

Abstract
Tumor necrosis factor-α (TNF-α) is elevated in obesity and in acute inflammatory states, and contributes to the elevated plasminogen activator inhibitor-1 (PAI-1) levels associated with these conditions. Mice genetically deficient in the p55 and p75 TNF-α receptors were used to study the roles of these receptors in the expression of PAI-1 in obese (ob/ob) mice, and in lean mice following acute stimulation with TNF-α. In ob/ob mice, p55 and p75 tumor necrosis factor-α receptors (TNFRs) act cooperatively to induce PAI-1 mRNA in most tissues, including the adipose tissue, kidney, heart, and liver. However, in lean mice, TNF-α-induced PAI-1 expression is mediated primarily by the p55 TNFR. Interestingly, PAI-1 mRNA expression in all tissues of the TNF-α-treated p75-deficient lean mice was significantly higher than that observed in TNF-α-treated wild-type mice. These observations suggest that the p75 TNFR may play a role in attenuating TNF-α-induced PAI-1 mRNA expression in acute inflammatory conditions. Our observation that soluble p75 TNFR was elevated in the plasma of TNF-α-treated mice in comparison to untreated mice supports this hypothesis. These studies thus provide insights into the TNF-α receptors involved in mediating and modulating the expression of PAI-1 in acute and chronic (eg, obesity) inflammatory states associated with elevated TNF-α.
Tumor necrosis factor-α (TNF-α) is a multifunctional cytokine involved in the pathogenesis of multiple disease states including septic shock, autoimmune disorders, inflammation, and insulin-resistance. 1-3 TNF-α protein is initially synthesized as a 26 kd bioactive membrane-bound protein (mTNF-α), 4 which is proteolytically cleaved to its 17-kd secreted/soluble form (sTNF-α) by the membrane metalloproteinase TNF-α converting enzyme (TACE). 5,6 Although sTNF-α appears to elicit most TNF-α-mediated responses, it is becoming increasingly evident that mTNF-α also is capable of mediating responses that are either similar to 7-11 or distinct from 11,12 its soluble counterpart. The biological activities of TNF-α are mediated by two structurally related, but functionally distinct receptors p55 and p75, which are co-expressed on the surface of most cell types. 13-16 Both receptors can also be proteolytically cleaved and released as soluble receptors capable of binding TNF-α. 17,18 The individual roles of the two receptors in signaling the different activities of TNF-α has been extensively investigated using receptor-specific antibodies which act as agonists or antagonists, 7,19-23 receptor-specific ligands, 22,24,25 and mice or cell lines genetically deficient in either the p55 or p75 TNF-α receptors. 26-31 Using these approaches it has been shown that the majority of inflammatory responses classically attributed to TNF-α are mediated by the p55 TNFR. For example, studies of TNF-α receptor-deficient mice reveal that p55 plays a critical role in mediating endotoxic shock, 32 LPS- and TNF-α-induced cytotoxicity, 32-34 development of lymphoid tissue, 35 and the pathology of collagen-induced arthritis. 36 TNFR p55 also was shown to play a protective role in L. monocytogenes-induced infection. 32,34 In contrast, p75 TNFR has been implicated in the suppression of TNF-α-induced inflammatory response 32 and other TNF-α-mediated responses, including thymocyte proliferation, skin necrosis, apoptosis of activated mature T lymphocytes, development of experimental cerebral malaria, and the up-regulation of NF-κ-dependent gene expression. 19,37-42
TNF-α is a potent inducer of the anti-fibrinolytic molecule plasminogen activator inhibitor-1 (PAI-1), and induces PAI-1 expression in many cell types in vitro and in most murine tissues in vivo. 43,44 PAI-1 plays an important role in the regulation of fibrinolysis by binding to and rapidly inactivating both tissue-type and urokinase-type plasminogen activators. 45 Additionally, PAI-1 influences neointima formation, cell migration, cell attachment/detachment, wound repair, and angiogenesis. 46-49 High levels of PAI-1 are associated with an increased risk for thrombotic diseases such as myocardial infarction, stroke, and venous thromboembolism. 50,51 Increased plasma PAI-1 levels have been detected in patients with gram-negative sepsis, 52 during the second and third trimesters of pregnancy, 53 as well as in obesity and type II diabetes and in insulin-resistance, 54-57 all of which are associated with an increased risk for thrombotic events. Interestingly, several of the disease states associated with elevated PAI-1, such as gram-negative sepsis, inflammatory conditions, and obesity, also are associated with increased TNF-α expression. 1-3,36 In fact, using ob/ob TNFR-deficient mice we demonstrated that TNF-α is a key component in the obesity-linked elevation of PAI-1. 58 Similarly, TNF-α contributes to the LPS-induced expression of PAI-1 in lean mice. 43
Although TNF-α was shown to induce PAI-1 in a variety of cells in vitro and in vivo, little is known regarding the role of TNFRs in mediating PAI-1 expression in various disease states. In this study, we used mice deficient in either the p55 or p75 TNFRs to begin to delineate the physiological roles of p55 and p75 in mediating and modulating PAI-1 gene expression in two different model systems. These include obese ob/ob mice characterized by chronically increased endogenous TNF-α levels, and lean mice treated acutely with high doses of TNF-α, a situation similar to a TNF-α-mediated acute inflammatory response. Our results show that in the obese mouse both TNFRs appear to act cooperatively to induce maximum PAI-1 mRNA in most tissues examined. However, during acute treatment with TNF-α, p55 TNFR is the dominant receptor for inducing PAI-1, while the p75 TNFR appears to play an antagonistic role in PAI-1 expression in this model. These data indicate that TNFRs are differentially engaged in inducing PAI-1 in a tissue and disease-specific fashion.
Materials and Methods
Animals
Adult male obese mice (C57BL/ob/ob; 20 to 24 weeks of age) and their lean counterparts (C57BL/6J +/?) were obtained from the Jackson Laboratory (Bar Harbor, ME). Age-matched male ob/ob mice deficient in either one (p55 or p75) or both TNFRs (p55 and p75) were generated by crossing and back-crossing lean mice deficient in these receptors to ob/ob mice as described 26,58 and genotyped using PCR-based assays. 26 In some experiments, lean mice lacking p55 TNFR, p75 TNFR, or both and wild-type controls were injected i.p. with recombinant murine TNF-α (4 μg per mouse in 100 μl of sterile saline; a kind gift of Richard Ulevitch, The Scripps Research Institute). Control animals were injected with 100 μl of saline alone. Three hours later, blood was collected and tissues were removed and processed for in situ hybridization (below) or the preparation of total RNA.
Determination of PAI-1, p75, and p55 TNFRs in Plasma
Active PAI-1 antigen in plasma was determined by using the t-PA binding assay as described. 59 The results were compared with a standard curve constructed by using recombinant mouse PAI-1. Plasma p75 and p55 TNFR levels were determined using the Quantikine M mouse sTNF R11 and Quantikine M mouse sTNF R1 ELISA assays, respectively, according to manufacturer’s instructions. (R & D Systems, Minneapolis, MN).
