

Abstract
Apoptosis or programmed cell death is a cellular suicide mechanism that frequently occurs in advanced human atherosclerotic plaques. Caspases, a family of cysteine proteases, have been identified as important effectors of the death machinery. In this study, we report strong caspase-2 immunoreactivity in foam cells of macrophage-origin around the necrotic core of advanced human atherosclerotic plaques. In contrast, smooth muscle cells (SMCs) and macrophages in the fibrous cap as well as endothelial cells, medial SMCs, and SMCs from mammary arteries are negative for caspase-2. Caspase-2-positive macrophages were isolated from human plaques by laser capture microdissection and were then analyzed by Western blotting. A single band of ∼35 kd corresponding with the precursor of the short, anti-apoptotic isoform of caspase-2 (caspase-2S) could be identified. Treatment of human U937 macrophages with the DNA strand-breaking agents etoposide or camptothecin stimulated caspase-2S expression. Since atherosclerotic plaques contain a high number of DNA strand breaks, our results provide evidence for a survival factor in macrophage-derived foam cells of human atherosclerotic plaques that might be up-regulated in response to DNA damage.
Several studies have shown in situ evidence for apoptotic cell death in both animal and human atherosclerotic plaques. 1-7 Apoptosis is absent or barely detectable in normal arteries and early atherosclerotic lesions (<0.1% TUNEL-positive nuclei), but is much more pronounced in advanced plaques (1 to 2%). 1 All cell types in the plaque are involved, including smooth muscle cells (SMCs), macrophages, and T lymphocytes. Despite many efforts in determining the potential cell death mechanisms, the significance of apoptosis in atherosclerosis remains unclear. 4 Recent evidence suggests, however, that apoptotic cell death is a major determinant of the thrombogenicity of the plaque lipid core and a potential contributor to plaque erosion and associated thrombosis. 8
The execution phase of apoptosis generally depends on cytoplasmic cysteinyl aspartate-specific proteinases, called caspases, which are synthesized as inactive zymogens. 9 The primary structure of procaspases consists of an N-terminal prodomain and two subunits of approximately 10 and 20 kd (p10 and p20 subunit, respectively) which undergo processing to constitute the active enzyme. Once activated, caspases induce intracellular signaling pathways and cleave specific subsets of proteins to evoke the stereotyped sequence of structural changes typical of apoptotic cells. Intracellular proteolytic caspase signaling pathways operate in a network-like fashion so that the initial activation of one caspase can lead to the activation of multiple other family members. 10 At present, 14 members of the mammalian caspase family have been identified, several of which display overlapping specificities and apparent redundancy. They can be subdivided into several groups based on phylogenetic analyses and substrate recognition. Human caspase-2, initially described as Ich-1, is unique among the different members of the caspase-family because it has features of both upstream caspases (long prodomain) and downstream caspases (DEXD substrate specificity). 11 The prodomain of caspase-2 is essential for oligomerization and autoactivation as well as for nuclear migration and interaction with the death adaptor protein, RAIDD, indicating that caspase-2 could act as an upstream activator of the intracellular caspase cascade. 12-14 However, in vitro studies showed that caspase-2 cannot initiate a wide procaspase activation cascade 10 and with the exception of golgin-160, 15 no downstream targets for caspase-2 have yet been found. Alternative splicing of caspase-2 mRNA generates at least two isoforms: procaspase-2L, whose overexpression induces cell death and a truncated variant, caspase-2S, that is devoid of the small subunit (p10) and that has anti-apoptotic potential. 11
Previous reports have shown that caspase-1 and -3 colocalize with apoptotic cells in advanced human atherosclerotic plaques. 16-17 Since caspase inhibitors have shown promise in preclinical animal models for disorders like traumatic brain injury, amyotrophic lateral sclerosis, and Parkinson’s disease, 18 identification of caspases in atherosclerotic plaques might open new perspectives for the development of therapeutic strategies to alter the progression of atherogenesis. In this study, we show overexpression of caspase-2S in foam cells of macrophage origin around the necrotic core of advanced human atherosclerotic plaques. We also demonstrate that caspase-2S expression can be stimulated in vitro by DNA strand-breaking agents. Since elevated levels of DNA strand breaks can be found in both human and experimental atherosclerosis, 19-20 overexpression of caspase-2S in human plaques might be associated with enhanced levels of oxidative DNA damage.
