

Abstract
Asbestos fibers up-regulate the extracellular signal-regulated kinase (ERK1/2) pathway in mesothelial and pulmonary epithelial cells in vitro, but the cell-type expression patterns and intracellular localization of activated, ie, phosphorylated, ERK in the lung after inhalation of asbestos are unclear. C57/BL6 mice were exposed to 7-mg/m3 air of crocidolite asbestos for 5 and 30 days, the times required for the development of epithelial cell hyperplasia and fibrotic lesions, respectively. Exposure to asbestos caused striking increases in both unphosphorylated and phosphorylated ERK (p-ERK), which were most marked at 30 days and co-localized in bronchiolar and alveolar epithelial cells using an antibody to cytokeratin. Alveolar macrophages, detected with an anti-macrophage antibody, did not express p-ERK. p-ERK was localized at the apical cell surface of bronchiolar and alveolar type II epithelial cells exposed to asbestos fibers, and was most marked in areas of epithelial hyperplasia in association with fibrotic lesions. Because translocation of p-ERK to the nucleus is associated with activation of early response genes and transcription factors, laser scanning cytometry was used to determine the kinetics of activation and nuclear translocation of p-ERK in an alveolar type II epithelial cell line in vitro after exposure to asbestos or the ERK stimuli, epidermal growth factor, or H2O2. Results showed that cytoplasmic to nuclear translocation of p-ERK occurred in a protracted manner in cells exposed to asbestos. The immunolocalization of p-ERK at the membrane surface, a site of initial exposure to asbestos fibers, and the chronic activation of p-ERK in epithelial cells at sites of fibrogenesis are consistent with the concept that epithelial cell signaling through the ERK pathway contributes to remodeling of the lung during the development of pulmonary fibrosis.
Asbestos is a family of ubiquitous, naturally occurring fibers used historically in more than 2000 industrial products. 1 Occupational exposures to asbestos induce nonmalignant fibroproliferative diseases such as asbestosis as well as a number of malignancies including lung cancer and malignant mesothelioma. 1-4 A common feature of both chrysotile [Mg6Si4O10(OH)8] and crocidolite [(Na2(Fe3+)2(Fe2+)3Si8O22(OH)2] asbestos fibers is their ability to cause epithelial cell proliferation in rodent lungs after inhalation. 4 This event may be critical in repair of lung injury and/or the initiation of lung cancers.
The extracellular signal-regulated kinases (ERK1/2) of the mitogen-associated protein kinase (MAPK) cascade have been linked causally to the advent of DNA synthesis in alveolar epithelial cells in vitro after exposure to asbestos. 5 Work to date suggests that crocidolite asbestos fibers cause activation of ERK1/2 via phosphorylation and aggregation of the epidermal growth factor receptor (EGFR). 5-9 In this regard, asbestos is one of several nonligands including polycations, 10 ultraviolet irradiation, 11 X-irradiation, 12 and H2O2, 13 that activate growth factor receptors and signaling cascades leading to cell injury and/or proliferation.
After inhalation of chrysotile fibers by mice, increased expression of phosphorylated or activated ERK1/2 (p-ERK) is observed by immunoperoxidase staining in cells at the alveolar duct junction where asbestos fibers initially deposit and accumulate. 14 However, fibrotic lesions, as evidenced by the development of Masson’s trichrome-positive cells and elevations in levels of hydroxyproline, do not develop throughout a 6-week period of exposure to asbestos. In work here, we used a murine model of asbestosis after chronic inhalation of crocidolite asbestos to demonstrate the patterns of expression of native and phosphorylated ERK1 and 2 proteins, the major mammalian forms of ERK, throughout time, and the cell types involved. Data show that p-ERK signaling is striking, protracted, and restricted to epithelial cells. Using confocal scanning laser microscopy (CSLM) and immunocytochemistry, we show p-ERK accumulation at the apical surface of bronchiolar epithelial cells, sites of direct contact with asbestos fibers. Because translocation of p-ERK from cytoplasm to nucleus is associated with activation and phosphorylation of the promoter regions of a number of key activator protein-1 (AP-1) family members such as c-fos, we also used laser scanning cytometry (LSC) to measure the kinetics of p-ERK translocation in alveolar type II epithelial cells (C10 line) in vitro after addition of asbestos, EGF, or H2O2. Results show that nuclear translocation of p-ERK is more delayed after exposure to asbestos in contrast to soluble stimuli, an observation consistent with the hypothesis that physical interactions of asbestos with epithelial cells, both in vitro and in vivo, initiate cell-signaling events leading to cell proliferation.
