

“Chance favors the prepared mind”
Louis Pasteur
Solid tumors are composed of two distinct but interdependent compartments: the malignant cells themselves (parenchyma) and the supporting connective tissue (stroma) that they induce and in which they are dispersed. 1,2 The quantity of stroma is highly variable from tumor to tumor but all solid tumors, regardless of type or cellular origin, require stroma for nutrition and waste disposal if they are to grow beyond minimal size. 3
The discrete separation of parenchymal cells from stroma is not unique to tumors. Normal tissues are also comprised of avascular parenchyma that abuts on vascularized connective tissue stroma. However, tumor stroma differs strikingly from normal connective tissue. Blood vessels offer one example. Tumor vessels differ from their normal counterparts with respect to organization, structure and function. 2,4-7 Unlike the normal vasculature, tumor vessels are not arranged in a hierarchical pattern but are instead irregularly spaced and structurally heterogeneous. They are also hyperpermeable to plasma and plasma proteins, may lack pericytes, and are lined by actively dividing endothelial cells. Other elements of tumor stroma are also abnormal: increased amounts of plasma protein-rich interstitial fluid; structural proteins not normally found in mature stroma such as fibrin, tenascin, and fetal forms of fibronectin; abnormal proteoglycans; variable numbers of inflammatory cells, etc.
Given these extensive differences in composition, it seemed likely to us that tumor and normal stroma were formed by different steps and mechanisms. Although normal stroma generation is not completely understood, it is known to require the cooperative activities of many different cytokines and inhibitors, each expressed in appropriate amounts and temporal sequence. 8-10 Among these are vascular permeability factor/vascular endothelial growth factor (VPF/VEGF, VEGF-A), other members of the VPF/VEGF family such as placenta growth factor (PlGF), and other cytokines such as TGF-β, PDGF, ephrins, FGFs, angiopoetins-1 and -2, and their respective receptors and inhibitors. By contrast, the imperfect nature of tumor stroma suggested to us that it was generated by a simpler process, one that deviated from normal developmental stromagenesis by having fewer steps and involving fewer cytokines.
The work reported here summarizes our progress over the past 25 years toward elucidating the steps and mechanisms of tumor angiogenesis and stromagenesis. A major conclusion is that a single cytokine, the 164/5 amino acid splicing variant of VEGF-A, is capable of inducing the formation of tumor-like blood vessels and connective tissue stroma. Further, VEGF-A164/5 is expressed not only by malignant tumors but also in healing wounds, in cellular immunity, and in chronic inflammatory diseases such as psoriasis and rheumatoid arthritis. 4,5 Therefore, VEGF-A164/5 provides a unifying principle that contributes importantly to many, if not all, examples of pathological angiogenesis and tissue repair.
Discovery of Vascular Permeability Factor/Vascular Endothelial Growth Factor (VPF, VPF/VEGF, VEGF-A)
VEGF-A was discovered as the result of microscopic studies of transplantable tumors at early stages after their implantation into syngeneic animals. 11,12 The critical observation was that tumors began as clumps of neoplastic cells dispersed in a fibrin gel stroma (Figure 1, a–d) ▶ . Fibrin results from the clotting of plasma fibrinogen. Fibrinogen, like other plasma proteins, is normally retained within blood vessels; therefore, the presence of extravascular fibrin deposits strongly implied that tumor blood vessels were hyperpermeable to fibrinogen and presumably therefore to other plasma proteins as well. To test this hypothesis, we injected various macromolecular tracers intravenously into tumor-bearing animals and found extensive tracer extravasation from tumor as compared with normal blood vessels (Figure 1, e and f) ▶ . Several earlier papers had also commented on the increased permeability of tumor blood vessels (reviewed in 5 ).
Figure 1.