RNA Analysis
The concentration of PAI-1, TNF-α, and β-actin mRNAs in tissues was determined by real time RT-PCR using the I cycler (BioRad). In this method, reactions are characterized by the point in time during cycling that amplification of the PCR product is first detected, rather than by the amount of PCR product accumulated after a fixed number of cycles. The higher the starting copy number of the target nucleic acid, the earlier a significant increase in fluorescence is observed. An increase in fluorescence above baseline indicates the presence of accumulated PCR product. A fixed fluorescence threshold is set above the baseline. The parameter CT (threshold cycle) is defined as the fractional cycle number at which the fluorescence exceeds the fixed threshold. A plot depicting the log of initial concentration for a set of standards versus the CT is a straight line (data not shown). Quantification of the amount of target mRNA in unknown samples is accomplished by measuring the CT of the sample and using the standard curve to determine starting concentration. In these experiments, a linearized synthetic plasmid containing the upstream and downstream primer sets for PAI-1 and β-actin was prepared and used to in vitro transcribe an RNA standard (cRNA) using the riboprobe Gemini 11 in vitro transcription system (Promega, Madison, WI) as previously described. 44,60 Various concentrations of the cRNA (in 2.5 μl) were analyzed by real time PCR using 150 nmol/L of 5[prime] and 3[prime] primers (for each of the specific genes) and the SYBR green PCR master mix (Perkin Elmer) in a total volume of 25 μl, under cycling conditions used previously. 44,60 mRNA from target tissues was reverse transcribed and subjected to PCR as above using the corresponding primer pairs for PAI-1 and β-actin, and the concentration of these specific RNAs was calculated from the respective standard curves.
In situ hybridization was performed as described by using 35S-labeled antisense or sense PAI-1 riboprobes. 44 Slides were exposed in the dark at 4°C for 4 to 8 weeks. After developing the slides they were counterstained with hematoxylin and eosin.
Statistical Analysis
Statistical comparisons of results were performed using the unpaired Student’s t-test.
Results
Effect of Deletion of TNFRs on PAI-1 Gene Expression in ob/ob Mice
TNF-α is elevated in the adipose tissue in both human and murine obesity 3,61-64 and may contribute to the abnormal levels of PAI-1 observed in obesity. We previously demonstrated a significant reduction in adipose tissue PAI-1 mRNA levels in ob/ob mice deficient in both p55 and p75 TNF-α receptors when compared with wild-type ob/ob mice. 58 These results suggested that TNF-α is a key component in the obesity-linked elevation of PAI-1 in adipose tissue. However, PAI-1 expression is known to be elevated in several other tissues in ob/ob mice, including the kidney, heart, liver, and lung. 60 To determine whether TNF-α contributes to the elevated PAI-1 expression outside the adipose tissue we initially determined whether TNF-α gene expression itself was elevated in these tissues in the ob/ob mice. Besides the adipose tissue, TNF-α mRNA was found to be significantly elevated in the kidney (P < 0.02), heart (P < 0.04), lung (P < 0.02), and to a lesser extent in the liver (P < 0.06) of ob/ob mice when compared to the lean counterparts (Figure 1 ▶ , top panel). To determine whether TNF-α contributes to increased PAI-1 levels observed in these tissues, we next measured PAI-1 mRNA expression in wild-type and TNFR-deficient ob/ob mice. Obese mice lacking both TNFRs showed a significant reduction in PAI-1 expression in the kidney (P < 0.02), heart (P < 0.009), liver (P < 0.005), and lung (P < 0.01) compared with the wild-type ob/ob mice (Figure 1 ▶ , bottom panel). Thus, TNF-α contributes not only to the elevated expression of PAI-1 in adipose tissue but also to the increased PAI-1 observed in the kidney, heart, liver, and lung of ob/ob mice.
Figure 1.
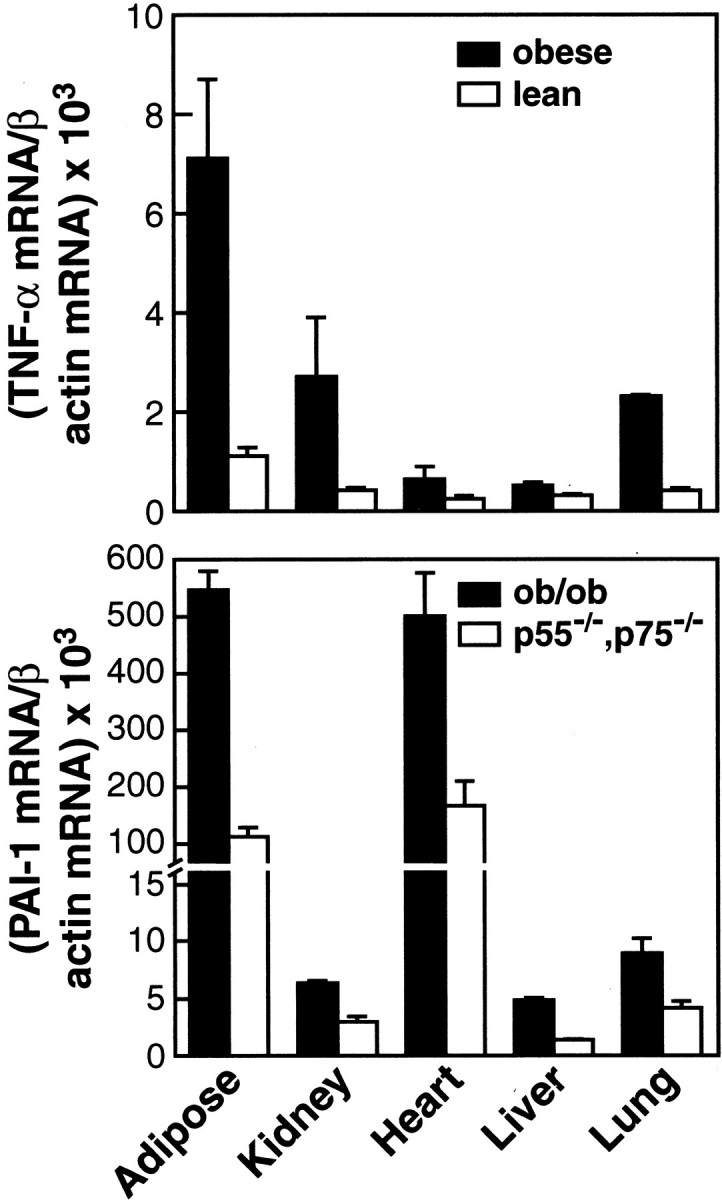
Top panel: Expression of TNF-α mRNA in tissues from obese (ob/ob) and lean mice. Total RNA was extracted from the adipose tissue, kidney, heart, liver, and lung of male ob/ob and lean mice. TNF-α mRNA was determined using quantitative real time RT-PCR analysis. N = 6 for each group; error bars represent mean ± SD. Bottom panel: Expression of PAI-1 mRNA in tissues from ob/ob and TNF-α receptor-deficient ob/ob mice. Total RNA was extracted from the adipose tissue, kidney, heart, liver, and lung of male ob/ob mice and ob/ob mice lacking both TNFRs (p55−/−, p75−/−). PAI-1 mRNA was determined using quantitative real time RT-PCR. N = 6 for each group, error bars represent ± SD.