Materials and Methods
Inhalation Exposures
Inhalation exposures were performed as previously described 14,15 using groups (n = 5 or 6 per group per time point) of 8- to 12-week-old C57/BL6 mice exposed to ambient air or the National Institute of Environmental Health Sciences (NIEHS) reference sample of crocidolite asbestos (7 mg/m3 air) for 6 hours per day. Mice were killed at 5 and 30 days after exposure via a lethal injection of pentobarbital.
Tissue Processing
The chest cavity was opened, and a polyurethane catheter was inserted into the trachea above the brachial branching point and the right lung lobes clamped off, excised, and frozen in liquid nitrogen. Left lung lobes were instilled at an atmospheric pressure of 25 cm for 5 minutes with 4% paraformaldehyde in phosphate-buffered saline (PBS), warmed to 37°C. The tissues were next placed in a tissue cassette overnight in 4% paraformaldehyde in PBS at 4°C before conventional embedding in paraffin. Lung sections were cut at a thickness of 7 to 8 μm and stained with Masson’s trichrome to delineate areas of fibrosis.
Cryosections
A small section of the paraformaldehyde-fixed upper left lung lobe was taken for use as frozen tissue sections. Tissue was transferred to vials containing 10 ml of PBS and rinsed for at least 1 hour before freezing. The tissue was cryoprotected in O.C.T. (Tissue-Tek O.C.T. compound; Sakura Finetek USA, Torrance, CA) and rapidly frozen in dry-ice-cooled 100% methanol. The frozen tissue blocks were transferred for storage at −80°C until the time of cryostat sectioning.
Conventional Bright-Field Microscopy
Trichrome-stained paraffin sections were imaged with an Olympus BX50 upright light microscope (Olympus America, Lake Success, NY) and images captured in digital format with an Optronics MagnaFire charge-coupled device camera operating with MagnaFire version 2.0 software (Optical Analysis, Nashua, NH).
Immunofluorescence Staining Protocols
Ten-μm thick frozen sections were cut with steel blades on a cryostat, and placed onto Superfrost +/+ slides. The sections on the slides were treated for 10 minutes with 100% methanol cooled to −20°C, followed by 1% Triton X-100 in PBS for 15 minutes at room temperature. The slides were then washed three times for 5 minutes each in PBS at room temperature. Slides were then treated for 5 minutes at room temperature with 1% sodium dodecyl sulfate in PBS, followed by rinses in PBS three times for 5 minutes at room temperature. Nonspecific antibody binding was blocked by treating the slides with normal goat serum (Vector Laboratories, Burlingame, CA) at a concentration of 1:100 in PBS, for three times at 20 minutes. The slides were then incubated with the primary antibody overnight at 4°C in a humid chamber. Two antibodies (Abs) were successfully used for the detection of p-ERK [p44/42 tyrosine kinase MAP Ab (New England Bio Labs, Beverly, MA), and anti-active MAPK rabbit polyclonal Ab (Promega Corp., Madison, WI)]. Both antibodies were used at a concentration of 1:250 diluted in normal goat serum. After incubation, the slides were washed three times for 5 minutes each in PBS and then incubated with the secondary antibody. A rabbit biotinylated IgG kit (Vector Laboratories) was used following the manufacturer’s instructions. The biotinylated secondary antibody was applied for 1 hour at room temperature in a humid chamber. Visualization of antibody-binding sites was achieved using a streptavidin-conjugated Alexa 568 fluorophore (5 μg/ml for 1 hour at room temperature; Molecular Probes, Eugene, OR). After washing three times for 5 minutes in PBS, a 15-μl drop of n-propyl gallate in PBS/glycerol was deposited onto the section followed by the addition of a coverslip. Samples were then stored at −20°C in the dark until microscopic evaluation.
Dual-Immunofluorescence Labeling
To determine the lung cell types expressing ERK and p-ERK immunoreactivity, cryostat sections were double stained with an anti-44/42 ERK or p-ERK antibody and either an anti-macrophage marker (Mac-3; PharMingen International, San Diego, CA) or an anti-epithelial cell marker (pan-cytokeratin; DAKO Corp., Carpinteria, CA). The ERK/p-ERK immunoreactivity was detected with a streptavidin-conjugated Alexa 568 as described above, the Mac3 antibody was detected with a goat anti-rat IgG conjugated to Alexa 488 (Molecular Probes), and the cytokeratin antibody was detected with a goat anti-mouse IgG conjugated to allophycocyanin (Molecular Probes).
