

Abstract
Objective:
The aim of this study was to assess the influence of severe steatosis with inflammation on hepatocellular recovery after 70% hepatectomy in a rat model of diet-induced steatosis.
Background:
Patients with steatosis have an increased risk of inflammatory complications after liver resection. This might be attributable to Kupffer cell-mediated inflammation in steatotic livers causing progressive injury.
Methods:
Male Wistar rats were fed a standard methionine- and choline-deficient diet for 1 or 5 weeks. A 70% partial hepatectomy (PH) was performed, after which rats were killed at 24, 48, or 72 hours. The extent of steatosis and inflammation was determined by assessment of hepatic triglycerides, cytokine content, and histopathology. Outcome parameters were: liver regeneration (MIB-5 proliferation rate, mitotic index, and regenerating liver mass), hepatocellular injury (plasma aminotransferases, lipid peroxidation, histopathology, and apoptosis), Kupffer cell-mediated proinflammatory response (TNF-α, IL-1β, IL-6, IL-10 in plasma and liver) and antioxidant content (total glutathione).
Results:
Methionine- and choline-deficient diet induced uncomplicated steatosis after 1 week (<30% hepatocytes affected without inflammation) and severe steatosis after 5 weeks (>60% hepatocytes affected, including prominent inflammation) as confirmed by histopathology. After PH, liver regeneration was impaired at all time points in the severe steatosis group as compared with the mild and control groups (P < 0.05). Hepatocellular injury was significantly increased in the severe steatosis group at all time points (P < 0.05). Kupffer cell-mediated inflammatory responses were aggravated in the severe steatosis group along with decreased antioxidant content (P < 0.05). Necrosis was the main type of cell death in severe steatotic livers compared with mainly apoptotic cell death in mild steatotic and normal livers.
Conclusion:
Steatosis with prominent inflammation impaired liver regeneration probably because of increased hepatocellular lipid peroxidation and damage in concert with Kupffer cell-mediated proinflammatory responses. These results suggest an increased risk of performing extensive liver resection in the presence of severe steatosis.
Steatosis is associated with an increased risk of complications after liver resection. The aim of this study was to assess the influence of severe steatosis on hepatocellular recovery after 70% hepatectomy in a rat model of diet-induced steatosis. Steatosis with prominent inflammation impaired liver regeneration and was related to increased hepatocellular lipid peroxidation and damage in concert with Kupffer cell-mediated proinflammatory responses.
Liver resections have become safer in the past 10 years, largely owing to improvements in preoperative diagnosis, surgical techniques, and postoperative care.1–3 Consequently, overall postoperative mortality has decreased to below 5% correlating directly with preoperative liver function and resected liver volume. Function of the remnant liver rapidly recovers in patients with normal liver parenchyma as hepatocytes proliferate to restore the loss of volume. However, in the presence of parenchymal liver disease, hepatocellular proliferation is impaired, exposing patients to liver dysfunction and associated complications, culminating into posthepatectomy liver failure, which has a high mortality (60%–90%).
Currently, hepatic fat accumulation, ie, steatosis, is the most common parenchymal liver disease in the Western world, affecting 20% of individuals in a general population and up to 95% among obese.4 As steatosis is closely related to obesity, dyslipidemia, and diabetes, the prevalence will increase as a consequence of the epidemic obesity in the Western population. Steatosis is classified by histopathology into 3 categories according to the extent of fat infiltration: mild/uncomplicated steatosis (<30% of hepatocytes affected, without inflammatory component), moderate (30%–60% of hepatocytes affected), and severe steatosis (>60% of hepatocytes affected).5 Furthermore, progressive inflammation and eventually fibrogenesis are observed throughout the development of steatosis.
Even though there are only few clinical studies evaluating the impact of steatosis on postoperative recovery after liver resection, any grade of steatosis is associated with increased postoperative morbidity. The steatosis severity grade has been correlated with increased infectious complications and increased postoperative mortality.6,7 Although the mechanisms of impaired liver recovery in steatotic patients are largely unclear, the underlying pathologic derangements together with disturbed liver regeneration have been implicated as potential factors.8 In fatty hepatocytes, intracellular fatty acids promote the generation of reactive oxygen species (ROS) increasing mitochondrial oxidative stress and lipid peroxidation.8a In addition, steatotic livers have impaired antioxidant scavenging of free radicals causing ROS accumulation and consequently the activation of hepatic macrophages, ie, the Kupffer cells (KC). KCs produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β, further exacerbating hepatocellular damage in steatotic livers.8a Specific knowledge of the mechanisms involved in the hepatocellular damage incurred in steatotic livers would enable development of interventional strategies to improve postoperative outcome of patients with liver steatosis.
We hypothesize that preoperatively increased lipid peroxidation and impaired antioxidant response aggravate KC-mediated inflammatory responses and hepatocellular injury in steatotic livers after partial hepatectomy (PH) and that liver regeneration under these circumstances is impaired. We therefore assessed the effects of mild and severe steatosis on hepatocellular recovery after PH in a rat model of diet-induced liver steatosis.