Identification of the TNF-α Receptors Involved in PAI-1 Expression
To investigate the role of p55 and p75 TNFRs, respectively, in mediating the induction of PAI-1 by TNF-α in obesity, we compared PAI-1 mRNA expression in various tissues of ob/ob mice and ob/ob mice deficient in each of the individual TNF-α receptors. PAI-1 mRNA was significantly reduced in the adipose tissue (P < 0.001), 58 kidney (P < 0.02), heart (P < 0.009), liver (P < 0.005), and lung (P < 0.004) of ob/ob mice that lacked both TNFRs when compared to wild-type ob/ob mice (Figure 1 ▶ , bottom panel; Figure 2 ▶ , top panels). Interestingly, in the adipose tissue, kidney, and heart, PAI-1 mRNA expression also was significantly reduced in ob/ob mice that lacked either the p55 (adipose, P < 0.0001; kidney, P < 0.005; heart, P < 0.006), or the p75 TNFR (adipose, P < 0.0001; kidney, P < 0.01; heart, P < 0.004) when compared with wild-type ob/ob mice (Figure 2 ▶ , top panels). These results suggest that the TNF-α-mediated signals leading to increased PAI-1 expression in the adipose tissue, kidney, and heart of ob/ob mice require the participation of both p55 and p75 TNFRs. PAI-1 mRNA also is elevated in the ob/ob liver when compared to its lean counterpart. 60 The absence of p55 in ob/ob mice caused a significant decrease in PAI-1 mRNA in the liver (P < 0.007), while the absence of p75 alone did not affect PAI-1 mRNA levels. PAI-1 mRNA expression in mice lacking both p55 and p75 TNFRs was lower than in mice lacking only the p55 TNFR. These results suggest that although the p75 TNFR itself does not induce PAI-1 mRNA in the liver, it appears to augment PAI-1 induction mediated by the p55 TNFR. In the lung, PAI-1 mRNA was significantly reduced in TNFR-deficient ob/ob mice when compared to wild-type ob/ob mice (P < 0.004; Figure 1 ▶ , bottom panel; Figure 2 ▶ , top panel: lung). Interestingly, mice lacking either the p55 or the p75 TNFR appeared to have levels of PAI-1 mRNA similar to wild-type ob/ob mice (wild-type versus p55-deficient ob/ob, P < 0.2). These results suggest that in the obese lung TNF-α can induce PAI-1 expression independently through either the p55 or the p75 TNFR.
Figure 2.
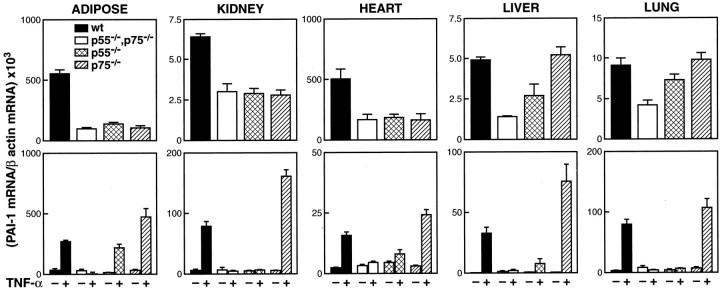
Top panels: Effect of deletion of TNFRs on PAI-1 expression in tissues from ob/ob mice. Total RNA was extracted from various tissues of wild-type ob/ob mice, ob/ob mice lacking both TNFRs (p55−/−, p75−/−), ob/ob mice lacking only the p55 TNFR (p55−/−), and from ob/ob mice lacking only the p75 TNFR (p75−/−). PAI-1 mRNA was determined using quantitative real time RT-PCR. N = 6 for each group; error bars represent ± SD. Bottom panels: Effect of exogenous TNF-α treatment on PAI-1 mRNA expression in tissues of lean wild-type and lean TNFR-deficient mice. Lean wild-type mice, lean mice lacking both TNFRs (p55−/−, p75−/−), lean mice lacking only the p55 TNFR (p55−/−), and lean mice lacking only the p75 TNFR (p75−/−) were injected i.p. with 4 μg of murine recombinant TNF-α or saline. Three hours later, tissues were collected and analyzed for PAI-1 mRNA using quantitative real time-RT-PCR as described in Materials and Methods. N = 6 for each group; error bars represent mean ± SD.
We next investigated the role of p55 and p75 TNFRs in mediating the induction of PAI-1 after acute administration of TNF-α to wild-type lean and TNFR-deficient lean mice (Figure 2 ▶ , bottom panels). In this model TNF-α significantly induced PAI-1 mRNA in all tissues of the wild-type (adipose tissue, P < 0.009; kidney, P < 0.01; heart, P < 0.0001; liver, P < 0.002; and lung, P < 0.002) and p75-deficient (adipose, P < 0.004; kidney, P < 0.004; heart, P < 0.0001; liver, P < 0.006; lung, P < 0.004) mice. However, a modest but significant induction of PAI-1 mRNA also was observed in the adipose tissue (P < 0.004), heart (P < 0.05), and liver (P < 0.02) of TNF-α-treated p55-deficient mice. These results suggest that while p55 TNFR is the dominant receptor for the TNF-α-induced expression of PAI-1 mRNA in this model, in some tissues (eg, adipose tissue, heart, and liver) PAI-1 mRNA also can be induced by the p75 TNFR. It is interesting that, in all of the tissues studied (Figure 2 ▶ , bottom panels), the PAI-1 mRNA response in TNF-α-treated p75-deficient mice was significantly higher than the PAI-1 response observed in TNF-α-treated wild-type mice (eg, adipose, P < 0.02; kidney, P < 0.02; heart, P < 0.03; liver, P < 0.04; and lung, P < 0.03). These data suggest that, in acute inflammation, p75 may play a key role in modulating the actions of TNF-α, by acting as an antagonist of TNF-α in the induction of PAI-1.
We were also interested in knowing the extent of correlation between tissue expression of PAI-1 mRNA and levels of plasma PAI-1 protein. Figure 3 ▶ shows active PAI-1 antigen in the plasma of wild-type and TNF-α receptor-deficient ob/ob mice (top panel) as well as wild-type and TNF-α receptor-deficient lean mice acutely treated with exogenous TNF-α (bottom panel). As we had demonstrated previously, 58 when compared to wild-type ob/ob mice, ob/ob mice deficient in both the p55 and p75 TNF-α receptors had significantly reduced levels of PAI-1 antigen in the plasma (52% reduction; P < 0.01; Figure 3 ▶ , top panel). PAI-1 antigen levels also were significantly reduced in ob/ob mice lacking either the p55 (P < 0.04) or the p75 (P < 0.05) TNF receptors when compared with the wild-type ob/ob mice. Thus PAI-1 antigen levels observed in the plasma of ob/ob and TNF-α receptor-deficient ob/ob mice most closely reflect PAI-1 mRNA levels observed for the adipose tissue, kidney, and heart in these animals (Figure 2 ▶ , top panel).