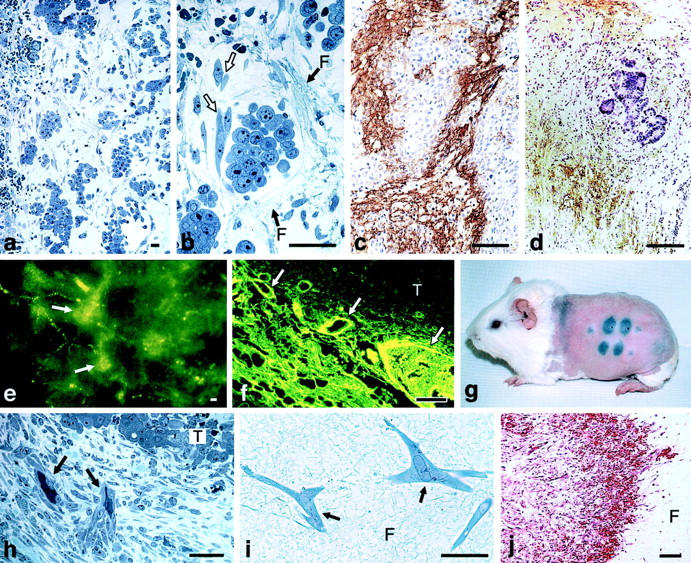
a and b: Line 10 tumor cells 48 hours after transplant into the subcutaneous space of syngeneic strain 2 guinea pigs. Fibrin forms a water-trapping gel (F) that serves as a provisional stroma that separates tumor cells into discrete islands and that provides a favorable matrix for fibroblast (white arrows) and endothelial cell migration. c and d: Immunohistochemical demonstration of fibrin (brown staining) in guinea pig line 1 and human colorectal adenocarcinoma, respectively. e and f: Blood vessels (arrows) supplying line 10 guinea pig tumors are hyperpermeable to circulating macromolecular fluoresceinated dextran. g: Miles assay illustrating permeability to Evans blue-albumin complex at sites of intradermal injections of the following: (top, left to right) neutralizing anti-VEGF-A antibody, line 10 guinea pig tumor ascites fluid, mix of line 10 tumor ascites fluid and control IgG, mix of tumor ascites fluid and neutralizing anti-VEGF-A antibody. Bottom: line 1 tumor ascites fluid, mix of line 1 tumor ascites fluid and control IgG, and mix of line 1 tumor ascites fluid and neutralizing anti-VEGF-A antibody. h: Fibroblasts and blood vessels (black arrows) invade line 1 tumor fibrin gel, replacing it with fibrous connective tissue. i: Fibroblasts (arrows) migrate through fibrin gel (F) in vitro. j: Implanted fibrin gel (F) in subcutaneous space is replaced by ingrowth of fibroblasts and new blood vessels, creating granulation-like vascular connective tissue. Scale bars, 25 μm (b,i); 50 μm (a,c,d,h); 100 μm (e,f,j). e–g: Reprinted 5 from Curr Top Microbiol Immunol 1999, 237:97–132 by copyright permission of Springer-Verlag GmbH & Co. KG; j reprinted 29 from Lab Invest 1987, 57:673–686 by permission of Lippincott Williams & Wilkins, copyright The United States and Canadian Academy of Pathology, Inc.
Hyperpermeable vessels were especially prominent at the tumor-host interface and it was therefore not certain from our studies whether tumor cells were permeabilizing normal host microvessels and/or were generating the formation of new, abnormal blood vessels that were intrinsically permeable. (Both of these possibilities were subsequently shown to be correct). To address the first possibility, we collected tumor cell culture supernatants and ascites fluids and found to our delight that both contained an activity that potently increased the permeability of microvessels in normal guinea pig skin (Figure 1g) ▶ . We called this activity vascular permeability factor (VPF).
VPF was found to be non-dialyzable, largely inactivated by heat, and both cold and puromycin profoundly depressed its appearance in culture medium. Also, the vascular permeabilizing activity of tumor culture supernatants was not inhibited by antihistamines nor were these supernatants able to degranulate or release histamine from basophilic leukocytes or mast cells. 12 We concluded from these studies that the VPF activity present in tumor culture supernatants and ascites fluids was a secreted protein and one that increased vascular permeability by acting directly on blood vessel endothelium.
Using the Miles assay to screen column fractions for their ability to induce vascular permeability, Donald Senger then purified VPF to homogeneity. 13,14 Sometime later, our colleagues at the Monsanto Company 15,16 made use of Senger’s N-terminal amino acid sequence data to clone VPF and show that it induced angiogenesis; independently, Napoleon Ferrara and his colleagues 17 at Genentech cloned VPF and reported that it was mitogenic for cultured endothelial cells. As a result of these studies, VPF has come to be known more widely as vascular endothelial growth factor (VEGF) and, more recently, as VEGF-A.