MATERIALS AND METHODS
Hepatic steatosis was induced using a standard MCD diet (Harlan Teklad, Madison WI). Male Wistar rats (250–300 g) (Harlan CPB, Zeist, The Netherlands) were acclimatized at least for 7 days to laboratory conditions, maintained at constant temperature of 24°C with 12 hours light-dark cycle, and fed a standard rodent chow (Harlan, Zeist, The Netherlands) and water ad libitum. After acclimatization, rats (n = 30) were fed the MCD diet for 1 or 5 weeks ad libitum. The control group (n = 15) received a standard chow with adequate methionine levels ad libitum. Two groups without surgery were used as baseline controls: a group (n = 5) fed with standard chow and a group (n = 5) fed with MCD diet. During all procedures, the animals were treated according to the guidelines of the Dutch legislation and international standards for animal care and handling. The protocol was approved by the Animal Ethics Committee of the University of Amsterdam, The Netherlands.
After a period of 1 or 5 weeks of diet, laparotomy was performed under inhalation anesthesia using a mixture of 02/N20 (1:1) and isoflurane (1%–2% Florene, Abbott Laboratories Ltd., Queensborough, UK) and pain medication (buprenorphine, Temgesic i.v. 0.033 mg/0.1 kg); 70% partial hepatectomy consisting of resection the median and left lateral liver lobes was performed according to the method described by Higgins and Anderson.9 After 24, 48, and 72 hours, respectively, rats were killed. Blood was collected by vena cava puncture, centrifuged (10 minutes, 3000g, 4°C) and plasma was stored at −80°C. Livers were removed and weighed, and thin slices of all lobes were immersed in 10% formalin and embedded in paraffin; 4-μm sections were routinely stained with hematoxylin and eosin for assessment of morphology and with Sirius red (0,1% Fast red in picric acid, Immunotech, Netherlands) for collagen deposition.
Assessment of Hepatic Steatosis
Liver samples were homogenized in phosphate buffered saline (pH 7.2) and centrifuged (4000g, 10 minutes, 4°C). Hepatic lipids were extracted by the chloroform-methanol extraction method and lipid levels were measured enzymatically using commercial kits (Trig/GB, Roche, Switzerland and Cholesterol, Biomerieux, Boxtel, Netherlands). Protein concentration was measured using BCA Protein Assay kit (Pierce, Rockford, IL) and lipid levels were expressed as mg/g total protein. Histopathologic features of hepatic steatosis were evaluated by 2 blinded investigators using a semi-quantitative, histopathologic score adapted from the recently accepted AASLD criteria for steatosis staging in hematoxylin and eosin-stained sections and for detection of fibrosis, in Sirius red stained sections using light microscopy in 30 high power fields (HPF) (magnification ×40).5
Assessment of Liver Regeneration
Three independent markers for hepatic regeneration were used as follows: restitution of liver weight was expressed as percentage of regenerated liver mass relative to total liver weight as previously described10; % = [C − (A − B)]/A × 100%, where A = estimated total liver weight at the time of partial hepatectomy, B = resected liver weight, and C = the weight of the regenerated liver at sacrifice.10 For assessment of hepatic proliferation, MIB-5, a rat equivalent of Ki-67 antibody was used, which detects all active parts of the cell cycle. The MIB-5 index has a strong positive correlation with proliferating antigen expression, bromodeoxyuridine incorporation, and thymidine incorporation.11 Briefly, sections were deparaffinized and pretreated (citric acid pH 6.0, 2 bar, 120°C, 20 minutes). For primary and secondary antibody, MIB-5 antibody (dilution 1:50, 60 minutes, DAKO Cytomation, Copenhagen, Denmark) and Poly-HRP (dilution 1:1, 30 minutes, Invitrogen, Carlsbad, CA), were used, respectively. The mitotic index was determined in hematoxylin and eosin-stained sections using previously reported criteria for mitosis as follows: complete absence of cell membrane, slight eosinophilic staining of nucleus, nuclear spindle matrix formation, absence of a nucleolus, and slight increase in cell size.12 All indexes were determined by 2 blinded investigators (magnification ×40, 30 HPF) and expressed as the rate of positive cells per 1000 hepatocytes/HPF.
Hepatocellular Necrosis and Apoptosis
Plasma samples were analyzed for aspartate aminotransferase (AST), alanine aminotransferase (ALT), and for albumin (the major hepatic plasma protein product) in the Department of Clinical Chemistry (AMC) using standard laboratory methods.