Alternate MAPK Antibodies
To confirm the staining results and cellular patterns seen with the p44/42 tyrosine kinase antibody a different antibody directed against the same epitope was used. In this case an anti-active MAPK (Promega Corp.) antibody was used. Following the same procedure described, this second MAP kinase antibody was used at a concentration of 1:250 overnight at 4°C in a humid chamber. All other aspects of the protocol were identical.
CSLM
The stained sections (from experiments done in triplicate) were examined with a Bio-Rad MRC 1024ES CSLM system (Bio-Rad Laboratories, Hercules, CA,). The system consists of a mixed gas krypton/argon laser (with lines of excitation at 488, 568, and 647 nm) with a scanning head mounted on an Olympus BX 50 upright light microscope. The system is controlled by Bio-Rad Laser Sharp software version 3.2. Immunostaining of the anti p-ERK and the anti-active MAPK antibodies were visualized with a streptavidin-conjugated Alexa 568 secondary antibody that is excited by the 568-nm wavelength line. Immunostaining of the anti-macrophage antibody was detected by excitation of the Alexa 488-conjugated secondary antibody with the 488-nm excitation line and the pan-cytokeratin antibody was visualized by the excitation of an allophycocyanin-conjugated secondary antibody with the 647-nm wavelength line.
All images were captured in a 512 × 512 pixel format directly onto a 100-MB Zip disk. Dual-labeled sections were imaged in sequential capture mode (to avoid fluorescence bleed-through), and the single channel gray-scale images electronically merged. Image montages were created and labeled in Microsoft PowerPoint (Microsoft, Redmond, WA) and printed with a Fujix Pictography 3500 digital image printer (Fuji Photo Film USA, Elmsford, NY).
Cell Cultures
A contact-inhibited, nontransformed murine alveolar type II epithelial cell line (C10), 16 was propagated in CMRL-1066 medium containing penicillin, streptomycin, l-glutamine, and 10% fetal bovine serum (Life Technologies, Inc., Grand Island, NY). For all experiments, cells were grown to near confluency, complete medium was removed, and serum-free medium was added 24 hours before exposures to agents. Mouse EGF (5 ng/ml medium; Upstate Biotechnology, Lake Placid, NY) or H2O2 (300 μmol/L; Sigma Chemical Co., St. Louis, MO), was added directly to medium at concentrations inducing ERK1/2 activation. 5 Crocidolite asbestos fibers (NIEHS reference sample) were suspended in Hanks’ balanced salt solution at 1 mg/ml, triturated 10 times through a 22-gauge needle, and added directly to medium at a final concentration of 5 μg/cm2 per dish. Sham control cultures received medium without agents and were treated identically. Groups in all experiments consisted of two to three determinations per time point.
Cell Immunostaining and LSC
Cultured C10 cells were treated with agents as described above, and fixed for 30 minutes with 4% paraformaldehyde in PBS, followed by permeabilization in 100% methanol for 10 minutes at −20°C and then 0.1% Triton X-100 in PBS for 15 minutes at room temperature. The cells were then rinsed for 30 minutes with PBS containing 0.1% Triton X-100 and 2% nonfat milk powder, followed by PBS containing 1% bovine serum albumin (BSA). The cells were incubated with anti-p-ERK antibody (1:250 dilution in PBS/BSA) overnight at 4°C, followed by incubation with Alexa 488 goat anti-rabbit IgG conjugate (1:200 dilution in PBS/BSA) for 1 hour at room temperature. After brief rinses with PBS, the cells were stained with propidium iodide solution (20 μg/ml with 0.2 mg/ml ribonuclease A and 0.5 mmol/L ethylenediaminetetraacetic acid) for 25 minutes at room temperature. Finally, the coverslips containing the cells were washed and inverted onto a drop of anti-fade mounting medium (KPL mounting medium; Kirkegaard and Perry Laboratories, Gaithersburg, MD) on a glass slide.
Nuclear translocation of activated ERK1/2 was assayed with a laser scanning cytometer (CompuCyte Corp., Cambridge, MA) essentially according to the method of Deptala and co-workers. 17 Instrument scan areas were set to include at least 5000 cells for analysis per coverslip. The slides were scanned with a ×20 objective lens using the 488-nm wavelength argon-ion laser and red and green detectors. The primary contouring parameter used to detect and quantify cells was set on the red fluorescence emitted by the propidium iodide as previously described. 5,18 Detector gain voltages were set so that the brightest pixels scanned were no more than 75% of saturated values. The threshold contour was set within the nucleus at a distance one-third from the nuclear border, using the instrument’s scan data display function. 5 The green fluorescence (integrated pixel value) associated with p-ERK was measured in the cytoplasmic and nuclear compartments as follows. The nuclear fraction of stain (Fn) included the triggering threshold plus four pixels toward the outside of the threshold. This area covered the entire nucleus, and the green fluorescence signal integrated within this area represented p-ERK fluorescence within the nuclear compartment. The cytoplasmic fraction of p-ERK staining (Fc) was determined using peripheral contouring. These contours were set to measure the integrated fluorescence within a cytoplasmic boundary eight pixels outside of the measured nuclear area. Background fluorescence signal was measured outside of the cell and automatically subtracted from the integrated nuclear and cytoplasmic values.