Figure 3.

Top panel: Plasma PAI-1 expression in wild-type and TNFR-deficient ob/ob mice. Plasma was collected from wild-type ob/ob mice, ob/ob mice lacking both TNFRs (p55−/−, p75−/−), ob/ob mice lacking only the p55 TNFR (p55−/−), and from ob/ob mice lacking only the p75 TNFR (p75−/−). Plasma PAI-1 antigen was analyzed as described in Materials and Methods. N = 6 for each group; error bars represent ± SD. Bottom panel: Plasma PAI-1 expression in TNF-α-treated wild-type and TNFR-deficient lean mice. Lean wild-type mice, lean mice lacking both TNFRs (p55−/−, p75−/−), lean mice lacking only the p55 TNFR (p55−/−), and lean mice lacking only the p75 TNFR (p75−/−) were injected i.p. with 4 μg of murine recombinant TNF-α or saline. Three hours later, plasma was collected and analyzed for PAI-1 antigen expression. N = 6 for each group; error bars represent mean ± SD.
In lean mice treated acutely with exogenous TNF-α (Figure 3 ▶ , bottom panel), active plasma PAI-1 antigen was significantly increased in wild-type (P < 0.0002) and p75-deficient mice (P < 0.0006) in response to stimulus. Plasma PAI-1 levels in TNF-α-treated p75-deficient mice was only modestly greater than that observed in TNF-α-treated wild-type mice, however this increase was highly significant (P < 0.0005). Although a moderate increase in PAI-1 mRNA expression had been observed in the adipose tissue, heart, and liver of TNF-α-treated p55-deficient mice, this was not reflected in the plasma PAI-1 levels. The origin of plasma PAI-1 is a complex process. Variations in the amount of PAI-1 synthesized by multiple tissues as well from cells in the blood probably contribute to the final plasma PAI-1 concentrations. Moreover, changes in the rate of clearance of PAI-1 in the two models tested also may be involved in the regulation of plasma PAI-1 levels. Discrepancy between tissue expression of PAI-1 mRNA and plasma protein levels may therefore depend on several factors, including tissue-specific post-transcriptional regulation and overall total contribution by a particular tissue to systemic circulating PAI-1. Thus PAI-1 mRNA expression observed locally in the tissues need not necessarily be reflected in PAI-1 antigen levels observed in the plasma.
Effect of Exogenous TNF-α Treatment on the Cellular Localization of PAI-1 in Wild-Type and TNFR-Deficient Lean Mice
Using in situ hybridization analysis we previously demonstrated that the adipocyte was the primary cell responding with elevated PAI-1 mRNA expression in adipose tissues of TNF-α-treated wild-type, p55−/−, and p75−/− mice. 58 To examine the cell-specific expression of PAI-1 mRNA in other tissues (eg, kidney, heart, liver, lung), tissues from saline or TNF-α-treated wild-type or TNFR-deficient (P55−/− or P75−/−) mice were removed, fixed in paraformaldehyde, and analyzed by in situ hybridization using a riboprobe specific for murine PAI-1. 44 Representative sections from these tissues are shown in Figure 4 ▶ . In saline-treated wild-type control mice no signals for PAI-1 mRNA could be detected in any of the tissues examined (A, kidney; B, heart; C, Liver; D, lung). In TNF-α-treated wild-type mice, strong hybridization signals were apparent in the glomeruli of the kidney (panel E), in cardiomyocytes of the heart (panel F), hepatocytes in the liver (panel G) and alveolar macrophages in the lung (panel H). In TNF-α-treated p55-deficient mice, positive signals for PAI-1 were observed in the heart (panel J) and the liver (panel K), but not in the kidney (panel I) and lung (L). The intensity of the hybridization signals observed in the heart and liver of TNF-treated p55-deficient mice were markedly less than that observed in TNF-treated wild-type mice (compare panels J and F; panels K and G). These observations agree with the data obtained by real time quantitative PCR (Figure 2 ▶ , bottom panels: heart, liver) and suggest that in the presence of high concentrations of soluble TNF-α, PAI-1 induction in the heart and liver also can be mediated by the p75 TNFR. In TNF-α-treated p75-deficient mice, very strong in situ hybridization signals were observed for PAI-1 in the glomeruli of the kidney (panel M), in cardiomyocytes of the heart (panel N), in hepatocytes of the liver (panel O), and in alveolar macrophages of the lung (panel P). The positive in situ hybridization signals for PAI-1 in these tissues from TNF-α-treated p75-deficient mice appear to be much stronger than the signals observed in TNF-α-treated wild-type mice (compare panels M, N, O, P and panels E, F, G, H). These results generally agree with data obtained by quantitative real time RT-PCR (Figure 2 ▶ , bottom panels), and suggest that in all tissues, p55 TNFR is the dominant receptor for the induction of PAI-1 in this model, while p75 TNFR may play an antagonistic role.
Figure 4.
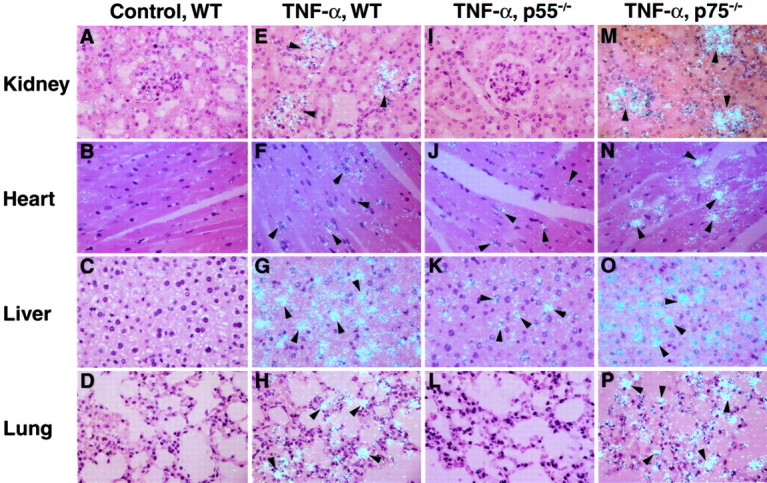
Effect of TNF-α on the cellular localization of PAI-1 mRNA in tissues of wild-type and TNFR-deficient mice. In situ hybridization was performed on paraffin sections of the kidney, heart, liver, and lung from lean wild-type untreated mice (A,B,C,D) and TNF-α-treated wild-type (E,F,G,H), p55 TNFR-deficient (I,J,K,L) and p75 TNFR-deficient (M,N,O,P) mice. Slides were exposed for 4 weeks at 4°C and then stained with hematoxylin and eosin. Arrowheads indicate positive signals for PAI-1 mRNA. Magnification, ×400 for all sections.