Materials and Methods
Human Carotid Endarterectomy Specimens
Human carotid endarterectomy specimens were obtained from patients with a carotid stenosis of >70%, as demonstrated by digital subtraction angiography and duplex ultrasonography. The specimens were opened along their longitudinal axis and submerged in an alcohol-based fixative within 2 minutes after surgical removal. Complete longitudinal sections of paraffin-embedded specimens contained the inner wall of the distal common carotid artery, the proximal part of the external carotid artery and the carotid sinus. 6 According to the adapted American Heart Association (AHA) classification scheme as modified by Virmani et al 21 thin fibrous cap atheromata alternated with other stages of atherosclerosis (fibrous cap atheromata and intimal xanthomata) in the same specimen. Non-atherosclerotic mammary arteries obtained during bypass surgery were used as negative control samples and were manipulated similarly. The study was approved by the Review Board of the University of Antwerp.
Antibodies
The following antibodies directed against human caspase-2 were used: anti-caspase-2 mouse monoclonal antibody (clone G310–1248) from PharMingen (San Diego, CA) recognizing both procaspase-2L and caspase-2S as well as the proteolytic fragments p33 and p20 of procaspase-2L on Western blots, anti-caspase-2 mouse monoclonal antibody (clone 47) from Immunotech (Marseille Cedex, France; antibody only applicable for immunohistochemistry), and anti-procaspase-2L (C-20) rabbit polyclonal antibody from Santa Cruz Biotechnology (Santa Cruz, CA; for Western blot analysis and immunohistochemistry). Other antibodies used in this study were anti-cleaved caspase-3 rabbit polyclonal antibody (Biosource, Camarillo, CA), anti-β-actin mouse monoclonal antibody (clone AC-15; Sigma, St. Louis, MO), anti-CD68 (clone PG-M1), and anti-SC-35 mouse monoclonal antibody (clone SC-35; Sigma). Goat anti-mouse and sheep anti-rabbit peroxidase-conjugated secondary antibodies were purchased from Jackson (West Grove, PA) and DAKO (Glostrup, Denmark), respectively.
Cell Culture
Human U937 monocytes (American Type Culture Collection, Manassas, VA) were grown in RPMI 1640 medium (Life Technologies, Paisley, UK) supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 units/ml), streptomycin (100 μg/ml) and gentamicin (50 μg/ml) in an atmosphere of 95% air and 5% CO2. Cells were differentiated into macrophages by addition of 0.2 μmol/L phorbol 12-myristate 13-acetate (Sigma) to the medium for 24 hours. U937 macrophages were then exposed to 50 μmol/L etoposide or 10 μmol/L camptothecin (Sigma) for 0, 2, 6, 12, or 24 hours. In some experiments a cocktail of human IFN-γ (1000 units/ml) and TNF-α (30 ng/ml) was added to the macrophages for 0, 1, 2, 3, or 4 days.
Immunohistochemistry and DNA in Situ End Labeling
The immunohistochemical reactions were carried out by an indirect peroxidase antibody conjugate method. 7 For the detection of oligonucleosomal DNA cleavage, a stringent terminal deoxynucleotidyl transferase end labeling (TUNEL)-technique was used. 22 TUNEL-staining was combined with an immunohistochemical stain for SC-35 to avoid aspecific labeling. 22
Laser Capture Microdissection (LCM)
Ten-μm thick sections of carotid endarterectomy specimens were mounted on Optiplus slides (Biogenex, San Ramon, CA) and deparaffinized in toluol (2 × 3 minutes). Slides were washed with isopropylalcohol (1 minute), 70% ethanol (1 minute each), water (1 minute), and rapidly stained with hematoxylin and eosin (15 seconds each). Next, sections were washed with water (1 minute), dehydrated with an ethanol gradient [70% ethanol (1 minute), 90% ethanol (1 minute), 100% ethanol (2 × 1 minute)], washed with xylene (5 minutes), and air dried (20 minutes). Caspase-2 immunoreactive areas were microdissected from 10 tissue sections using the Pixcell II LCM system (Arcturus Engineering Inc., Mountain View, CA). An adjacent immunostained section was used as a guide for the microdissection (navigated-LCM).