VEGF-A and Its Activities
Over the past two decades much has been learned about VEGF-A. 4,5,18 It is the founding member of a family of structurally related proteins with varying degrees of vasculogenic, angiogenic, and lymphangiogenic activity. 4,5,18,19,20 The VEGF-A gene is alternatively spliced to yield major isoforms of 189, 165, and 121 amino acids; the murine homologues are in each case one amino acid shorter. Of these, the 164/5 isoform is generally expressed most abundantly. However, all three isoforms are essential for normal development and inactivation of even a single copy of the VEGF-A gene results in embryonic death from failure of vascular development. 18,21,22 VEGF-A164/5 is expressed by the vast majority of malignant animal and human tumors, 4,5 and interference with its activity, as by blocking antibodies or soluble receptors, inhibits tumor angiogenesis and tumor growth and may cause pre-existing tumors to regress. 18
VEGF-A164/5 is a multifunctional cytokine with a vascular permeabilizing activity some 50,000 times more potent than histamine. It also stimulates endothelial cell migration, is mitogenic for endothelial cells and is active in standard angiogenesis assays. Important for these activities is its ability to reprogram endothelial cell gene expression, leading to the up-regulation of many proteins including various proteases, tissue factor, a glucose transporter, etc. VEGF-A164/5 is also an endothelial cell survival factor, able to avert both endothelial cell apoptosis and senescence. 23,24 As far as is known, all of the activities of VEGF-A164/5 are mediated through two receptor tyrosine kinases (VEGFR-1, flt-1) and VEGFR-2 (flk-1, KDR), and through a more recently described, non-tyrosine kinase receptor, neuropilin. 19,25
Expression of VEGF-A164 in Vivo
To investigate its capacity to induce tumor-like blood vessels and stroma, we sought to express VEGF-A164 in normal mouse tissues at a constant rate for a sufficient period of time to induce angiogenesis and new stroma. To that end we made use of a non-replicating adenoviral expression vector. 26 Such vectors are readily engineered and offer a number of advantages for investigating the potential functions of angiogenic cytokines. 1) They can be injected in any accessible tissue of adult or embryonic mice where they direct infected host cells to express gene inserts. 2) Protein expression levels are readily adjusted by varying the dose of injected virus. 3) Control vectors engineered to express non-inflammatory proteins give insignificant backgrounds. 4) Combinations of cytokines can be studied, together or in sequence, by injecting vectors engineered to express different genes. 5) Adenoviral vectors are not integrated into the host cell genome and so the genes they introduce are not expressed indefinitely; this was actually an advantage for our purposes because it permitted us to study the effects of cytokine withdrawal on newly formed blood vessels and stroma.
The Vascular Response to VEGF-A164 Expression
Within hours of injection into any of several different tissues of nude mice or rats, infected host cells began to express VEGF-A164 mRNA and continued to do so at steady levels for 10 to 14 days, generating a strong angiogenic response (Figure 2) ▶ ; 26 thereafter, VEGF-A164 expression declined gradually and, by 4 to 6 weeks was no longer detectable by in situ hybridization.
Figure 2.

Angiogenic response to Ad-VEGF-A164 in the ears of nude mice at the indicated times and magnifications. (Reprinted 20 from Cold Spring Harbor Symp Quant Biol 2002, 67:227–237 by copyright permission of Cold Spring Harbor Laboratory Press.).
The Initial Response to Ad-VEGF-A164: Fibrin Deposition and Edema
The initial response (1 to 3 days) was similar in all tissues studied and consisted of vascular hyperpermeability, fibrin deposition, and edema. 26 Generation of an extravascular fibrin gel requires not only leaky blood vessels but also activation of the clotting system. Clotting is triggered in most tissues within minutes of plasma leakage by tissue factor, a procoagulant expressed on tumor cells and many normal tissue cells. 27,28 Tissue factor reacts with clotting factor VIIa and subsequently with other extravasated plasma-clotting proteins, culminating in thrombin generation, clotting of fibrinogen to fibrin, and subsequent cross-linking of fibrin by activated factor XIII. Once deposited, cross-linked fibrin behaves as a gel that causes edema by trapping extravasated plasma and provides a proangiogenic provisional stroma similar to that observed in transplanted tumors and healing wounds. 1,11,12,29
The fibrin deposited in tissues by tumors or by Ad-VEGF-A164 represents a net balance, at any moment of time, between fibrinogen influx, clotting and fibrinolysis. Fibrinogen influx results from leakage of plasma from permeabilized vessels and clotting from activation of the tissue factor pathway. However, the plasma protein plasminogen also extravasates from leaky blood vessels where plasminogen activators cleave it to generate plasmin, a potent fibrinolytic protease. VEGF-A164 selectively up-regulates plasminogen activator activity in endothelial cells, and tumors commonly express this enzyme so that deposited fibrin is continually being remodeled. 30
VVOs: The Principle Pathway for Macromolecule Extravasation from Hyperpermeable Blood Vessels
To investigate the anatomical pathway(s) by which macromolecules extravasated from Ad-VEGF-A164 permeabilized vessels, we injected anionic ferritin intravenously as a tracer. Ferritin is an iron-rich plasma protein that can be visualized directly by electron microscopy. In agreement with earlier studies of tumor vessels 31 and normal skin following local injection of VEGF-A165 protein, 32,33 ferritin extravasated from venules by a trans-endothelial cell pathway, that provided by the vesiculo-vacuolar organelle (VVO). VVOs are grape-like clusters of uncoated, largely para-junctional, cytoplasmic vesicles and vacuoles that traverse endothelial cytoplasm from lumen to ablumen (Figure 3) ▶ . 31-35 The individual vesicles and vacuoles that comprise VVOs are linked to each other, and to the luminal and abluminal plasma membranes, by stomata that are normally closed by thin diaphragms. By some as yet unknown mechanism, VEGF-A164/5 (and other vasoactive mediators) causes these stomata to open, providing a pathway for plasma and plasma protein extravasation. By contrast, interendothelial cell junctions remained tightly closed and did not admit tracer ferritin, even in the presence of abundant VEGF-A164/5.