Histopathologic injury was evaluated by 2 blinded investigators in hematoxylin and eosin-stained sections in 10 HPF at magnification ×40, by a point-counting method using an ordinal scale as reported previously: grade 0, minimal or no evidence of injury; grade 1, mild injury consisting of cytoplasmic vacuolization and focal nuclear pyknosis; grade 2, moderate to severe injury with extensive nuclear pyknosis, cytoplasmic hypereosinophilia, and loss of intercellular borders; and grade 3, severe necrosis with disintegration of hepatic cords, hemorrhage, and neutrophil infiltration.13,14 Also, for determination of apoptotic cells, cleaved caspase-3 antibody detecting endogenous large fragment of activated caspase-3 was used. Briefly, 4-μm sections were deparaffinized and pretreated (citric acid, pH 6.0, 2 bar, 120°C, 10 minutes). For primary and secondary antibody, a cleaved caspase-3 (dilution 1:200, 60 minutes, Cell Signaling Technology, Frankfurt, Germany) and Poly-HRP (dilution 1:1, 30 minutes, Invitrogen), respectively, were used and positive cells were determined in 20 HPF at magnification of ×80 by 2 blinded investigators.
Hepatic and Systemic Proinflammatory Cytokine Response
Sections were incubated with an ED-1 antibody against the lysosomal membrane glycoprotein on resident KC and circulatory macrophages (dilution 1:100, 60 minutes, Serotec, Humbeek, Belgium). After incubation with a secondary antibody (1:100, 60 minutes, GAM-IgG1, Southern Biotechnologies, Birmingham, AL), Fast DAB (Sigma chemical, Munich, Germany) was used to visualize the peroxidase complexes. Macrophages were counted (30 HPF, magnification of 25) by 2 blinded investigators.
A bioplex kit for determination of plasma and hepatic levels of KC-mediated cytokine's TNF-α, IL-1β, IL-6, and IL-10 was used (Biorad, Hercules). Samples of all regenerating liver lobes were homogenized (NaPi buffer 5 mmol/L, pH 6.0), centrifuged (10,000g, 4°C, 10 minutes) and the supernatants were used in the cytokine assay. The samples were analyzed according to manufacturer's instructions and concentrations were expressed as pg/mg protein.
Assessment of Hepatic Lipid Peroxidation and Antioxidant Response
Antioxidant response was assessed by measurement of hepatic total glutathione (tGSH) concentrations as follows: samples were homogenized in buffer (MPA, pH 6.0), centrifuged (4000g, 10 minutes, 4°C) and supernatants were analyzed for tGSH. Lipid peroxidation was assessed by hepatic malondialdehyde (MDA) levels. MDA was measured as free thiobarbituric acid reactive substances (TBARS) in the presence of the antioxidant butylated hydrotoluene that limits the generation of new TBARS during the assay. The hepatic tGSH and TBARS concentrations were expressed as mmol/mg total protein.
Statistical Analysis
Data analysis was performed with GraphPad Prism 3.02 for Windows (GraphPad Software Inc., San Diego, CA). The results are presented as mean ± SEM. Significant differences between groups were tested using ANOVA one-way analysis and Mann-Whitney's U test followed by Bonferroni post test. P values less than 0.05 were considered significant.
RESULTS
Induction of Steatosis
In control rats, no pathologic changes were seen (Fig. 1A). After 1 week of MCD diet, steatosis mainly consisted of mild microvesicular steatosis with scattered macrovesicular hepatocytes (Fig. 1B). After 5 weeks of MCD diet, steatosis consisted of extensive macrovesicular steatosis (>60% hepatocytes affected) with foci of inflammatory cells along with increased fibrosis in the portal areas (Fig. 1C). Corresponding with these histopathologic features of steatosis, a 10-fold increase in hepatic triglyceride content was seen after 1 week and a 20-fold increase after 5 weeks of MCD diet (vs. controls, P < 0.01, Fig. 1D).

FIGURE 1. Micrograph of liver tissue of control rat (A) showing no pathologic changes. After a 1-week MCD diet (B), mainly microvesicular steatosis with occasional macrovesicular foci and no inflammatory cells were observed. After 5 weeks of MCD diet (C), mainly macrovesicular steatosis and prominent inflammation were observed. All slides stained with hematoxylin and eosin (original magnification ×20). The histopathologic changes corresponded with increased hepatic triglycerides content (D). Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatosis group.
Liver regeneration is impaired in the presence of severe steatosis after 70% PH. After 70% PH, the increase in regenerating liver mass was lowest in the severe steatosis group at 24, 48, and 72 hours, as compared with the mild steatosis and control groups (P < 0.05) (Fig. 2A). Also, hepatocyte proliferation (MIB-5 index) was lower at all time points in the severe steatosis group, as compared with the mild steatosis and control groups (P < 0.05) (Fig. 2B). The mitotic index results followed the pattern of MIB-5 index in all groups at all time points (data not shown). Between the control and mild steatosis groups, no differences were observed at all time points. There were no differences in preoperative mitotic or MIB-5 indexes between the groups (data not shown).