Plasma p75 and p55 Levels in TNF-α-Treated Lean Mice
In many tissues of TNF-α-treated p75-deficient lean mice, PAI-1 mRNA expression was found to be increased over that observed for TNF-α-treated wild-type controls (Figure 2 ▶ , bottom panels). These results raise the possibility that p75 may act as an antagonist of TNF-α signaling for PAI-1 induction. A variety of inflammatory stimuli are known to trigger shedding of p55 and p75 TNFRs from the cell surface into the circulation. 17,18 Such a process could conceivably regulate the effects of TNF-α by binding to and neutralizing TNF-α bioactivity. We therefore initially measured soluble p75 and p55 levels in the plasma of control (saline-treated) and TNF-α-treated wild-type lean mice. Soluble p75 TNFR was increased in the plasma after TNF-α treatment of wild-type mice (Figure 5) ▶ , while there was no change in plasma levels of p55 after TNF treatment (data not shown). Although a modest, but significant, increase in soluble p75 also was observed in TNF-α-treated p55-deficient mice, this expression was significantly lower than what was observed in TNF-α-treated wild-type mice. These results suggest that while the increase of plasma p75 TNFR in response to TNF-α is mediated primarily by the p55 TNFR, the p75 TNFR also can mediate this event to a smaller extent. The increase in plasma p75 TNFR in response to an acute inflammatory stimulus such as TNF-α may function as an antagonist and offer a certain degree of protection in disease states associated with acute increases in TNF-α.
Figure 5.

Expression of TNFR p75 in the plasma of TNF-α-treated lean wild-type and p55−/− mice. Wild-type or p55−/− lean mice were injected i.p. with 4 μg of murine recombinant TNF-α or saline. TNFR p75 in the plasma was determined using an ELISA assay (R & D Systems).
Discussion
In this study we used mice genetically deficient in the p55 and p75 TNFRs to elucidate the roles of these receptors in mediating TNF-α-induced PAI-1 mRNA expression in tissues of ob/ob mice and lean mice treated acutely with exogenous TNF-α. While several studies have demonstrated the involvement of the p55 TNFR in various TNF-α-mediated responses, 13,33,65,66 the role of the p75 TNFR remains less clear. The data presented here reveal the importance of both the p55 and p75 TNFRs in the induction of PAI-1 mRNA in ob/ob mice, a model for elevated endogenous TNF-α or chronic inflammation. We find convincing evidence for the involvement of p75 in PAI-1 mRNA induction in several tissues which exhibited elevated endogenous TNF-α. These include the adipose tissue, kidney, liver, heart, and lung of ob/ob mice (Figure 2 ▶ , top panels). In ob/ob mice, p55 and p75 TNFRs are both required for PAI-1 gene induction and may act cooperatively to achieve this. In this respect, it is interesting to note that a recent study demonstrated that PAI-1 antigen levels in adipose tissues from obese humans were strongly correlated with both TNFR1 and TNFR11. 67 In studies relating to TNF-α-mediated insulin-resistance in the same ob/ob model, it was demonstrated that while p55 deficiency caused a significant improvement of insulin sensitivity and p75 deficiency did not affect insulin sensitivity, it appeared to potentiate the effect of p55. 29 The fast association/dissociation kinetics of soluble TNF-α with the p75 receptor is thought to create an increase in the local concentration of the cytokine in the proximity of the cell membrane, thus potentially augmenting soluble TNF-α binding to the p55 receptor. 7,13 Such a mechanism may be expected to be important in physiological and/or pathological situations in which the circulating concentrations of TNF-α are low. Membrane-associated TNF-α can bind both receptors, but is reported to bind more efficiently to the p75 TNFR. 7,27,68 In adipocytes, however, p55 appears to be the dominant receptor for mTNF-α. 61 In this regard, a recent study demonstrated that membrane-bound TNF-α also was increased in adipose tissues in both human and murine obesity. 69 The scenario therefore exists in obesity in which circulating TNF-α concentrations are low and TNF-α may be increased locally at the sites of production to act in an autocrine and/or paracrine manner. A ligand passing mechanism or a cooperation between signals transmitted independently by p55 and p75 may explain the requirement for both these receptors to generate maximum PAI-1 mRNA induction in several tissues of the obese mouse. In this regard, both TNFRs were involved in signaling endothelial tissue factor expression in endothelial cells. 68 Similarly, a murine transmembrane TNF-α transgene induces arthritis by cooperative p55/p75 TNFR signaling. 70 A functional role for both TNFRs and mTNF-α also was demonstrated in experimental hepatitis. 71 Ligand-induced formation of p55 and p75 heterocomplexes have been reported on intact cells. 72 It is not known yet whether a similar phenomenon occurs in obesity.
In lean mice that we treated acutely with a high dose (4 μg/mouse) of exogenous TNF-α, providing a model for acute inflammation, the p55 TNFR appeared to be the major receptor for transmitting the TNF-α signal leading to the induction of PAI-1 mRNA in all tissues examined. In these mice TNF-α increased PAI-1 mRNA expression in the adipose tissue ( 58 adipocytes), liver (hepatocytes), lung (alveolar macrophages), heart (cardiomyocytes), and kidney (glomerular cells). In the adipose tissue, liver, and heart, p75 also independently induced a modest but significant increase in PAI-1 mRNA. Although the majority of biological responses classically attributed to TNF-α are mediated by the p55 TNFR, it is becoming increasingly evident that the p75 TNFR is capable of delivering a TNF-α signal under certain circumstances. For example, it has been demonstrated that activation-induced apoptosis of T cells is mediated by p75. 40 Additionally, p75 is involved in the development of TNF-α-induced necrosis of the skin, in the migrations of epidermal Langerhans cells, initiation of cutaneous immune responses, and in the development of experimental cerebral malaria. 37-41
Interestingly, the induction of PAI-1 mRNA in all tissues of the TNF-α-treated p75-deficient lean mice was higher than that observed in TNF-α-treated wild-type mice (Figure 2 ▶ , bottom panels). These results suggest an antagonistic role for p75 TNFR in TNF-α-induced expression of PAI-1 mRNA in this acute in vivo model. A variety of inflammatory stimuli are known to trigger shedding of cell surface-associated p55 and p75 into the circulation. 18,73,74 Soluble p55 and p75 receptors have been reported to function both as TNF-α agonists by stabilizing TNF-α, 32 as well as effective TNF-α antagonists by neutralizing TNF-α. 75-79 An antagonistic role for solubleTNFRs is supported by the observation that exogenously administered recombinant soluble p55 or p75 Ig fusion proteins are effective TNF-α antagonists in several models of inflammation known to involve TNF-α. 75,76,78,79 Also, infusion of a soluble TNFR-IgG chimeric protein to obese fa/fa rats reduced TNF-α-mediated insulin-resistance in this model. 80 Although inflammatory stimuli induce proteolytic shedding of both p55 and p75 receptors, levels of soluble p75 greatly exceed those of soluble p55, suggesting a dominant role for endogenously produced soluble p75 in the down-regulation of TNF-α driven responses. 18,73,81 In this study we demonstrate that TNF-α-treatment of wild-type lean mice leads to an increase in plasma p75 TNFR (Figure 5) ▶ . This soluble receptor could conceivably neutralize circulating TNF-α, thereby attenuating TNF-α-mediated responses. This phenomenon might help explain the exacerbated PAI-1 mRNA response we observed in TNF-α-treated p75-deficient mice when compared with TNF-α-treated wild-type mice. TNFR p75 may be important in attenuating the harmful effects of TNF-α in such an acute inflammatory model. Our results are similar to studies by Peschon et al, 82 who used an inflammatory mouse model and demonstrated an exacerbated pulmonary inflammation and dramatically increased endotoxin-induced serum TNF-α levels in mice lacking p75, suggesting an important role for p75 in suppressing TNF-α-mediated inflammatory responses. It is also possible that p75 signaling dampens the TNF-α response, thereby explaining the exacerbated PAI-1 mRNA induction in p75-deficient mice, and providing an alternative mechanism of p75 antagonism.