Figure 3.
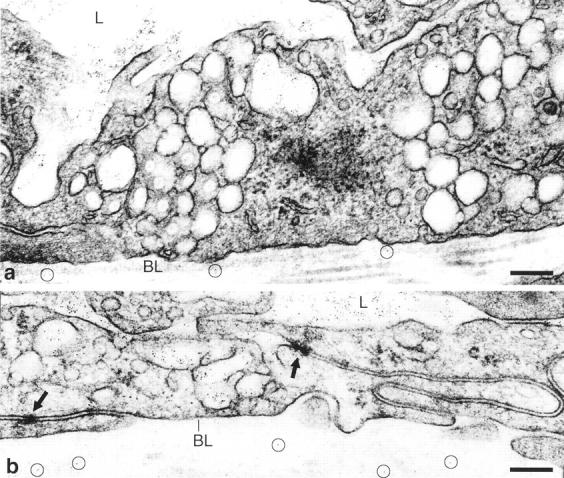
Electron micrographs 3 days after local Ad-VEGF-A164 injection and 20 minutes after i.v. injection of ferritin tracer. a: Hyperpermeable venule lined by endothelial cells of normal height, illustrating two prominent VVOs (collections of vesicles/vacuoles) that span endothelial cell cytoplasm from lumen (L) to abluminal basal lamina (BL). Note ferritin particles in vascular lumen, in VVO vesicles and vacuoles, and beneath BL (black dots, encircled). b: Mother vessel with greatly thinned endothelium spanned by four or fewer VVO vesicles/vacuoles. Note ferritin in these vesicles and vacuoles but tight junctions (arrows) remain closed and do not admit tracer. Bars, 200 nm. (Reprinted 26 from Lab Invest 2000, 80:99–115 by permission of Lippincott Williams & Wilkins, copyright The United States and Canadian Academy of Pathology, Inc.)
Formation of “Mother” Vessels from Pre-Existing Venules
Within 18 hours of Ad-VEGF-A164 injection, enlarged, thin-walled, serpentine, pericyte-poor, hyperpermeable, and strongly VEGFR-2-positive sinusoids appeared, structures to which we have given the name “mother” vessels (Figure 4) ▶ . Vessels of this description are commonly found in human and animal tumors (Figure 4k) ▶ 35 and are also transiently present in healing wounds (Figure 4l) ▶ . 36 They continued to develop for ∼5 days following injection of Ad-VEGF-A164 into tissues.
Figure 4.