Protein Isolation and Immunoblot Assays of Microdissected Cells
Microdissected cells were lysed by adding 20 μl of Laemmli sample buffer (BioRad, Richmond, CA) to the LCM caps. Cell lysates were heat denatured for 5 minutes and loaded on a 12.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel. Western blotting to Hybond-enhanced chemiluminescence membranes (Amersham Pharmacia Biotech, Rainham, UK) was performed according to standard procedures. Antibody detection was accomplished with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL). Signals were visualized using a Lumi-imager (Roche Diagnostics, Mannheim, Germany). Long-term exposures (30 minutes) and 2 × 2 binning settings (ie, conversion of four adjacent pixels to one “super” pixel) were applied to enhance sensitivity.
Results
Overexpression of Caspase-2S in Human Atherosclerotic Plaques
Strong immunoreactivity for caspase-2 was found in foam cells of macrophage-origin (CD68-positive cells) around the necrotic core of advanced human atherosclerotic plaques (thin fibrous cap atheromata of carotid endarterectomy specimens) (Figure 1) ▶ . Caspase-2 immunoreactivity was found both in the cytoplasm and nucleus of the labeled cells. Cells from the fibrous cap including macrophages and SMCs as well as endothelial cells, medial SMCs, and SMCs from mammary arteries were negative for caspase-2. We also found that some CD68-positive cells in the necrotic core were caspase-2-negative. To determine whether the proapoptotic isoform caspase-2L or the truncated anti-apoptotic variant caspase-2S was up-regulated, caspase-2-positive cells were isolated from the plaque by laser capture microdissection (Figure 2, A and B) ▶ . After SDS-PAGE and Western blotting, we were able to detect a single band of ∼35 kd which corresponds with caspase-2S (Figure 2C) ▶ . The procaspase-2L precursor (48 kd) or cleaved fragments of procaspase-2L could not be identified. Extracts from microdissected regions of the plaque that were negative for caspase-2 did not reveal detectable amounts of caspase-2 on Western blots, neither caspase-2S nor procaspase-2L. When carotid endarterectomy specimens were stained for procaspase-2L, macrophages around the necrotic core were negative, suggesting that the anti-apoptotic isoform is up-regulated. Moreover, caspase-2S immunoreactive macrophages did not show signs of apoptotic cell death (cleaved caspase-3-negative, Figure 2C ▶ ) and were not labeled by the TUNEL technique.
Figure 1.