Results
We first used our murine model of asbestosis to characterize the time frame of ERK1/2 expression in relationship to the development of epithelial cell proliferation and fibrosis. At 5 days, a time point when increased numbers of bronchiolar and alveolar epithelial cells express proliferating cell nuclear antigen, 19 expression of nonphosphorylated and phosphorylated ERKs appeared comparable in sham and asbestos-exposed mice (Figure 1; A to D) ▶ . In contrast, increases in both forms were noted at 30 days after exposures to asbestos, a time period when inflammation and fibrosis occurred in lungs (Figure 1; E to H) ▶ . The localization of p-ERKs was particularly dramatic on the apical surface of distal bronchiolar epithelial cells where it appeared to be concentrated at the plasma membrane. Immunoreactivity was most pronounced in bronchiolar epithelial cells in areas of hyperplasia and in what appeared to be alveolar type II epithelial cells in the peripheral lung, although all bronchioles examined showed a similar staining pattern.
Figure 1.
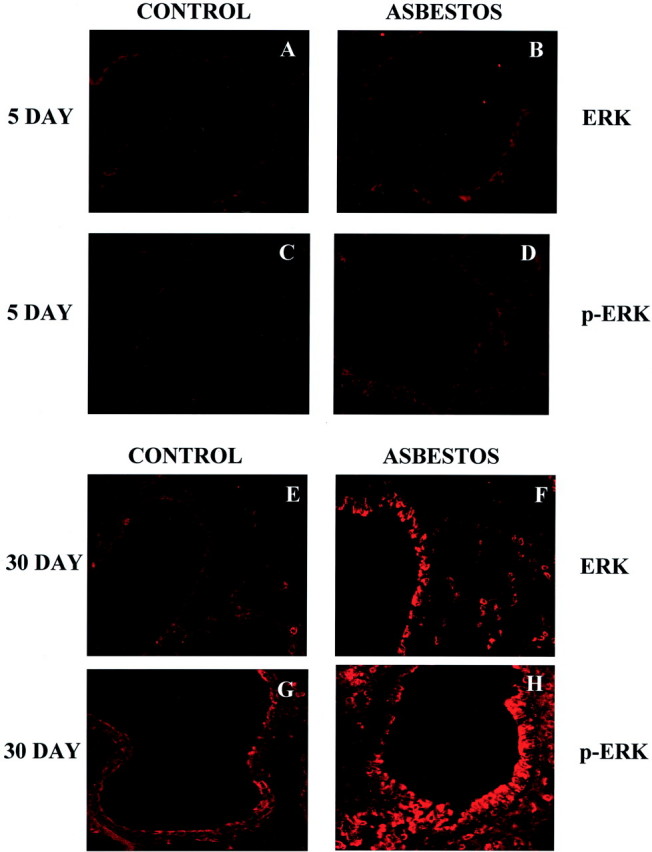
CSLM of ERK and p-ERK immunoreactivity in cryosections from mouse lung. At both 5 days of air exposure (A, C) and crocidolite exposure (B, D) little ERK (A, B) and p-ERK (C, D) immunoreactivity is observed. After 30 days of crocidolite exposure, a dramatic increase in both ERK (F) and p-ERK (H) immunoreactivity is observed as compared to sham air-exposed animals (E, G). All images were captured with a ×40 objective lens and represent experiments done in triplicate.
To confirm the epithelial cell specificity of p-ERK localization, we used co-labeling with cell-type-specific antibodies. Because accumulation of alveolar macrophages is an initial response to asbestos, we first used an anti-macrophage antibody in combination with the antibody detecting p-ERK and examined dual-labeled sections using CSLM. As shown in Figure 2A ▶ , there was no overlap of signals in asbestos-exposed mouse lungs, and in comparison to the green signal (anti-mac 3), p-ERK (red stain) is restricted to the bronchiolar luminal cells. In contrast, use of an anti-cytokeratin antibody showed co-localization of p-ERK with epithelial cells in asbestos-exposed lungs, including prominent alveolar epithelial cells (Figure 2B) ▶ .