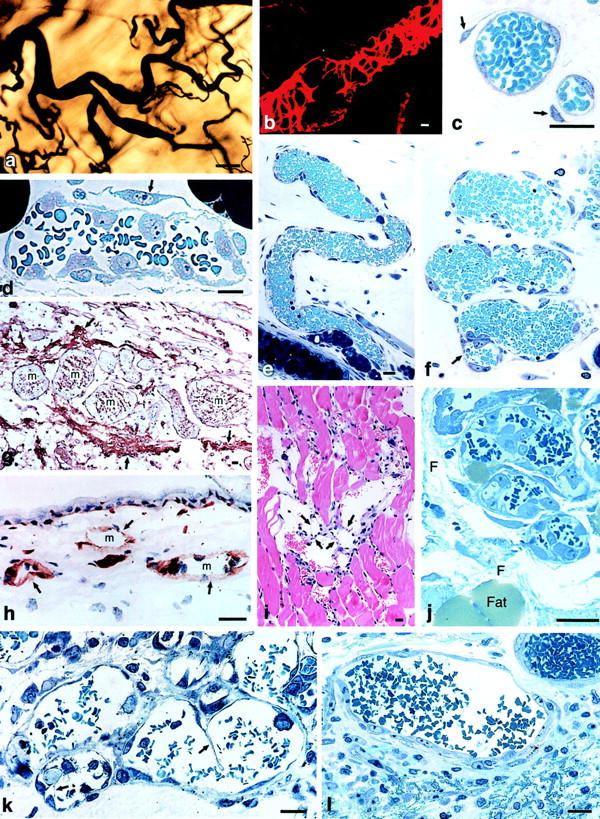
Mother vessels induced by Ad-VEGF-A164 (a–j), MOT tumor (k) and healing skin wound (l). a: Whole mount of colloidal carbon-perfused vascular bed. Mother vessels appear as enlarged segments of much smaller, normal venules. b: Confocal microscopic image of mother vessel stained for pericytes with an antibody to α-smooth-muscle actin. Note incomplete pericyte covering, especially over segments of greatest vessel enlargement. c: Developing mother vessels illustrating pericyte detachment (arrows). d: Mother vessel with detached pericyte (arrow) and activated endothelial cells whose large nuclei bulge into the vascular lumen, creating a highly irregular surface. e and f: Serpentine mother vessels with irregular luminal surface and endothelial cell bridging (f, arrow). g: Mother vessels (m) embedded in fibrin (arrows) provisional stroma. h: Loss of laminin staining (arrows) in developing mother vessels (m). i and j: Endothelial cell bridging (i, arrows) in mother vessels in skeletal muscle and ear skin, respectively. k and l: Mother vessels in mouse ovarian tumor and healing rat skin wound, respectively. Note bridging in k (arrows) and fibrin in l (lower, between cells). Scale bars, 20 μm (b–l); 100 μm (a). a and b: Reprinted 20 from Cold Spring Harbor Symp Quant Biol 2002, 67:227–237 by copyright permission of Cold Spring Harbor Laboratory Press; c,e,f,h–j reprinted 26 from Lab Invest 2000, 80:99–115 by permission of Lippincott Williams & Wilkins, copyright The United States and Canadian Academy of Pathology, Inc.
Mother vessels arose from the enlargement of pre-existing microvessels, primarily venules. By 18 to 24 hours, mean cross-sectional microvessel area had increased by ∼4-fold, mean vessel perimeter by ∼2-fold, and the percent of dermis occupied by microvessels by 4- to 7-fold. 26 Mother vessels formed by a multistage process that involved proteolytic digestion of vascular basement membranes, pericyte detachment, and spreading and thinning of endothelial cells to cover a greatly expanded surface area (Figure 3b and Figure 4) ▶ . For the first 48 hours, mother vessels formed without significant endothelial cell or pericyte division; thereafter, both types of cells proliferated extensively.
The vascular basement membrane is an especially important impediment to microvessel enlargement. Intact basement membranes are non-compliant (inelastic) structures that limit vascular enlargement to an ∼30% increase in cross-sectional area. 37 Therefore, structural changes in the vascular basement membrane were necessary to accommodate the dramatic increases in microvessel area that were associated with VEGF-A164-induced mother vessel formation. VEGF-A is known to up-regulate the expression of endothelial cell proteases that degrade vascular basement membranes 4,18 and reduced or absent staining for laminin and other basement membrane components, indicative of basement membrane degradation, was demonstrated by immunohistochemistry in developing mother vessels (Figure 4h) ▶ . 26
A Role for VVOs in Mother Vessel Formation
In addition to providing a pathway for macromolecule extravasation, VVOs also had a critical role in mother vessel formation. They provided an intracellular store of membrane that could be rapidly mobilized to the plasma membrane to increase cell surface area and permit vessel enlargement. Normal venular endothelium stores an amount of membrane in VVOs that is equivalent to 2.25 times that of plasma membrane. 20 Therefore, transfer of VVO membranes to the cell surface has the potential to provide a >3-fold increase in endothelial cell perimeter without new membrane synthesis. Consistent with this possibility, mother vessel formation was accompanied by progressive endothelial cell thinning and reduction in VVO vesicles and vacuoles (Figure 3b ▶ , unpublished data). Our findings, therefore, indicate that the endothelial cell spreading and thinning that accompany mother vessel formation can be accommodated, at least initially, by transfer of intracellular VVO membrane to the cell surface.