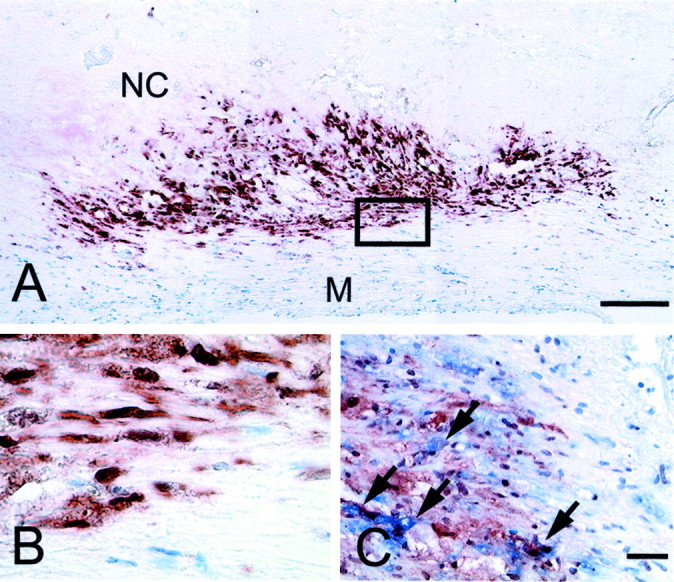
Overexpression of caspase-2 in advanced atherosclerotic plaques of human carotid endarterectomy specimens. A: Low-power photomicrograph of an advanced plaque stained for caspase-2. The plaque necrotic core (NC) is surrounded by a layer of foam cells that show strong caspase-2 expression. Smooth muscle cells from the underlying media (M) are caspase-2-negative. Bar, 200 μm. B: High-power photomicrograph of the boxed area of panel A. Caspase-2 could be detected both in the nucleus and the cytoplasm of the immunoreactive cells. C: Double immunohistochemical stain for caspase-2 (brown) and CD68 (blue). CD68-positive cells show both nuclear and cytoplasmic caspase-2 colocalization (arrows). Bar, 50 μm.
Figure 2.
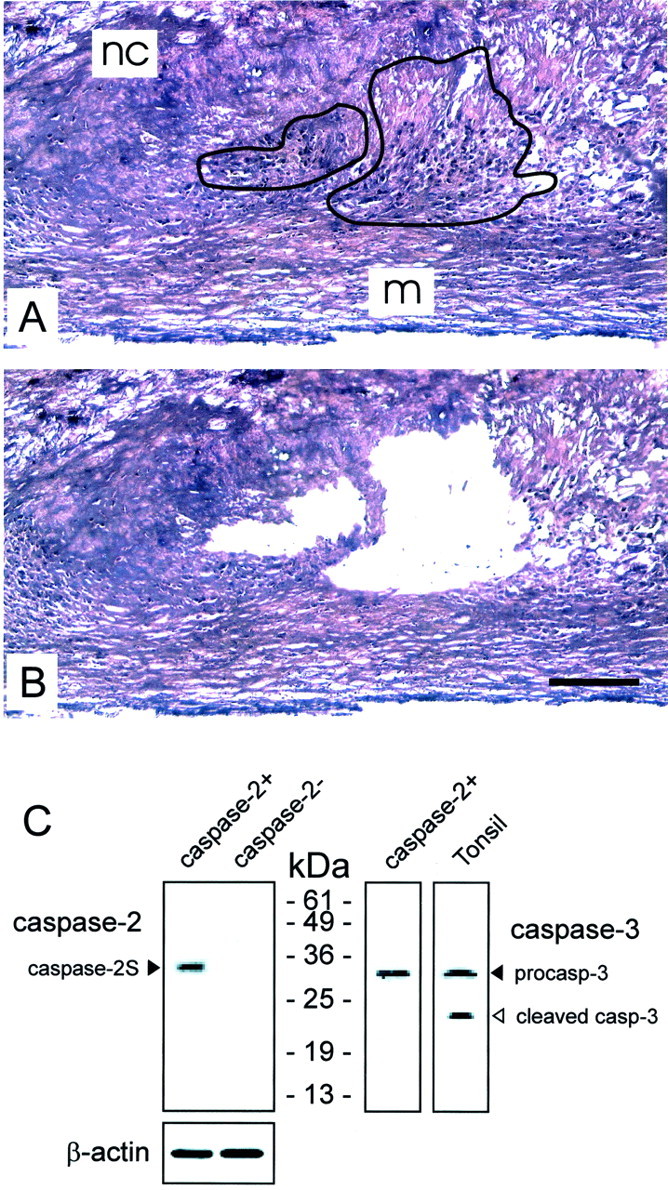
Laser capture microdissection (LCM) of caspase-2 immunoreactive areas in carotid endarterectomy specimens. A–B: Tissue section of the same carotid endarterectomy specimen as shown in Figure 1 ▶ before (A) and after (B) LCM. Sections were stained with hematoxylin and eosin. A serial section was immunostained for caspase-2 to guide the microdissection. Caspase-2 immunoreactive areas (delineated in panel A) were localized around the necrotic core (nc) but not in normal media (m). Less advanced stages of atherosclerosis without necrotic core formation in the same specimen were used as negative control tissue. Bar, 200 μm. C: Western blot analysis of caspase-2 and -3 in caspase-2 immunoreactive cells (caspase-2+) and caspase-2-negative cells (caspase-2−) that were isolated by LCM (see panels A and B). Microdissected cells from hyperplastic tonsils were used as a positive control for cleavage of caspase-3.