Figure 2.
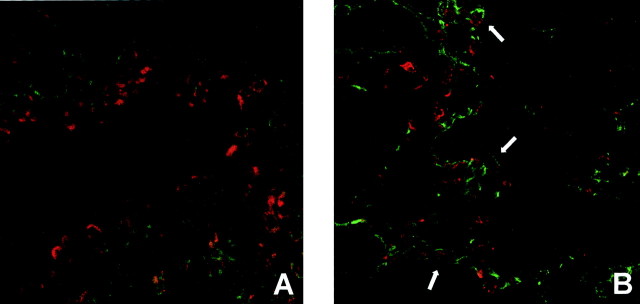
CSLM of dual-stained cryosections from lungs of mice exposed to crocidolite asbestos for 30 days. A: p-ERK immunoreactivity (red signal) does not overlap with the anti-macrophage Mac-3 antibody staining (green signal). B: p-ERK immunoreactivity (red signal) is observed in the same lung cells (white arrows) also expressing the epithelial cell type marker cytokeratin (green signal). All images were captured with a ×40 objective lens and represent experiments done in triplicate.
We also examined the localization of p-ERK immunoreactivity in asbestos-associated trichrome-positive fibrotic lesions observed at 30 days (Figure 3A) ▶ . These studies showed the punctate localization of p-ERK-positive epithelial cells within a lesion (Figure 3B) ▶ . Such results suggest that chronic activation of ERKs occurs in epithelial cells during the pathogenesis of fibrosis, presumably because of their interactions with asbestos fibers distributed throughout the lesions.
Figure 3.
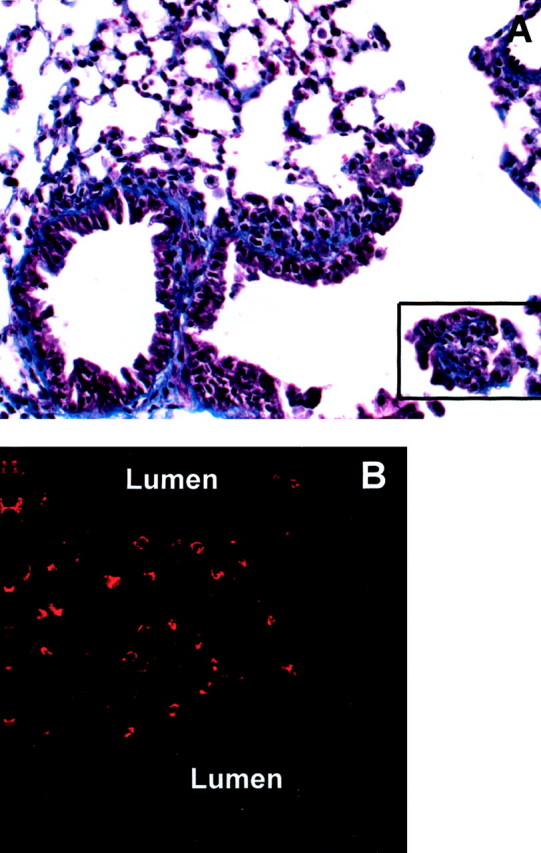
p-ERK immunoreactivity is observed at the site of parenchymal lesions in animals exposed to crocidolite asbestos for 30 days. A: Trichrome-stained paraffin section showing a peribronchiolar lesion (boxed area) with blue stain indicating areas of fibrosis. B: p-ERK immunostaining in a large lesion in the parenchyma of the distal lung. Images captured with a ×20 objective lens in conventional bright-field microscopy (A), and a ×40 objective lens with CSLM (B).
LSC was then used to document the nuclear translocation of p-ERK in alveolar epithelial cells in vitro in comparison to the soluble ERK stimuli, EGF, and H2O2. In C10 cells exposed to asbestos, no nuclear translocation, in comparison to untreated control cells, occurred until 4 hours after addition of fibers to cultures (Figure 4A) ▶ . At 24 hours, nuclear translocation of p-ERK still persisted in a small fraction of cells. Similar patterns were observed with H2O2, although in comparison to asbestos effects, a smaller proportion of cells exhibited increased nuclear to cytoplasmic ratios of p-ERK at all time points (Figure 4B) ▶ . In contrast, the kinetics of nuclear translocation of p-ERK were more rapid in EGF-treated cells (Figure 4C) ▶ . Maximum translocation was observed by 30 minutes and had decreased by 4 hours. The patterns of translocation observed with each agent resembled the time frame of phosphorylation of p-ERK1 and p-ERK2 observed previously in C10 cells by Western blot analyses. 5
Figure 4.