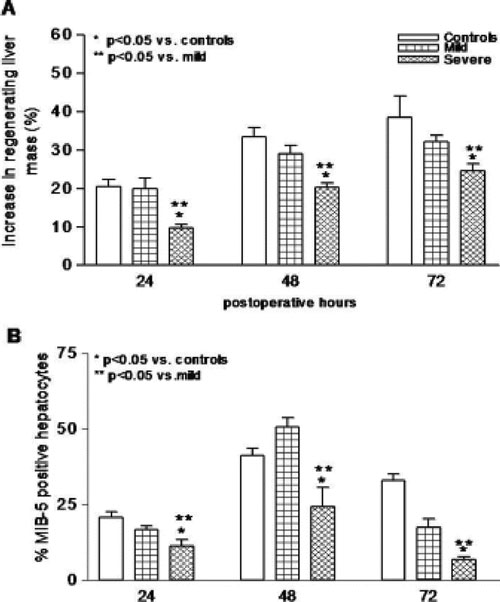
FIGURE 2. Liver regeneration after PH was evaluated by measuring increase in postoperative liver mass (A) and hepatocyte proliferation index (B). A decreased response was seen in the severe steatotic rats with respect to both parameters in contrast to no differences between the mild steatotic and control rats. Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatosis group.
Hepatocellular damage is increased in severe steatosis after 70% PH. Plasma ALT and AST were significantly increased preoperatively in the severe steatosis group (P < 0.05), indicating increased hepatocellular damage. After PH, AST, and ALT were significantly elevated in the severe steatosis group at all time points as compared with the mild steatosis and control groups (P < 0.05) (Fig. 3A, B). Concerning the mild steatosis group, AST and ALT were increased at 24 hours as compared with the controls (P < 0.05), but thereafter no differences were observed.
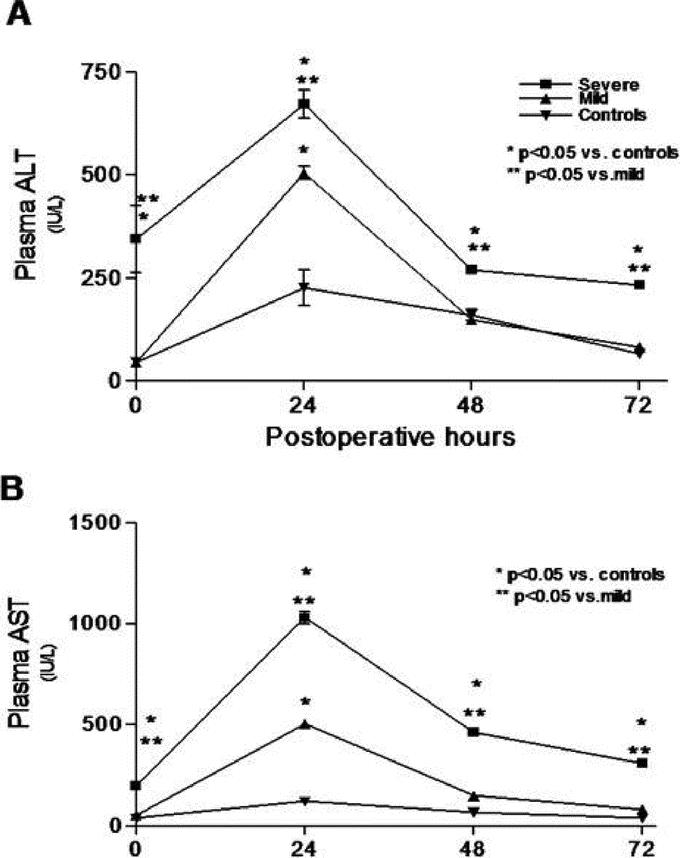
FIGURE 3. Hepatocellular damage after PH assessed by plasma ALT (A) and AST (B) was most increased in the severe steatotic rats. Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatosis group.
The histopathology scores corresponded with the plasma markers and was increased at all time points in the severe steatosis group compared with the mild steatosis and control groups (P < 0.01) (Fig. 4A). The initial hepatocellular damage in the severe steatosis group was mainly due to necrosis as no increase in the apoptotic cells was seen at 24 hours after PH (Fig. 4B). However, the rate of apoptotic hepatocytes remained increased at 48 hours and 72 hours in the severe steatosis group compared with the control and mild steatosis groups (P < 0.01). In the mild steatosis group, apoptosis was slightly increased at 48 hours compared with the controls (P < 0.05).