Up-Regulation of Caspase-2S in U937 Macrophages
Because the DNA strand-breaking agent etoposide can induce expression of caspase-2 and -3 genes in human tumor cells, 23 U937 macrophages were treated with etoposide and with a related DNA-damaging compound, camptothecin, for up to 24 hours. In both cases, apoptotic cell death occurred within 6 hours of treatment as shown by the activation of caspase-3, cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) (Figure 3, A and B) ▶ and the appearance of apoptotic bodies (not shown). Expression of PARP-1 was up-regulated, especially in cells that were treated with etoposide. Apoptotic cell death was associated with cleavage of procaspase-2L into active p20 fragments (Figure 3, A and B) ▶ . This resulted in a progressive loss of procaspase-2L levels during treatment. We also observed the intermediate proteolytic fragment p33 consisting of the prodomain and p18 subunit (Figure 3, A and B) ▶ . In contrast to procaspase-2L, caspase-2S was up-regulated in response to the cytotoxic effects of etoposide and camptothecin. Elevated levels of caspase-2S protein were detectable after 2 hours of treatment (Figure 3, A and B) ▶ . At this time point, the majority of cells were not in the execution phase of apoptosis as cleavage of caspase-3 and its substrate PARP-1 did not occur. Due to rapid cell death, caspase-2S could not be detected when cells were incubated for longer than 2 hours. U937 cells treated with a cocktail of the pro-inflammatory cytokines TNF-α and IFN-γ initiated apoptotic cell death, but did not up-regulate caspase-2 isoforms, even after 4 days of treatment (data not shown). This suggests that apoptotic U937 cells do not necessarily up-regulate caspase-2S expression to prevent cell death.
Figure 3.

Up-regulation of caspase-2S in U937 cells after treatment with DNA-damaging agents. U937 cells were treated with 50 μmol/L etoposide (A) or 10 μmol/L camptothecin (B) up to 24 hours. Caspase-2 expression was verified by Western blotting using an antibody that recognizes both caspase-2L and caspase-2S (top panel) and an antibody specific for procaspase-2L. Part of the upper blot was enlarged to distinguish caspase-2S (35 kd) from the intermediate pro/p18 fragment (33 kd) of caspase-2L. Apoptotic cell death was demonstrated by evaluating cleavage of both caspase-3 and poly(ADP-ribose) polymerase 1 (PARP-1). β-actin expression was used as a control for protein loading.
Discussion
Caspases are cysteine proteases that play an essential role in apoptosis by cleaving several key cellular proteins. 9 In this study, we found overexpression of the short isoform of caspase-2 (caspase-2S) in foam cells of macrophage origin around the necrotic core of advanced human atherosclerotic plaques. Both cytoplasmic and nuclear staining could be observed. This is in accordance with subcellular localization studies of procaspase-2L which has been found in the Golgi apparatus and intermembrane space of mitochondria as well as in the nucleus of viable cells. 12,24,25 The intracellular localization of caspase-2S is currently unknown but could be similar.