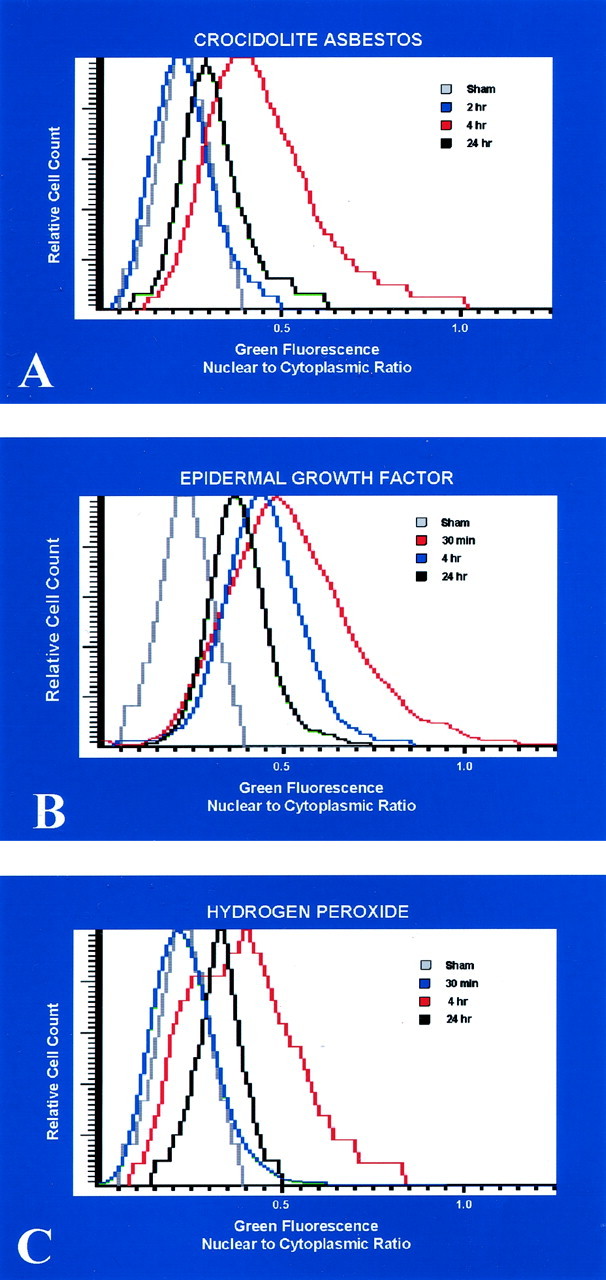
Histograms from LSC demonstrating nuclear to cytoplasmic ratios for p-ERK immunoreactivity in response to cultured lung epithelial cells exposed to various agents known to induce ERK expression. In cells exposed to crocidolite asbestos (A) and H2O2 (B), nuclear translocation of p-ERK occurs only after 4 hours of exposure. In cells treated with EGF (C), nuclear translocation occurs more rapidly, with maximum translocation by 30 minutes.
Discussion
The role of the ERK pathway in cell injury and proliferation has been well established in various in vitro systems, but localization of ERKs and their activation in models of disease is poorly defined. We show here that increased expression of native ERKs as well as their activation, as measured by a phosphospecific antibody, are epithelial cell-specific in a murine model of asbestos-induced fibrosis. To our knowledge, this is the first demonstration that p-ERKs are localized subcellularly in epithelial cells at sites of initial injury by asbestos fibers. This may be of broad relevance to ERK activation by a variety of inhaled pathogenic agents. Moreover, results show that ERK activation in epithelial cells persists during the development of asbestosis.
The punctate appearance of the p-ERK immunostaining in bronchiolar epithelial cells is intriguing. We observed a similar punctate staining previously in an in vitro model of injury using mouse C10 cells. 5 In that study, the punctate cytoplasmic staining for p-ERK was observed in C10 cells after exposure to H2O2, or in cells directly in contact with crocidolite asbestos fibers, as detected by light microscopy. 5 Thus, although the association of asbestos fibers with enhanced punctate staining for p-ERK could be directly visualized in a cultured cell model, asbestos fibers are difficult to observe in lung cells in an in vivo inhalation model. Nevertheless, the similarities between the p-ERK staining patterns in both in vivo and in vitro models exposed to crocidolite asbestos is noteworthy and deserves further investigation to determine its physiological significance.