Use of p55 and p75 receptors for TNF-α-mediated PAI-1 mRNA tissue expression may depend on various factors, including tissue-specific receptor/ligand availability and the state of inflammation, namely chronic versus acute. Soluble TNF-α primarily appears to stimulate p55 TNFR whereas mTNF-α, while able to bind both receptors, strongly activates TNFR p75. 7 Moreover, like TNF-α itself, both TNF-α receptors were shown to be cleaved by TACE. 17,18 Such mechanisms and the regulation of ligand and receptor expression in the various tissues may explain the complexity of the transduction of the TNF-α signal by either both or exclusively one of the two TNF-α receptor subtypes observed in these studies.
In summary, these data provide insights into the biological role of the p55 and p75 TNFRs in mediating as well as modulating TNF-α-induced PAI-1 gene expression in chronic (obese) and acute (TNF-α-treated) inflammatory states. Our studies provide genetic evidence for a cooperative role for p55 and p75 TNFRs in the obese model for the induction of PAI-1 by endogenous TNF-α. In contrast, in lean mice acutely treated with high doses of TNF-α, TNFR p55 was the dominant mediator of TNF-α action while TNFR p75 appeared to play an antagonistic role. These studies additionally suggest that, in vivo, TNF-α receptor selectivity and the ultimate biological outcome depend on the tissue/cell type as well as the underlying pathophysiology.
Acknowledgments
We thank T. Thinnes for her excellent technical assistance and Marcia McRae for her expert secretarial skills.
Footnotes
Address reprint requests to Fahumiya Samad, Ph.D., The Scripps Research Institute, Department of Cell Biology, Division of Vascular Biology, VB-3, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail: fsamad@scripps.edu.
Supported by grant numbers HL 59549 and AHA 00230054N.
This is the Scripps Research Institute manuscript number 15305-CB.
References
- 1.Beutler B, Cerami A: The biology of cachectin/TNF-α primary mediator of the host response. Annu Rev Immunol 1989, 7:625-655 [DOI] [PubMed] [Google Scholar]
- 2.Barbara JA, van ostade X, Lopez A: Tumor necrosis factor-α (TNF-α): the good, the bad and potentially very effective. Immunol Cell Biol 1996, 74:434-443 [DOI] [PubMed] [Google Scholar]
- 3.Sethi JK, Hotamisligil GS: The role of TNFα in adipocyte metabolism. Cell Dev Biol 1999, 10:19-29 [DOI] [PubMed] [Google Scholar]
- 4.Wajant H, Pfeffer K, Pfizenmaier K, Scheurich P: Tumor necrosis factors in 1998. Cytokine Growth Factor Rev 1998, 9:297-302 [DOI] [PubMed] [Google Scholar]
- 5.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP: A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385:729-733 [DOI] [PubMed] [Google Scholar]
- 6.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD: Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385:733-736 [DOI] [PubMed] [Google Scholar]
- 7.Grell M: Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J Inflam 1995, 47:8-17 [PubMed] [Google Scholar]
- 8.Decoster E, Vanhaesebroeck B, Vandenabeele P, Grooten J, Fiers W: Generation and biological characterization of membrane-bound, uncleavable murine tumor necrosis factor. J Biol Chem 1995, 31:18473-18478 [DOI] [PubMed] [Google Scholar]
- 9.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K: The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 KDa tumor necrosis factor receptor. Cell 1995, 83:793-802 [DOI] [PubMed] [Google Scholar]
- 10.Perez C, Albert I, DeFay K, Zachariades N, Gooding L, Kriegler M: A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell 1990, 63:251-258 [DOI] [PubMed] [Google Scholar]
- 11.Probert L, Akassoglou K, Alexopoulou L, Douni E, Haralambous S, Hill S, Kassiotis G, Kontoyiannis D, Pasparakis M, Plows D, Kollias G: Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice. J Leukoc Biol 1996, 59:518-525 [DOI] [PubMed] [Google Scholar]
- 12.Mueller C, Corazza N, Trachsel-Loseth S, Eugster HP, Buhler-Jungo M, Brunner T, Imboden MA: Noncleavable transmembrane mouse tumor necrosis factor-α (TNFα) mediates effects distinct from those of wild-type TNF-α in vitro and in vivo. J Biol Chem 1999, 274:38112-38118 [DOI] [PubMed] [Google Scholar]
- 13.Bazzoni F, Beutler B: How do tumor necrosis factor receptors work? J Inflam 1995, 45:221-238 [PubMed] [Google Scholar]
- 14.Vandenabeele P, Declercq W, Beyaert R, Fiers W: Two tumor necrosis factor receptors: structure and function. Trends Cell Biol 1995, 5:392-399 [DOI] [PubMed] [Google Scholar]
- 15.Rothe J, Gehr G, Loetscher H, Lesslauer W: Tumor necrosis factor receptors: structure and function. Immunol Res 1992, 11:81-90 [DOI] [PubMed] [Google Scholar]
- 16.Bigda J, Holtmann H: TNF receptors: how they function and interact. Arch Immunol Ther Exp 1997, 45:263-270 [PubMed] [Google Scholar]
- 17.Solomon KA, Pesti N, Wu G, Newton RC: Cutting edge: a dominant negative form of TNF-α converting enzyme inhibits proTNF and TNFRII secretion. J Immunol 1999, 163:4105-4108 [PubMed] [Google Scholar]
- 18.Crowe PD, Walter BN, Mohler KM, Otten-Evans C, Black RA, Ware CF: A metalloprotease inhibitor blocks shedding of 80-kD TNF receptor and TNF processing in T lymphocytes. J Exp Med 1995, 181:1205-1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheehan KC, Pinckard JK, Arthur CD, Dehner LP, Goeddel DV, Schreiber RD: Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J Exp Med 1995, 181:607-617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shalaby MR, Sundan A, Loetscher H, Brockhaus M, Lesslauer W, Espevik T: Binding and regulation of cellular functions by monoclonal antibodies against human tumor necrosis factor receptors. J Exp Med 1990, 172:1517-1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu LS, Spelleken M, Röhrig K, Hauner H, Eckel J: Tumor necrosis factor-α acutely inhibits insulin signaling in human adipocytes: implication of the p80 tumor necrosis factor receptor. Diabetes 1998, 47:515-522 [DOI] [PubMed] [Google Scholar]
- 22.Clauss M, Grell M, Fangmann C, Fiers W, Scheurich P, Risau W: Synergistic induction of endothelial tissue factor by tumor necrosis factor and vascular endothelial growth factor: functional analysis of the tumor necrosis factor receptors. FEBS Lett 1996, 390:334-338 [DOI] [PubMed] [Google Scholar]