In contrast to procaspase-2L, caspase-2S behaves like an endogenous inhibitor of apoptotic cell death. 11,26 Indeed, overexpression of caspase-2S can inhibit nuclear changes associated with apoptotic cell death. 26 Caspase-2S also prevents the maturation of apoptotic bodies, delays phosphatidylserine externalization on the plasma membrane of dying cells and prevents cleavage and activation of procaspase-2L. 26 Therefore, it is conceivable that caspase-2 immunoreactive cells in human atherosclerotic plaques are TUNEL-negative and do not contain active caspase-3. However, cleavage of procaspase-3, procaspase-7, and poly(ADP-ribose) polymerase as well as the fragmentation of nuclear DNA are not affected in caspase-2S overexpressing cells, suggesting that caspase-2S only interferes with selective features of apoptosis. 26
In comparison with monocytes, activated macrophages are resistant to numerous death stimuli, including death receptor ligation, anti-neoplastic agents, and ionizing radiation. This suggests up-regulation of survival factors during macrophage differentiation. At present, the mechanisms responsible for macrophage resistance to apoptosis and persistance in pathological conditions are poorly understood. Perlman et al 27 demonstrated that overexpression of FADD-like ICE (FLICE)-inhibitory protein in macrophages confers resistance to Fas-mediated apoptosis. Other studies suggest that macrophages develop several anti-apoptotic mechanisms by uptake of oxLDL or agLDL. 5 Importantly, enhanced caspase gene expression is not a frequent event because caspases are regulated predominantly via post-translational mechanisms such as cleavage of the precursor enzyme and subcellular relocalization. 9,25 However, the DNA-damaging agent etoposide can increase caspase-2 and -3 expression in various human tumor cells. 23 Etoposide is a cytotoxic drug that complexes with topoisomerase II and DNA to enhance double-strand and single-strand DNA breaks. 28 According to recent evidence, caspase-2L seems to provide an important link between etoposide-induced DNA damage, cytochrome C release from the mitochondria, and cleavage of procaspase-9 and -3. 29,30 Since procaspase-2L needs an adaptor molecule for activation, 14,31,32 it is possible that etoposide-induced activation of procaspase-2L triggers the formation of a nuclear signaling complex. In the present study, we could demonstrate overexpression of caspase-2S when U937 macrophages were treated with the DNA-damaging agents, etoposide and camptothecin. Up-regulation of caspase-2S was only evident in the early stages of treatment, ie, before cells entered the execution phase of apoptosis. This implicates that caspase-2S is rapidly degraded during cell death. Recently, we observed an extensive formation of DNA strand breaks in both human and experimental atherosclerotic plaques using single cell gel electrophoresis assays. 19,20 Since this method requires cell lysis and removal of the cytoplasmic content, a direct relation between DNA strand breaks and caspase-2S overexpression in human plaques cannot be determined. However, DNA strand break formation was associated with the up-regulation of several DNA repair enzymes including poly(ADP-ribose) polymerase 1 (PARP-1). The latter enzyme is activated by DNA strand breaks to participate in DNA repair 33 and is predominantly expressed in macrophage-derived foam cells of advanced atherosclerotic lesions. 19,20 Around the necrotic core of human plaques, macrophages also express inducible nitric oxide synthase and contain large amounts of nitrotyrosine and oxidized lipids. 34 Therefore, overexpression of caspase-2S and oxidative DNA damage in human plaques may be coherent events, especially in macrophages that are subjected to high levels of oxidative stress. It is important to note that intracellular reactive oxygen species (ROS) are key players in oxidative tissue injury of human plaques, including the formation of DNA strand breaks, so that free radical attack of DNA may be a potential underlying cause of caspase-2S overexpression. However, in contrast to etoposide and camptothecin, a direct in vitro link between ROS and caspase-2S gene expression remains difficult to prove as ROS modulate a variety of signaling pathways in which many gene products are involved. 35 It is currently unknown how DNA strand breaks induce alternative splicing yielding high levels of caspase-2S transcript. Caspase-2L is derived from the skipping of alternative exon 9 in the pre-mRNA whereas caspase-2S is a truncated version of the protein due to inclusion of exon 9 (containing a premature termination codon). Recent evidence suggests that the polypyrimidine track-binding protein (PTB/hnRNP I) binds downstream of the alternative exon 9, thereby preventing exon 9 inclusion and caspase-2S formation. 36 Possibly, DNA strand breaks affect the activity of PTB/hnRNP I or modulate its subcellular localization.