The increased expression of p-ERK in epithelial cells at 30 days may reflect several possibilities. First, a critical minimal concentration or lung burden of asbestos fibers may be necessary to elicit the response. Secondly, inflammation in this model occurs after 5 days of exposure and also increases with time, suggesting a need for accumulation of inflammatory mediators or other cell types to invoke an ERK response.
The epithelial cell specificity of ERK activation has been suggested in other organs. For example, in normal gastric mucosa, ERK1 and ERK2 are localized in surface epithelial cells and in some glandular epithelial cells. 20 In control tissues, epithelial ERK localization is cytoplasmic, but after the induction of gastric ulcers, ERK immunoreactivity is nuclear in some cells and attributed to wound healing. ERK phosphorylation is also increased in a number of malignant epithelial cells including prostate cancers 21 and in alveolar epithelial cells during the development of urethane-induced lung tumors. 22 Although the subcellular distribution of ERKs have not been characterized in vivo, our results are consistent with in vitro studies showing serum-induced translocation of ERKs to cell surface membranes. 23
Little is known about the intracellular transport of native or phosphorylated ERKs. In activated cells, ERK2 enters the nucleus of cells in vitro by a carrier-independent mechanism, 24 and evidence also exists for its nuclear export. These studies, which have also revealed the direct interaction of ERK2 with nuclear pore complex proteins, have been facilitated by labeling of ERK2 with green fluorescent protein and an in vitro import assay. 24 Osmotic stress in vitro induces the translocation of ERK from the cytoplasm to the nucleus, which is dependent on specific docking and phosphorylation sites. 25 Our studies are the first to use LSC to define the kinetics of p-ERK translocation in epithelial cells by diverse ERK-activating agents inducing different biological outcomes. It should be stressed that we chose concentrations of the agents known to produce a striking increase in p-ERK. However, it is not clear what nuclear/cytoplasmic ratio of p-ERK is required to foster a functional biological response. Exposure to asbestos or H2O2 at concentrations causing apoptosis in C10 cells, 5 caused protracted nuclear translocation of p-ERK. In contrast, exposure to the mitogenic agent, EGF, caused a more rapid and transient nuclear translocation pattern. These results demonstrate that LSC, which has been used previously to measure the kinetics of translocation of nuclear factor-κB, 17 may be useful as a functional assay to measure p-ERK translocation in relationship to the development of various endpoints, ie, cell injury and proliferation associated with ERK activation. The potential for determining the subcellular transport of phosphospecific proteins using both LSC and cell imaging techniques is exciting and of relevance to the broad field of signal transduction.
Footnotes
Address reprint requests to Douglas J. Taatjes, Ph.D., Department of Pathology, University of Vermont, 89 Beaumont Ave., Burlington, VT 05405. E-mail: douglas.taatjes@uvm.edu.
Supported by grant PO1 HL67004 from the National Institutes of Health.
References
- 1.Mossman B, Bignon J, Corn M, Seaton A, Gee J: Asbestos: scientific developments and implications for public policy. Science 1990, 247:294-301 [DOI] [PubMed] [Google Scholar]
- 2.Mossman B, Gee J: Asbestos related disease. N Engl J Med 1989, 320:1721-1730 [DOI] [PubMed] [Google Scholar]
- 3.Mossman B, Kamp D, Weitzman S: Mechanisms of carcinogenesis and clinical features of asbestos-associated cancers. Cancer Invest 1996, 14:466-480 [DOI] [PubMed] [Google Scholar]
- 4.Mossman B, Churg A: State-of-the-art: mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 1998, 157:1666-1680 [DOI] [PubMed] [Google Scholar]
- 5.Buder-Hoffmann S, Palmer C, Vacek P, Taatjes D, Mossman B: Different accumulation of activated extracellular signal-regulated kinases (ERK 1/2) and role in cell-cycle alterations by epidermal growth factor, hydrogen peroxide, or asbestos in pulmonary epithelial cells. Am J Respir Cell Mol Biol 2001, 24:405-413 [DOI] [PubMed] [Google Scholar]
- 6.Jimenez L, Zanella C, Fung H, Janssen Y, Vacek P, Charland C, Goldberg J, Mossman B: Role of extracellular signal-regulated protein kinases in apoptosis by asbestos and H2O2. Am J Physiol 1997, 273:L1029-L1035 [DOI] [PubMed] [Google Scholar]