- 23.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W: Tumor necrosis factor α (TNF-α)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med 1993, 177:1277-1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Poll T, Jansen PM, Van Zee KJ, Welborn MB3, de Jong I, Hack CE, Loetscher H, Lesslauer W, Lowry SF, Moldawer LL: Tumor necrosis factor-α induces activation of coagulation and fibrinolysis in baboons through an exclusive effect on the p55 receptor. Blood 1996, 88:922-927 [PubMed] [Google Scholar]
- 25.Hube F, Hauner H: The two tumor necrosis factor receptors mediate opposite effects on differentiation and glucose metabolism in human adipocytes in primary culture. Endocrinol 2000, 141:2582-2588 [DOI] [PubMed] [Google Scholar]
- 26.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS: Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389:610-614 [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Sethi JK, Hotamisligil GS: Transmembrane tumor necrosis factor (TNF)-α inhibits adipocyte differentiation by selectively activating TNF receptor 1. J Biol Chem 1999, 274:26287-26295 [DOI] [PubMed] [Google Scholar]
- 28.Kalb A, Bluethmann H, Moore MW, Lesslauer W: Tumor necrosis factor receptors (Tnfr) in mouse fibroblasts deficient in Tnfr1 or Tnfr2 are signaling competent and activate the mitogen-activated protein kinase pathway with differential kinetics. J Biol Chem 1996, 271:28097-28104 [DOI] [PubMed] [Google Scholar]
- 29.Uysal KT, Wiesbrock SM, Hotamisligil GS: Functional analysis of tumor necrosis factor (TNF) receptors in TNF-α-mediated insulin resistance in genetic obesity. Endocrinol 1998, 139:4832-4838 [DOI] [PubMed] [Google Scholar]
- 30.Sethi JK, Xu H, Uysal KT, Wiesbrock SM, Scheja L, Hotamisligil GS: Characterisation of receptor-specific TNFα functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. FEBS Lett 2000, 469:77-82 [DOI] [PubMed] [Google Scholar]
- 31.Yamada Y, Kirillova I, Peschon JJ, Fausto N: Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 1997, 94:1441-1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW: Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993, 73:457-467 [DOI] [PubMed] [Google Scholar]
- 33.Tartaglia LA, Rothe M, Hu YF, Goeddel DV: Tumor necrosis factor’s cytotoxic activity is signaled by the p55 TNF receptor. Cell 1993, 73:213-216 [DOI] [PubMed] [Google Scholar]
- 34.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H: Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364:798-802 [DOI] [PubMed] [Google Scholar]
- 35.Pasparakis M, Alexopoulou L, Grell M, Pfizenmaier K, Bluethmann H, Kollias G: Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc Natl Acad Sci USA 1997, 94:6319-6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rink L, Kirchner H: Recent progress in the tumor necrosis factor-α field. Int Arch Allergy Immunol 1996, 111:199-209 [DOI] [PubMed] [Google Scholar]
- 37.Erickson SL, De Sauvage F, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan KCF, Schreiber RD, Goeddel DV, Moore MW: Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372:560-563 [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Fujisawa H, Zhuang L, Kondo S, Shivji GM, Kim CS, Mak TW, Sauder DN: Depressed Langerhans cell migration and reduced contact hypersensitivity response in mice lacking TNF receptor p75. J Immunol 1997, 159:6148-6155 [PubMed] [Google Scholar]
- 39.Lucas R, Juillard P, Decoster E, Redard M, Burger D, Donati Y, Giroud C, Monso-Hinard C, De Kesel T, Buurman WA, Moore MW, Dayer JM, Fiers W, Bluethmann H, Grau GE: Crucial role of tumor necrsosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur J Immunol 1997, 27:1719-1725 [DOI] [PubMed] [Google Scholar]
- 40.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ: Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995, 377:348-351 [DOI] [PubMed] [Google Scholar]
- 41.Tartaglia LA, Goeddel DV, Reynolds C, Figari IS, Weber RF, Fendly BM, Palladino MA, Jr: Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor. J Immunol 1993, 151:4637-4641 [PubMed] [Google Scholar]
- 42.Rao P, Hsu KC, Chao MV: Up-regulation of NF-κ B-dependent gene expression mediated by the p75 tumor necrosis factor receptor. J Interferon Cytokine Res 1995, 15:171-177 [DOI] [PubMed] [Google Scholar]
- 43.Samad F, Yamamoto K, Pandey M, Loskutoff D: Elevated expression of transforming growth factor-β in adipose tissue from obese mice. Mol Med 1997, 3:37-48 [PMC free article] [PubMed] [Google Scholar]
- 44.Samad F, Yamamoto K, Loskutoff DJ: Distribution and regulation of plasminogen activator inhibitor-1 in murine adipose tissue in vivo: induction by tumor necrosis factor-α and lipopolysaccharide. J Clin Invest 1996, 97:37-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fearns C, Samad F, Loskutoff DJ: Synthesis and localization of PAI-1 in the vessel wall. van Hinsbergh VWM eds. Vascular Control of Hemostasis. 1995:pp 207-226 The Netherlands, Harwood Academic Publishers Amsterdam
- 46.Noël A, Bajou K, Masson V, Devy L, Frankenne F, Rakic JM, Lambert V, Carmeliet P, Foidart JM: Regulation of cancer invasion and vascularization by plasminogen activator inhibitor-1. Fibrinolysis Proteolysis 1999, 13:220-225 [Google Scholar]
- 47.Devy L, Blacher S, Grignet-Debrus C, Bajou K, Masson V, Gerard RD, Gils A, Carmeliet G, Carmeliet P, Declerck PJ, Noël A, Foidart J-M: The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. EMBO J 2002, 16:47-154 [DOI] [PubMed] [Google Scholar]
- 48.Loskutoff DJ: PAI-1 inhibits neointimal formation after arterial injury in mice: a new target for controlling restenosis? Circulation 1997, 96:2772-2774 [PubMed] [Google Scholar]
- 49.Deng G, Curriden SA, Hu G, Czekay R-P, Loskutoff DJ: Plasminogen activator inhibitor-1 regulates cell adhesion by binding to the somatomedin B domain of vitronect. J Cell Physiol 2001, 189:23-33 [DOI] [PubMed] [Google Scholar]
- 50.Huber K, Christ G, Wojta J, Gulba D: Plasminogen activator inhibitor type-1 in cardiovascular disease: status report 2001. Thromb Res 2001, 103:S7-S19 [DOI] [PubMed] [Google Scholar]
- 51.Nordt TK, Peter K, Ruef J, Kubler W, Bode C: Plasminogen activator inhibitor type-1 (PAI-1) and its role in cardiovascular disease. Thromb Haemost 1999, 82:14-18 [PubMed] [Google Scholar]
- 52.Westendorp RG, Hottenga JJ, Slagboom PE: Variation in plasminogen-activator-inhibitor-1 gene and risk of meningococcal septic shock. Lancet 1999, 354:561-563 [DOI] [PubMed] [Google Scholar]