Taken together, our data indicate that the short anti-apoptotic isoform of caspase-2 is up-regulated in macrophage-derived foam cells around the necrotic core of human atherosclerotic plaques. Since DNA-damaging agents cause caspase-2S up-regulation in vitro, caspase-2S may act as a nuclear sensor to detect DNA strand breaks and may help macrophages to survive increased levels of oxidative DNA damage. This hypothesis supports the concept that macrophages in the interior of atheroma lesions need mechanisms to cope with various cytotoxic plaque components such as oxidized lipids and free radicals that may cause dysfunction and, possibly, death of the inflammatory cells.
Acknowledgments
We thank Martine De Bie and Jeff Thielemans for their excellent technical assistance.
Footnotes
Address reprint requests to Dr. Mark M. Kockx, Department of Pathology, A.Z. Middelheim, Lindendreef 1, B-2020 Antwerp, Belgium. E-mail: mark.kockx@uia.ua.ac.be.
Supported by the Fund for Scientific Research-Flanders (project numbers 1.5.206.00, G.0080.98, and G.0180.01). M.M. Kockx is holder of a fund for fundamental clinical research of the Fund for Scientific Research-Flanders.
References
- 1.Kockx MM: Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol 1998, 18:1519-1522 [DOI] [PubMed] [Google Scholar]
- 2.McCarthy NJ, Bennett MR: The regulation of vascular smooth muscle cell apoptosis. Cardiovasc Res 2000, 45:747-755 [DOI] [PubMed] [Google Scholar]
- 3.Mallat Z, Tedgui A: Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol 2000, 130:947-962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kockx MM, Herman AG: Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res 2000, 45:736-746 [DOI] [PubMed] [Google Scholar]
- 5.Martinet W, Kockx MM: Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol 2001, 12:535-541 [DOI] [PubMed] [Google Scholar]
- 6.Kockx MM, De Meyer GRY, Muhring J, Jacob W, Bult H, Herman AG: Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 1998, 97:2307-2315 [DOI] [PubMed] [Google Scholar]
- 7.Kockx MM, De Meyer GRY, Buyssens N, Knaapen MWM, Bult H, Herman AG: Cell composition, replication, and apoptosis in atherosclerotic plaques after 6 months of cholesterol withdrawal. Circ Res 1998, 83:378-387 [DOI] [PubMed] [Google Scholar]
- 8.Mallat Z, Tedgui A: Current perspective on the role of apoptosis in atherothrombotic disease. Circ Res 2001, 88:998-1003 [DOI] [PubMed] [Google Scholar]
- 9.Nicholson DW: Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 1999, 6:1028-1042 [DOI] [PubMed] [Google Scholar]
- 10.Van de Craen M, Declercq W, Van den brande I, Fiers W, Vandenabeele P: The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ 1999, 6:1117-1124 [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Miura M, Bergeron L, Zhu H, Yuan J: Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994, 78:739-750 [DOI] [PubMed] [Google Scholar]
- 12.Colussi PA, Harvey NL, Kumar S: Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor: a novel function for a caspase prodomain. J Biol Chem 1998, 273:24535-24542 [DOI] [PubMed] [Google Scholar]
- 13.Butt AJ, Harvey NL, Parasivam G, Kumar S: Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J Biol Chem 1998, 273:6763-6768 [DOI] [PubMed] [Google Scholar]
- 14.Duan H, Dixit VM: RAIDD is a new “death” adaptor molecule. Nature 1997, 385:86-89 [DOI] [PubMed] [Google Scholar]
- 15.Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA, Rosen A: Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol 2000, 149:603-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mallat Z, Ohan J, Lesèche G, Tedgui A: Colocalization of CPP-32 with apoptotic cells in human atherosclerotic plaques. Circulation 1997, 96:424-428 [DOI] [PubMed] [Google Scholar]