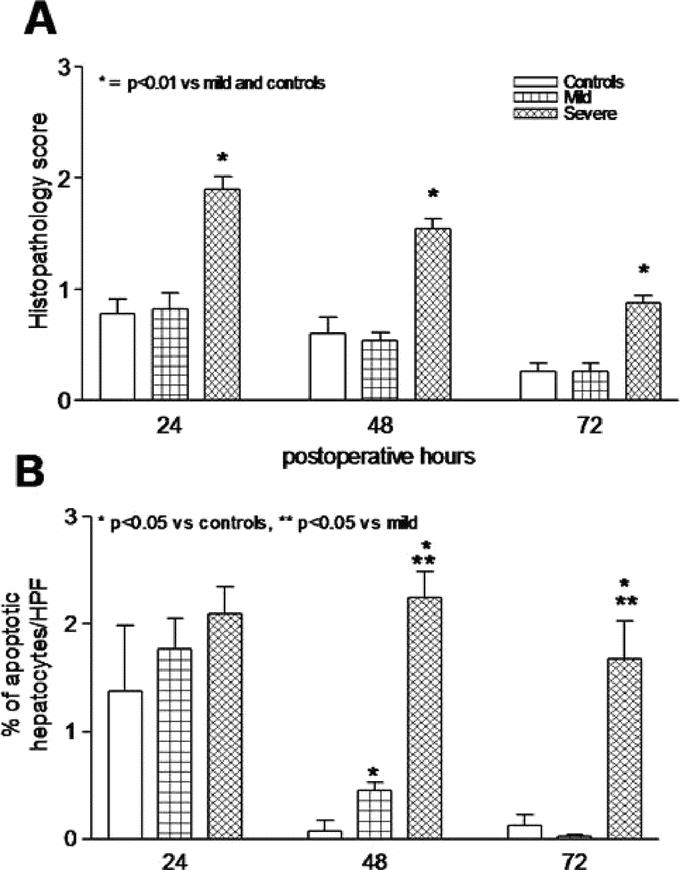
FIGURE 4. Histopathology scores in hematoxylin and eosin-stained liver tissue (A) after PH was significantly increased in severe steatotic rats compared with no changes between the control and mild steatotic rats (P < 0.01). The rate of apoptotic cells (B) remained increased in the severe steatotic rats compared with decreasing rates in the control and mild steatotic rats after PH. Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatosis group.
Severe steatosis increases lipid peroxidation and decreases antioxidant response after 70% PH. Hepatic TBARS was significantly increased preoperatively in the severe steatosis group compared with the mild steatosis and control groups and remained increased postoperatively as compared with the control group. When compared with the mild steatosis group, hepatic TBARS was significantly higher at 24 hours and 48 hours in the severe steatosis group (P < 0.01) (Fig. 5A). Hepatic levels of the antioxidant, total glutathione, were significantly increased in the mild steatosis group as compared with the control and severe steatosis groups at 24 hours and 48 hours postoperatively (P < 0.01). In the control group, no significant changes were detected postoperatively (Fig. 5B).
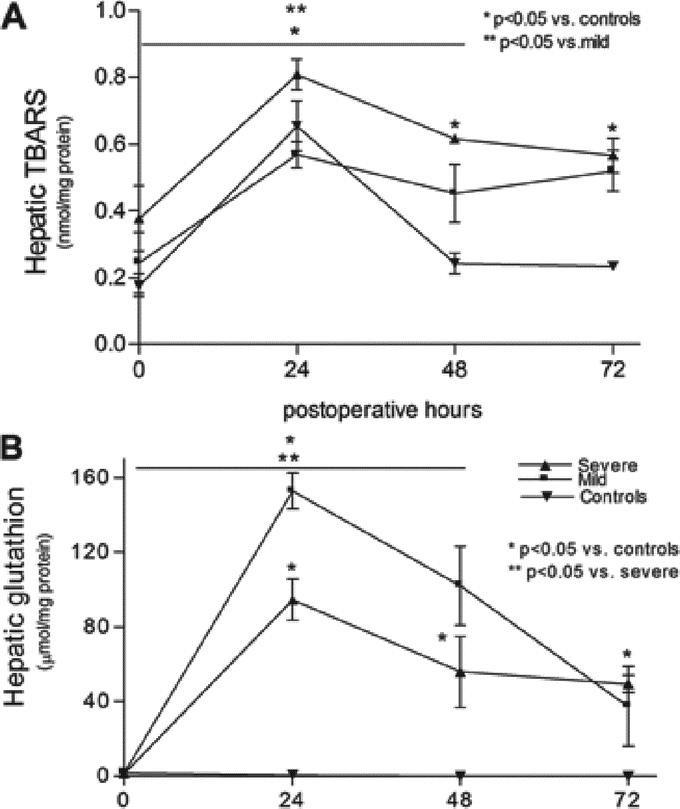
FIGURE 5. Effect of severe and mild steatosis on hepatic lipid peroxidation after PH as measured by free TBARS (A) and hepatic total GSH response (B). Hepatic TBARS was increased in the severe steatotic rats after PH and hepatic GSH was increased in the mild steatotic rats compared with severe steatotic and control rats. Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatotic group.