- 7.Pache J, Janssen Y, Walsh E, Quinlan T, Zanella C, Low R, Taatjes D, Mossman B: Increased epidermal growth factor-receptor (EGF-R) protein in a human mesothelial cell line in response to long asbestos fibers. Am J Pathol 1998, 152:333-340 [PMC free article] [PubMed] [Google Scholar]
- 8.Zanella C, Posada J, Tritton T, Mossman B: Asbestos causes stimulation of the ERK-1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res 1996, 56:5334-5338 [PubMed] [Google Scholar]
- 9.Zanella C, Timblin C, Cummins A, Jung M, Goldberg J, Raabe R, Tritton T, Mossman BT: Asbestos-induced phosphorylation of epidermal growth factor receptor is linked to c-fos expression and apoptosis. Am J Physiol 1999, 277:L684-L693 [DOI] [PubMed] [Google Scholar]
- 10.Mohammadi M, Honegger A, Sorokin A, Ullrich A, Schlessinger J, Hurwitz D: Aggregation-induced activation of the epidermal growth factor receptor protein tyrosine kinase. Biochemistry 1993, 32:8742-8748 [DOI] [PubMed] [Google Scholar]
- 11.Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf H: Involvement of growth factor receptors in the mammalian UVC response. Cell 1994, 78:963-972 [DOI] [PubMed] [Google Scholar]
- 12.Stevenson M, Pollock S, Coleman C, Calderwood S: X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res 1994, 54:12-15 [PubMed] [Google Scholar]
- 13.Abe M, Chao T, Solway J, Rosner M, Hershenson M: Hydrogen peroxide stimulates mitogen-activated protein kinase in bovine tracheal myocytes: implications for human airway disease. Am J Respir Cell Mol Biol 1994, 11:577-585 [DOI] [PubMed] [Google Scholar]
- 14.Robledo R, Buder-Hoffmann S, Cummins A, Walsh E, Taatjes D, Mossman B: Increased phosphorylated ERK immunoreactivity associated with proliferative and morphologic lung alterations following chrysotile asbestos inhalation in mice. Am J Pathol 2000, 156:1307-1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan T, Marsh J, Janssen Y, Leslie K, Hemenway D, Vacek P, Mossman B: Dose responsive increases in pulmonary fibrosis after inhalation of asbestos. Am J Respir Crit Care Med 1994, 149:795-802 [DOI] [PubMed] [Google Scholar]
- 16.Malkinson A, Dwyer-Nield L, Rice P, Dinsdale D: Mouse lung epithelial cell lines—tools for the study of differentiation and the neoplastic phenotype. Toxicology 1997, 123:53-100 [DOI] [PubMed] [Google Scholar]
- 17.Deptala A, Bedner E, Gorczyca W, Darzynkiewicz Z: Activation of nuclear factor kappa B (NF-kappaB) assayed by laser scanning cytometry (LSC). Cytometry 1998, 33:376-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taatjes DJ, Palmer CJ, Pantano C, Hoffmann SB, Cummins A, Mossman BT: Laser-based microscopic approaches: application to cell signaling in environmental lung disease. Biotechniques 2001, 31:880-892 [DOI] [PubMed] [Google Scholar]
- 19.Manning C, Vallyathan V, Mossman B: Diseases caused by asbestos: mechanisms of injury and disease development. Int Immunopharmacol 2002, 2:191-200 [DOI] [PubMed] [Google Scholar]
- 20.Tarnawski A, Pai R, Wang H, Tomikawa M: Translocation of MAPK (ERK-1 and -2) kinases to cell nuclei and activation of c-fos gene during healing of experimental gastric ulcers. J Physiol Pharmacol 1998, 49:479-488 [PubMed] [Google Scholar]
- 21.Price D, Della Rocca G, Guo C, Ballo M, Schwinn D, Luttrell L: Activation of extracellular signal-regulated kinase in human prostate cancer. J Urol 1999, 162:1537-1542 [PubMed] [Google Scholar]
- 22.Yano T, Yano Y, Nagashima Y, Yuasa M, Yajima S, Horikawa S, Hagiwara K, Kishimoto M, Ichikawa T, Otani S: Activation of extracellular signal-regulated kinase in lung tissue of mice treated with carcinogen. Life Sci 1999, 64:229-236 [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez F, Seth A, Raden D, Bowman D, Fay F, Davis R: Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J Cell Biol 1993, 122:1089-1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehurst A, Wilsbacher J, You Y, Luby-Phelps K, Moore M, Cobb M: ERK2 enters the nucleus by a carrier-independent mechanism. Proc Natl Acad Sci USA 2002, 99:7496-7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanoue T, Meada R, Adachi M, Nishida E: Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interaction. EMBO J 2001, 20:466-479 [DOI] [PMC free article] [PubMed] [Google Scholar]