Mother Vessel Evolution
Mother vessels are transitional forms that evolved along several different pathways (Figure 5) ▶ . 26 Many underwent a process of bridging, in which cells expressing endothelial markers projected processes into and across mother vessel lumens (Figure 4, f, i, j) ▶ . It remains to be determined whether these cells represent locally-activated vascular endothelium or were instead derived from circulating bone marrow precursors. 38,39 Whatever their source, they formed transluminal bridges that divided blood flow into multiple smaller-sized channels. Similar bridging takes place in tumor mother vessels (Figure 4k) ▶ , and we have postulated that at least some of the smaller channels separated from each other to form individual smaller-caliber daughter capillaries. 35,40
Figure 5.

Schematic diagram of mother vessel formation and evolution into daughter capillaries, vascular malformations and glomeruloid bodies. (Modified 26 from Lab Invest 2000, 80:99–115 by permission of Lippincott Williams & Wilkins, copyright The United States and Canadian Academy of Pathology, Inc.)
Glomeruloid Bodies (GB)
GB are poorly organized vascular structures that resemble renal glomeruli (hence the name) and are a common feature of several human tumors, among them glioblastoma multiforme. 41 GB precursors were first recognized at ∼3 days as focal collections of large, primitive cells in the endothelial lining of mother vessels (Figures 5 and 6) ▶ . 42 These cells bore endothelial cell markers, proliferated rapidly, and extended both into the vascular lumen and outwards into the extravascular connective tissue. By 7 to 10 days, pericytes were prominently included and organized themselves around endothelial cells to form primitive microvessels. As they expanded, GB severely compromised the mother vessels in which they had arisen, reducing and dividing originally large single vascular lumens into multiple, smaller, tortuous channels.
After about 14 days, as VEGF-A164 expression declined, GB progressively devolved by a process that involved both apoptosis and reorganization of endothelial cells and pericytes to form normal appearing capillaries (Figure 6i) ▶ . By ∼8 weeks, VEGF-A164 expression was no longer detectable and the angiogenic response had largely resolved with a return of microvascular density to near-normal levels. Taken together, these experiments provided the first animal model for generating GB, offered important insights into the mechanisms of GB formation and devolution, and demonstrated that VEGF-A164 is both sufficient for GB generation and necessary for their maintenance.
Figure 6.
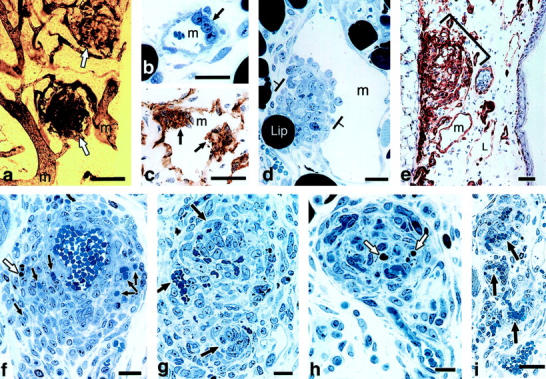
GB formation and devolution. a: Whole mount illustrates two GBs in ear of mouse perfused with colloidal carbon (white arrows), supplied by afferent and efferent mother vessels (m). b: 1-μm Epon section illustrates focal accumulation of large primitive cells (arrow) in a developing mother vessel (m). c: Immunohistochemistry demonstrates that such cells are CD31-positive. d: Primitive GB develops as focal nodule (between brackets) of proliferating cells in the wall of a mother vessel (m), extending both into the lumen and out into the extravascular connective tissue. Lip, adipocyte. e: Immunohistochemical staining for entactin demonstrates abundant basement membrane in a large GB (bracket) as well as in a mother vessel (m) and, conversely, very little in a lymphatic (L). f and g: Maturing GBs divide mother vessels into multiple, much smaller vascular channels (arrows). White arrows in f and h indicate apoptotic bodies. h and i: Devolving GBs reorganize into more normal-appearing microvessels (black arrows). Scale bars, 25 μm (c–f, h–i); 50 μm (a,b); 100 μm (g). (Reprinted 42 from Am J Pathol 2001, 158:1145–1160 by copyright permission of the American Society for Investigative Pathology.)
Vascular Malformations and Arteriogenesis
Thin-walled mother vessels lacking pericyte and basement membrane support are subject to thrombosis or collapse. While many mother vessels avoid this fate by evolving into smaller daughter vessels or GB, others maintained their large size by acquiring a coat of smooth muscle cells and/or collagen-expressing fibroblasts. 25 Such vessels were readily distinguished from normal arteries and veins by their inappropriately large size, by their thinner and often asymmetric muscular coat, and by the presence of perivascular fibrosis (Figure 7, a and b) ▶ . Vessels of this description closely resemble those found in vascular malformations. 43 This finding is of interest because it provides an animal model for generating vascular malformations and suggests that the vascular malformations observed in patients may be induced by VEGF-A overexpression. Once formed, these malformations persisted indefinitely (for more than a year), long after VEGF-A164 expression had ceased, indicating that they had achieved VEGF-A164 independence. In parallel with the formation of vascular malformations, small arteries and arterioles also responded to VEGF-A164 with modest replication of endothelial and smooth muscle cells, leading to the formation of larger, well-differentiated arteries (Figure 7, c–f) ▶ . 20
Figure 7.