Severe steatosis increases Kupffer cell release of proinflammatory cytokines after 70% PH. Plasma IL-6, IL-10, and TNF-α were significantly higher in the severe steatosis group at 24 hours after PH compared with the control and mild steatosis groups (P < 0.01) (Fig. 6A), whereas plasma IL-10 levels were increased in the mild steatosis group compared with controls (P < 0.05). Also at 24 hours, plasma IL-1β was significantly increased in the severe steatosis group (30.4 ± 6.5 ng/mL, P < 0.01) compared with undetectable levels in the mild steatosis and control groups (P < 0.01, detection limit 5 ng/mL, data not shown). There were no differences in preoperative plasma cytokine levels between the groups (data not shown).

FIGURE 6. Kupffer cell-mediated inflammatory response was increased in severe steatotic rats as assessed by plasma (A) and hepatic cytokines (B) and by the amount of hepatic resident and circulatory macrophages (C–E, ED-1-positive macrophages in the control, mild steatosis, and severe steatosis groups, respectively). Values are mean ± SEM. *P < 0.05 versus controls. **P < 0.05 versus the mild steatotic group.
Also hepatic IL-1β, IL-6, Il-10, and TNF-α were significantly increased the severe steatosis group compared with the mild and control groups at all time points (P < 0.001, Fig. 6B, IL-10 and IL-1β, data not shown). The number of hepatic macrophages was significantly increased at all time points in the severe steatosis group compared with mild and control groups (macrophages per HPF: in the severe at 24, 48, and 72 hours, respectively; 90 ± 8, 218 ± 51, and 162 ± 53, in mild 46 ± 8, 20 ± 5 and 10 ± 3, P < 0.05) (Fig. 6C–E).
DISCUSSION
Severe steatosis had a negative impact on hepatocyte proliferation and on regeneration of remnant liver mass after 70% PH. In rats with normal liver parenchyma or mild steatosis without inflammation, no differences in the regenerative capacity of the remnant liver were detected. In contrast, rats with severe steatosis showed an excessive proinflammatory cytokine response and insufficient antioxidant response affecting liver regeneration and increasing hepatocellular damage after PH.
In experimental models of parenchymal liver injury and KC-mediated inflammation, liver regeneration is impaired after resection.15–17 However, the influence of chronic KC-activation in combination with steatosis is unclear. To date, experimental studies have mainly applied steatosis models based either on genetic leptin mutation (Zucker rats, ob/ob mice) or on choline deficiency in which the development of prominent and progressive inflammation as seen in human steatosis lacks.18,19 A period of 1 week MCD diet was chosen to induce mild steatosis without inflammatory components since activation of hepatic inflammation is already observed after 2 weeks of this diet.20 A 5-week diet period was applied to induce severe steatosis with prominent inflammation, however, without extensive fibrosis.20 We speculate that these changes can resolve if the rats would be returned to normal chow. However, this might not be the case if irreversible changes, ie, severe fibrosis or cirrhosis have developed. This model, because of its inflammatory component, might also be useful for investigating the upcoming, major clinical problem of chemotherapy induced steatohepatitis. However, only little is still known about this subject and more studies are urgently needed.21
Mild steatosis had no effect on liver regeneration as has been reported in previous studies.22,23 However, in severely steatotic rats, both the restoration of liver mass and hepatocyte proliferation were impaired.22,23 In contrast to our results, Rao et al22 described an unaffected regenerative response in steatotic rats with inflammation.19 However, in their study only histopathology was used to assess hepatic changes and the steatosis model applied in their study was based on a choline-deficient diet, which is considered mainly a model of uncomplicated steatosis. In similar studies using MCD diet, a delayed but not impaired liver regeneration in the presence of moderate-to-severe steatosis has been reported. This discrepancy is most likely explained by the shorter diet period of 4 weeks applied in these studies in contrast to the 5 weeks in our study.22,23 During the MCD diet, hepatic inflammation progresses rapidly as a result of increased lipid peroxidation triggering hepatocellular necroinflammation.
After PH in the presence of severe inflammation, steatosis and increased oxidative stress, as measured indirectly by the amount of lipid peroxidation (TBARS) and by decreased levels of the hepatocellular antioxidant, total GSH, was observed. The intracellular lipids induce cytochrome p-450 microsomal lipoxygenases and generate ROS oxidants that promote cell membrane damage via activation of lipid peroxidation and production of reactive lipid aldehyde, MDA.24–26 In addition, the decreased antioxidant scavenger capacity in severe steatotic rats contributes to the amplified hepatic ROS levels and predisposes hepatocytes to extensive necrosis. In contrast, in mild steatotic rats, sufficient antioxidant response together with attenuated hepatocellular necrosis was observed. The main type of cell death in livers with severe steatosis was necrosis in contrast to apoptosis as seen in normal and mildly steatotic livers. In steatotic livers, both the direct inhibition of caspase-3 and the inhibition of Jnk kinase family that activate caspase-3 are observed in the presence of moderate to severe steatosis.27 These protective antiapoptotic mechanisms help to explain why the severely steatotic hepatocytes in our study did not undergo massive apoptosis but showed necrosis when progression of the cell cycle was disrupted.
