis of intraocular uveal melanomas identifies distinctive imbalances associated with loss of chromosome 3. Br J Cancer. 2005;93:1191–1196. doi: 10.1038/sj.bjc.6602834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada H, Imoto I, Tsuda H, Nakanishi Y, Ichikura T, Mochizuki H, Mitsufuji S, Hosoda F, Hirohashi S, Ohki M, Inazawa J. ADAM23, a possible tumor suppressor gene, is frequently silenced in gastric cancers by homozygous deletion or aberrant promoter hypermethylation. Oncogene. 2005;24:8051–8060. doi: 10.1038/sj.onc.1208952. [DOI] [PubMed] [Google Scholar]
- Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RC, Albertson DG. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene. 2005;24:4232–4242. doi: 10.1038/sj.onc.1208601. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Hollanders K, Badisco L, Melotte C, Van Hummelen P, Vermeesch JR, Devriendt K, Fryns JP, Marynen P, Froyen G. Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum Mol Genet. 2005;14:1795–1803. doi: 10.1093/hmg/ddi186. [DOI] [PubMed] [Google Scholar]
- Davison EJ, Tarpey PS, Fiegler H, Tomlinson IP, Carter NP. Deletion at chromosome band 20p12.1 in colorectal cancer revealed by high resolution array comparative genomic hybridization. Genes Chromosom Cancer. 2005;44:384–391. doi: 10.1002/gcc.20252. [DOI] [PubMed] [Google Scholar]
- Rubio-Moscardo F, Climent J, Siebert R, Piris MA, Martin-Subero JI, Nielander I, Garcia-Conde J, Dyer MJ, Terol MJ, Pinkel D, Martinez-Climent JA. Mantle-cell lymphoma genotypes identified with CGH to BAC microarrays define a leukemic subgroup of disease and predict patient outcome. Blood. 2005;105:4445–4454. doi: 10.1182/blood-2004-10-3907. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Reardon W, Rosser EM, Lacombe D, Hurst JA, Law CJ, Bongers EM, van Ravenswaaij-Arts CM, Leisink MA, van Kessel AG, Veltman JA, de Vries BB. Molecular characterisation of patients with subtelomeric 22q abnormalities using chromosome specific array-based comparative genomic hybridisation. Eur J Hum Genet. 2005;13:1019–1024. doi: 10.1038/sj.ejhg.5201456. [DOI] [PubMed] [Google Scholar]
- de Stahl TD, Hartmann C, de Bustos C, Piotrowski A, Benetkiewicz M, Mantripragada KK, Tykwinski T, von Deimling A, Dumanski JP. Chromosome 22 tiling-path array-CGH analysis identifies germ-line- and tumor-specific aberrations in patients with glioblastoma multiforme. Genes Chromosom Cancer. 2005;44:161–169. doi: 10.1002/gcc.20226. [DOI] [PubMed] [Google Scholar]
- Ammerlaan AC, de Bustos C, Ararou A, Buckley PG, Mantripragada KK, Verstegen MJ, Hulsebos TJ, Dumanski JP. Localization of a putative low-penetrance ependymoma susceptibility locus to 22q11 using a chromosome 22 tiling-path genomic microarray. Genes Chromosom Cancer. 2005;43:329–338. doi: 10.1002/gcc.20207. [DOI] [PubMed] [Google Scholar]
- Callagy G, Pharoah P, Chin SF, Sangan T, Daigo Y, Jackson L, Caldas C. Identification and validation of prognostic markers in breast cancer with the complementary use of array-CGH and tissue microarrays. J Pathol. 2005;205:388–396. doi: 10.1002/path.1694. [DOI] [PubMed] [Google Scholar]
- Tsubosa Y, Sugihara H, Mukaisho K, Kamitani S, Peng DF, Ling ZQ, Tani T, Hattori T. Effects of degenerate oligonucleotide-primed polymerase chain reaction amplification and labeling methods on the sensitivity and specificity of metaphase- and array-based comparative genomic hybridization. Cancer Genet Cytogenet. 2005;158:156–166. doi: 10.1016/j.cancergencyto.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Schleiermacher G, Bourdeaut F, Combaret V, Picrron G, Raynal V, Aurias A, Ribeiro A, Janoueix-Lerosey I, Delattre O. Stepwise occurrence of a complex unbalanced translocation in neuroblastoma leading to insertion of a telomere sequence and late chromosome 17q gain. Oncogene. 2005;24:3377–3384. doi: 10.1038/sj.onc.1208486. [DOI] [PubMed] [Google Scholar]
- Le Caignec C, De Mas P, Vincent MC, Boceno M, Bourrouillou G, Rival JM, David A. Subtelomeric 6p deletion: clinical, FISH, and array CGH characterization of two cases. Am J Med Genet A. 2005;132:175–180. doi: 10.1002/ajmg.a.30409. [DOI] [PubMed] [Google Scholar]
- Wakui K, Gregato G, Ballif BC, Glotzbach CD, Bailey KA, Kuo PL, Sue WC, Sheffield LJ, Irons M, Gomez EG, Hecht JT, Potocki L, Shaffer LG. Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki-Shaffer syndrome. Eur J Hum Genet. 2005;13:528–540. doi: 10.1038/sj.ejhg.5201366. [DOI] [PubMed] [Google Scholar]
- Kawaguchi K, Honda M, Yamashita T, Shirota Y, Kaneko S. Differential gene alteration among hepatoma cell lines demonstrated by cDNA microarray-based comparative genomic hybridization. Biochem Biophys Res Commun. 2005;329:370–380. doi: 10.1016/j.bbrc.2005.01.128. [DOI] [PubMed] [Google Scholar]