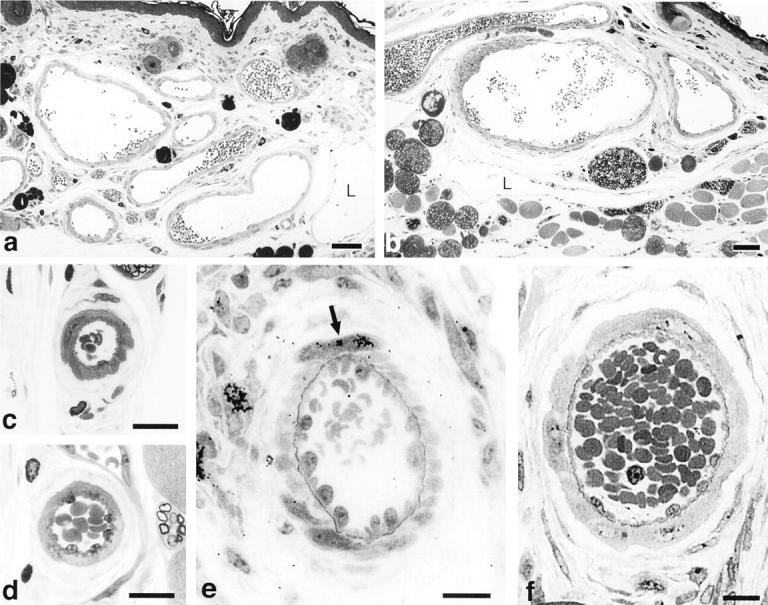
a and b: Vascular malformations induced by Ad-VEGF-A164 in ear skin at 35 and 131 days, respectively. L, giant lymphatics. c–f: Arteriogenesis following administration of Ad-VEGF-A164 in ear skin. Small arteries (c,d) enlarge by 6 to 7 days (e,f) accompanied by replication of endothelium and smooth muscle cells. Arrow (e), [3H]thymidine incorporated by a smooth-muscle cell. Scale bars, 50 μm (a,b); 15 μm (c–f). (Modified 20 from Cold Spring Harbor Symp Quant Biol 2002, 67:227–237 by copyright permission of Cold Spring Harbor Laboratory Press.)
Lymphangiogenesis
Unexpectedly, VEGF-A164 also stimulated the proliferation of lymphatic endothelium, leading to the formation of a dense meshwork of tortuous, irregularly shaped, giant lymphatic channels (Figure 8) ▶ . 44 These giant lymphatics were functional but poorly so, clearing tracers over a period of many hours rather than in a few minutes as in normal lymphatics.
Figure 8.
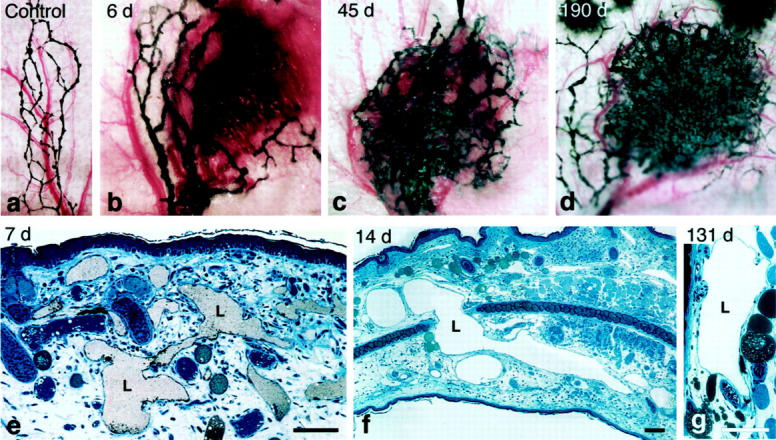
a–d: Giant lymphatics in mouse ear skin at indicated times after injection of Ad-VEGF-A164. Lymphatics were perfused with colloidal carbon and viewed macroscopically. e–g: 1-μm Epon sections of giant lymphatics at indicated times after Ad-VEGF-A164. Lymphatics were injected with colloidal carbon (e). Scale bars, 50 μm. (Reprinted 44 form J Exp Med 2002, 196:1497–1506 by copyright permission of The Rockefeller University Press.)