Increased ROS formation after PH in rats with severe steatosis triggered the release of proinflammatory cytokines by KC, such as TNF-α, IL-1β, and IL-6. Increased TNF-α and IL-6 levels were observed preoperatively in steatotic rats indicating chronic KC activation as has also been reported in other studies.28,29 Extensive studies by a number of groups have proven that liver injury after PH in normal rodents is mediated by TNF-α.30,31 In addition, KCs also produce cytotoxins such as hydrogen peroxidase, which further activate TNF-α, subsequently creating a vicious circle.32 Therefore, KC dysfunction can be considered one of the key features contributing to the increased hepatocellular damage seen after PH in severe steatosis. These mechanisms of injury may also play a role in steatotic donor livers in the setting of cadaveric and particularly, living donor liver transplantation.
The role of KCs is controversial, as after PH, KCs reduce endotoxin-induced hepatocyte damage and are also essential triggers inducing liver regeneration.33 Conversely, factors enhancing TNF-α production generally exacerbate liver injury, whereas TNF-α inhibitors such as IL-10 are hepatoprotective.34 Although a significant increase in IL-10 levels in severe steatotic rats was observed, it was not sufficient enough to attenuate hepatocellular injury. Increased IL-10 levels together with an increased antioxidant response in the mildly steatotic rats potentially attenuated hepatocellular damage as no increased damage was observed compared with controls. It is noteworthy that control animals had no significant increase in hepatic glutathione levels and had unchanged plasma IL-10 levels. It seems that, in normal livers after PH, the actual hepatocellular injury is so limited that the protective mechanisms that become active in steatotic livers are unnecessary.
CONCLUSION
Steatosis with prominent inflammation impaired liver regeneration with a significant role of increased lipid peroxidation and KC-mediated hepatocellular injury after PH. These results suggest an increased risk when performing extensive liver resection in the presence of severe steatosis.
ACKNOWLEDGMENTS
The authors thank Adrie Maas for his assistance in the animal experiments, Chris van der Loos (Department of Pathology) for his assistance in the immunohistochemical assays, and Nikol Snoeren for her assistance in the histopathology assessment.
Footnotes
Reprints: Thomas M. van Gulik, MD, Department of Surgery, Academic Medical Center, Meibergdreef 9, IWO-1, 1105AZ Amsterdam, The Netherlands. E-mail: t.m.vangulik@amc.uva.nl.
REFERENCES
- 1.Scheele J, Stangl R, Altendorf-Hofmann A. Hepatic metastases from colorectal carcinoma: impact of surgical resection on the natural history. Br J Surg. 1990;77:1241–1246. [DOI] [PubMed] [Google Scholar]
- 2.Paquet KJ, Koussouris P, Mercado MA, et al. Limited hepatic resection for selected cirrhotic patients with hepatocellular or cholangiocellular carcinoma: a prospective study. Br J Surg. 1991;78459–78462. [DOI] [PubMed] [Google Scholar]
- 3.Wu CC, Yang MD, Liu TJ. Improvements in hepatocellular carcinoma resection by intraoperative ultrasonography or intermittent hepatic inflow blood occlusion. Jpn Clin Oncol. 1992;22:107–111. [PubMed] [Google Scholar]
- 4.Hilden M, Christofferson P, Juhl E. Liver histology in a ‘normal population’: examination of 503 consecutive fatal traffic casualties. Scand J Gastroenterol. 1977;12:593–597. [DOI] [PubMed] [Google Scholar]
- 5.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. [DOI] [PubMed] [Google Scholar]
- 6.Kooby DA, Fong Y, Suriawanata A. Impact of steatosis on perioperative outcome following hepatic resection. J Gastrointest Surg. 2004;7:1034–1044. [DOI] [PubMed] [Google Scholar]
- 7.Behrns KE, Tsiotos GG, DeSouza NF, et al. Hepatic steatosis as a potential risk factor for major hepatic resection. J Gastrointest Surg. 1998;2:292–298. [DOI] [PubMed] [Google Scholar]
- 8.Marcos A, Fisher R, Ham JM, et al. Liver regeneration and function in donor and recipient after right lobe adult to adult living donor liver transplantation. Transplant. 2000;69:1375–1379. [DOI] [PubMed] [Google Scholar]
- 8a.Angulo P. Nonalcoholic fatty liver disease. N Eng J Med. 2002;346:1221–1231. [DOI] [PubMed] [Google Scholar]
- 9.Higgins GM, Anderson RM. Experimental pathology of the liver: restoration of the liver of a white rat following surgical removal. Arch Pathol. 1931;12:186–206. [Google Scholar]