- 53.Kruithof EKO, Tran-Thang C, Gudinchet A, Hauert J, Nicoloso G, Genton C, Welti H, Bachmann FW: Fibrinolysis in pregnancy: a study of plasminogen activator inhibitors. Blood 1987, 69:460-466 [PubMed] [Google Scholar]
- 54.McGill JB, Schneider DJ, Arfken CL, Lucore CL, Sobel BE: Factors responsible for impaired fibrinolysis in obese subjects and NIDDM patients. Diabetes 1994, 43:104-109 [DOI] [PubMed] [Google Scholar]
- 55.Juhan-Vague I, Alessi MC: PAI-1, obesity, insulin resistance, and risk of cardiovascular events. Thromb Haemost 1997, 78:656-660 [PubMed] [Google Scholar]
- 56.Legnani C, Maccaferri M, Tonini P, Cassio A, Cacciari E, Coccheri S: Reduced fibrinolytic response in obese children: association with high baseline activity of the fast acting plasminogen activator inhibitor (PAI-1). Fibrinolysis 1988, 2:211-214 [Google Scholar]
- 57.Potter van Loon BJ, Kluft C, Radder JK, Blankenstein MA, Meinders AE: The cardiovascular risk factor plasminogen activator inhibitor type 1 is related to insulin resistance. Metabolism 1993, 42:945-949 [DOI] [PubMed] [Google Scholar]
- 58.Samad F, Uysal KT, Wiesbrock SM, Pandey M, Hotamisligil GS, Loskutoff DJ: Tumor necrosis factor α is a key component in the obesity-linked elevation of plasminogen activator inhibitor-1. Proc Natl Acad Sci USA 1999, 96:6902-6907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schleef RR, Sinha M, Loskutoff DJ: Immunoradiometric assay to measure the binding of a specific inhibitor to tissue-type plasminogen activator. J Lab Clin Med 1985, 106:408-415 [PubMed] [Google Scholar]
- 60.Samad F, Loskutoff DJ: Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol Med 1996, 2:568-582 [PMC free article] [PubMed] [Google Scholar]
- 61.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM: Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J Clin Invest 1995, 95:2409-2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hotamisligil GS, Spiegelman BM: Tumor necrosis factor α: a key component of the obesity-diabetes link. Diabetes 1994, 43:1271-1278 [DOI] [PubMed] [Google Scholar]
- 63.Peraldi P, Spiegelman B: TNF-α and insulin resistance: summary and future prospects. Mol Cell Biochem 1998, 182:169-175 [PubMed] [Google Scholar]
- 64.Hube F, Hauner H: The role of TNF-α in human adipose tissue: prevention of weight gain at the expense of insulin resistance? Horm Metab Res 1999, 31:626-631 [DOI] [PubMed] [Google Scholar]
- 65.Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MA, Jr, Goeddel DV: The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci USA 1991, 88:9292-9296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tartaglia LA, Ayres TM, Wong GH, Goeddel DV: A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74:845-853 [DOI] [PubMed] [Google Scholar]
- 67.Bastelica D, Mavri A, Verdier M, Berthet B, Juhan-Vague I, Alessi M-C: Relationships between fibrinolytic and inflammatory parameters in human adipose tissue: strong contribution of TNFα receptors to PAI-1 levels. Thromb Haemost 2002, 88:481-487 [PubMed] [Google Scholar]
- 68.Schmid EF, Binder K, Grell M, Scheurich P, Pfizenmaier K: Both tumor necrosis factor receptors, TNFR60 and TNFR80, are involved in signaling endothelial tissue factor expression by juxtacrine tumor necrosis factor α. Blood 1995, 86:1836-1841 [PubMed] [Google Scholar]
- 69.Xu H, Uysal KT, Becherer JD, Arner P, Hotamisligil G: Altered tumor necrosis factor-α (TNF-α) processing in adipocytes and increased expression of transmembrane TNFα in obesity. Diabetes 2002, 51:1876-1883 [DOI] [PubMed] [Google Scholar]
- 70.Alexopoulou L, Pasparakis M, Kollias G: A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol 1997, 27:2588-2592 [DOI] [PubMed] [Google Scholar]
- 71.Kusters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E, Kunstle G, Bluethmann H, Wendel A, Pfizenmaier K, Kollias G, Grell M: In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol 1997, :2870-2875 [DOI] [PubMed] [Google Scholar]
- 72.Pinckard JK, Sheehan KCF, Schreiber RD: Ligand-induced formation of p55 and p75 tumor necrosis factor receptor heterocomplexes on intact cells. J Biol Chem 1997, 272:10784-10789 [DOI] [PubMed] [Google Scholar]
- 73.Carpenter A, Evans TJ, Buurman WA, Bemelmans MH, Moyes D, Cohen J: Differences in the shedding of soluble TNF receptors between endotoxin-sensitive and endotoxin-resistant mice in response to lipopolysaccharide or live bacterial challenge. J Immunol 1995, 155:2005-2012 [PubMed] [Google Scholar]
- 74.Aderka D: The potential biologial and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev 1996, 7:231-240 [DOI] [PubMed] [Google Scholar]
- 75.McFadden G, Graham K, Ellison K, Barry M, Macen J, Schreiber M, Mossman K, Nash P, Lalani A, Everett H: Interruption of cytokine networks by poxviruses: lessons from myxoma virus. J Leukoc Biol 1995, 57:731-738 [DOI] [PubMed] [Google Scholar]
- 76.Lesslauer W, Tabuchi H, Gentz R, Brockhaus M, Schlaeger EJ, Grau G, Piguet PF, Pointaire P, Vassalli P, Loetscher H: Recombinant soluble tumor necrosis factor receptor proteins protect mice from lipopolysaccharide-induced lethality. Eur J Immunol 1991, 21:2883-2886 [DOI] [PubMed] [Google Scholar]
- 77.Hunger RE, Carnaud C, Garcia I, Vassalli P, Mueller C: Prevention of autoimmune diabetes mellitus in NOD mice by transgenic expression of soluble tumor necrosis. Eur J Immunol 1997, 27:255-261 [DOI] [PubMed] [Google Scholar]
- 78.Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB: Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol 1993, 151:1548-1561 [PubMed] [Google Scholar]
- 79.Wooley PH, Dutcher J, Widmer MB, Gillis S: Influence of a recombinant human soluble tumor necrosis factor receptor FC fusion protein on type II collagen-induced arthritis in mice. J Immunol 1993, 151:6602-6607 [PubMed] [Google Scholar]
- 80.Hotamisligil GS, Shargill NS, Spiegelman BM: Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 1993, 259:87-91 [DOI] [PubMed] [Google Scholar]
- 81.Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D: Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med 1992, 175:323-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM: TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 1998, 160:943-952 [PubMed] [Google Scholar]