- 17.Geng Y-J, Libby P: Evidence for apoptosis in advanced human atheroma: colocalization with interleukin-1β-converting enzyme. Am J Pathol 1995, 147:251-266 [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholson DW: From bench to clinic with apoptosis-based therapeutic agents. Nature 2000, 407:810-816 [DOI] [PubMed] [Google Scholar]
- 19.Martinet W, Knaapen MWM, De Meyer GRY, Herman AG, Kockx MM: Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106:927-932 [DOI] [PubMed] [Google Scholar]
- 20.Martinet W, Knaapen MWM, De Meyer GRY, Herman AG, Kockx MM: Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 2001, 88:733-739 [DOI] [PubMed] [Google Scholar]
- 21.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM: Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000, 20:1262-1275 [DOI] [PubMed] [Google Scholar]
- 22.Kockx MM, Muhring J, Knaapen MWM, De Meyer GRY: RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am J Pathol 1998, 152:885-888 [PMC free article] [PubMed] [Google Scholar]
- 23.Droin N, Dubrez L, Eymin B, Renvoizé C, Bréard J, Dimanche-Boitrel MT, Solary E: Up-regulation of casp genes in human tumor cells undergoing etoposide-induced apoptosis. Oncogene 1998, 16:2885-2894 [DOI] [PubMed] [Google Scholar]
- 24.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prévost M-C, Alzari PM, Kroemer G: Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med 1999, 189:381-393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhivotovsky B, Samali A, Gahm A, Orrenius S: Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ 1999, 6:644-651 [DOI] [PubMed] [Google Scholar]
- 26.Droin N, Rébé C, Bichat F, Hammann A, Bertrand R, Solary E: Modulation of apoptosis by procaspase-2 short isoform: selective inhibition of chromatin condensation, apoptotic body formation, and phosphatidylserine externalization. Oncogene 2001, 20:260-269 [DOI] [PubMed] [Google Scholar]
- 27.Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM: FLICE-inhibitory protein expression during macrophage differentiation confers resistance to Fas-mediated apoptosis. J Exp Med 1999, 190:1679-1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wozniak AJ, Ross WE: DNA damage as a basis for 4′-demethylepipodophyllotoxin-9-(4, 6-O-ethylidene-β-D-glucopyranoside) (etoposide) cytotoxicity. Cancer Res 1983, 43:120-124 [PubMed] [Google Scholar]
- 29.Roberston JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S: Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem 2002, 277:29803-29809 [DOI] [PubMed] [Google Scholar]
- 30.Lassus P, Opitz-Araya X, Lazebnik Y: Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002, 297:1352-1354 [DOI] [PubMed] [Google Scholar]
- 31.Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, Alnemri ES: CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res 1997, 57:615-619 [PubMed] [Google Scholar]
- 32.Shearwin-Whyatt LM, Harvey NL, Kumar S: Subcellular localization and CARD-dependent oligomerization of the death adaptor RAIDD. Cell Death Differ 2000, 7:155-165 [DOI] [PubMed] [Google Scholar]
- 33.Pieper AA, Verma A, Zhang J, Snyder SH: Poly (ADP-ribose) polymerase, nitric oxide, and cell death. Trends Pharmacol Sci 1999, 20:171-181 [DOI] [PubMed] [Google Scholar]
- 34.Cromheeke KM, Kockx MM, De Meyer GRY, Bosmans JM, Bult H, Beelaerts WJF, Vrints CJ, Herman AG: Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res 1999, 43:744-754 [DOI] [PubMed] [Google Scholar]
- 35.Kunsch C, Medford RM: Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 1999, 85:753-766 [DOI] [PubMed] [Google Scholar]
- 36.Côté J, Dupuis S, Wu JY: Polypyrimidine track-binding protein binding downstream of caspase-2 alternative exon 9 represses its inclusion. J Biol Chem 2001, 276:8535-8543 [DOI] [PMC free article] [PubMed] [Google Scholar]