- 10.Selzner M, Clavien PA. Failure of regeneration of the steatotic rat liver: disruption at two different levels in the regeneration pathway. Hepatology. 2000;31:35–42. [DOI] [PubMed] [Google Scholar]
- 11.Gerlach C, Sakkab DY, Scholzen T, et al. Ki-67 expression during rat liver regeneration after partial hepatectomy. Hepatology. 1997;26:573–578. [DOI] [PubMed] [Google Scholar]
- 12.Fabricant JI. The kinetics of cellular proliferation in regenerating liver. J Cell Biol. 1968;38:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camargo CA Jr, Madden JF, Gao W, et al. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology. 1997;26:1513–1520. [DOI] [PubMed] [Google Scholar]
- 14.Serafín A, Roselló-Catafau J, Prats N, et al. Ischemic preconditioning increases the tolerance of fatty liver to hepatic ischemia-reperfusion injury in the rat. Am J Path. 2002;161:587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haney A, Peacock EE Jr, Madden JW. Liver regeneration and hepatic collagen deposition in rats with dethylnitrosamine-induced cirrhosis. Morphol Embryol. 1975;21:213–217. [Google Scholar]
- 16.Bismuth H, Chiche L, Adam R. Surgical treatment of hepatocellular carcinoma in cirrhosis: liver resection or transplantation. Transplant Proc. 1993;25:1066–1067. [PubMed] [Google Scholar]
- 17.Fan ST, Lai EC, Lo CM, et al. Hospital mortality of major hepatectomy for hepatocellular carcinoma associated with cirrhosis. Arch Surg. 1995;130:198–203. [DOI] [PubMed] [Google Scholar]
- 18.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. [DOI] [PubMed] [Google Scholar]
- 19.Caraceni P, Nardo B, Domenicali M, et al. Ischemia-reperfusion injury in the rat fatty liver: the role of nutritional status. Hepatology. 1999;29:1139–1146. [DOI] [PubMed] [Google Scholar]
- 20.Koppe SW, Sahai A, Malladi P, et al. Pentoxifylline attenuates steatohepatitis induced by the methionine choline deficient diet. J Hepatol. 2004;4:592–598. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez FG, Ritter J, Goodwin JW, et al. Effect of steatohepatitis associated with irinotecan or oxaplatin pretreatment on respectability of hepatic colorectal metastases. J Am Coll Surg. 2005;200:845–853. [DOI] [PubMed] [Google Scholar]
- 22.Rao MS, Pafreddy K, Abecassis M, et al. Regeneration of fatty liver with marked fatty change following partial hepatectomy in rats. Dig Dis Dis. 2001;9:1821–1826. [DOI] [PubMed] [Google Scholar]
- 23.Picard C, Lambotte L, Starkel P, et al. Steatosis is not sufficient to cause an impaired regenerative response after partial hepatectomy in rats. J Hepatol. 2002;32:645–652. [DOI] [PubMed] [Google Scholar]
- 24.Pessayre D, Mansouri A, Fromety B. Nonalcoholic steatosis and steatohepatitis: V. Mitochondrial dysfunction in steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G139–G199. [DOI] [PubMed] [Google Scholar]
- 25.Leclerq I, Farrell G, Field J, et al. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxidation in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaeschke H, Gores GJ, Cederbaum AI, et al. Mechanisms of hepatotoxicity. Toxic Sci. 2002;65:166–176. [DOI] [PubMed] [Google Scholar]
- 27.Yang SQ, Lin HZ, Mandal AK, et al. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathology. Hepatology. 2003;34:694–706. [DOI] [PubMed] [Google Scholar]
- 28.McCuskey RS, Ito Y, Robertson GR, et al. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology. 2004;40:386–393. [DOI] [PubMed] [Google Scholar]
- 29.Yang SQ, Lin HZ, Lane MD, et al. Obesity increases sensitivity to endotoxin liver injury: implications for pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yokoyama H, Fukuda M, Okamura Y, et al. Superoxide anion release into the hepatic sinusoid after an acute ethanol challenge and its attenuation by Kupffer cell depletion. Alcohol Clin Exp Res. 1999;123(suppl):71. [DOI] [PubMed] [Google Scholar]
- 31.Iimuro Y, Gallucci YM, Luster MI, et al. Antibodies to tumor necrosis factor alpha attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26:1530–1538. [DOI] [PubMed] [Google Scholar]
- 32.Enomoto N, Ikejima K, Yamashima S, et al. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res. 2001;25(suppl):51–54. [DOI] [PubMed] [Google Scholar]
- 33.Prins HA, Meijer C, Boelens PG. Kupffer-cell-depleted rats have a diminished acute-phase response following major liver resection. Shock. 2004;21:561–565. [DOI] [PubMed] [Google Scholar]
- 34.Mayeux PR. Pathobiology of lipopolysaccharide. J Toxicol Environ Health. 1997;51:415–435. [DOI] [PubMed] [Google Scholar]