Lymphangiogenesis developed in parallel with angiogenesis but at a slower pace. Enlarged lymphatics were first recognized at ∼3 days after injection of Ad-VEGF-A164, increased in size and number over the course of several weeks, and persisted indefinitely, in some instances for >1 year. Unlike venules, normal lymphatics lack pericytes and a well-developed basement membrane and are able to enlarge in response to edema. 45,46 Therefore, formation of giant lymphatics was largely attributable to replication of lymphatic endothelial cells without the need for basement membrane degradation, pericyte detachment, or endothelial cell thinning as was necessary for the formation of mother vessels.
Transformation of Provisional Fibrin Gel into Mature Connective Tissue Stroma
As was noted earlier, fibrin deposited in tumors and wounds imposes structure (Figure 1, a and b) ▶ . It also provides an environment that supports cell migration (Figure 1, h–j) ▶ . Endothelial cells, fibroblasts, and inflammatory cells express adhesion molecules (integrins) whose interaction with fibrin and fibronectin allows them to move freely in tumor and wound stroma. 29,47 As a consequence, fibrin facilitates both angiogenesis and the inward migration of fibroblasts. Over time, as VEGF-A164 expression declined, vascular permeability returned to normally low levels and the balance shifted from fibrin accumulation to fibrinolysis. Eventually, fibrin was digested, small blood vessels underwent apoptosis, and fibroblasts synthesized collagen, resulting finally in dense, relatively avascular connective tissue. 1,48
Commentary
A number of conclusions can be drawn from these studies. First, VEGF-A164 sets in motion a complicated chain of events that leads to the generation of a vascular and connective tissue stroma that closely resembles that found in animal and human tumors. Because nearly all malignant tumors express VEGF-A164/5, there is every reason to believe that tumors use this cytokine to generate stroma. Further, the steps and mechanisms by which tumors generate stroma closely mimic the events of wound healing in which VEGF-A164/5 is also expressed. 36 Both tumor stroma generation and wound healing are characterized by vascular hyperpermeability, leakage of plasma, and clotting of extravasated fibrinogen to form a fibrin gel that serves as a provisional stroma for blood vessel and fibroblast migration. In both, the fibrin gel is degraded and the vascular connective tissue subsequently replaced by desmoplasia (tumors) or scar (wounds). However, whereas in healing wounds VEGF-A expression ceases with restoration of tissue normoxia, in tumors VEGF-A is expressed indefinitely. Thus, tumors may be likened to wounds that do not heal. 1
Another important point is that the new blood vessels induced by Ad-VEGF-A164 and by tumors are not of a single kind but rather can be classified into at least several morphologically distinct types, ie, mother vessels, glomeruloid bodies, and vascular malformations. This heterogeneity may be expected to complicate anti-angiogenic and anti-vascular tumor therapy because not one but several distinct types of blood vessels must be targeted. Also, whereas mother vessels and glomeruloid bodies required continued VEGF-A164 expression for maintenance, vascular malformations did not and therefore would not be expected to respond to therapies that neutralized VEGF-A164 or its receptors. Consistent with this line of thinking, Benjamin et al 23 have shown that withdrawal of VEGF-A leads to the selective apoptosis of only those tumor microvessels that lack a pericyte coating.
In addition to inducing angiogenesis (formation of small blood vessels) VEGF-A164 also induced vascular malformations and arteries. These large vessels could have clinical significance quite apart from cancer. Administration of VEGF-A164/5 and other angiogenic cytokines has been proposed as a novel approach for the treatment of coronary heart disease and other examples of tissue ischemia. 49 However, the angiogenesis induced by VEGF-A164/5 would not be expected to offer much clinical benefit, first because the response is transient and second because the small vessels that form in response to VEGF-A supply a newly induced stroma, not the native tissue. Instead, relief of tissue ischemia requires large patent arterial blood vessels that are located proximal to, not within, the ischemic tissue. The vascular malformations and arteries induced by VEGF-A164 might satisfy this criterion because they are of appropriate size and persist indefinitely, independent of VEGF-A. It is possible, therefore, that some of the clinical benefits that have been reported for gene therapy with VEGF-A 49 resulted not from angiogenesis but from unrecognized formation of these larger vessels.
The finding that VEGF-A induced lymphangiogenesis was unanticipated. Other members of the VEGF-A family, VEGF-C and VEGF-D, have been thought to be responsible for lymphatic development. 50 However, we found no evidence for VEGF-C or VEGF-D expression in our studies of Ad-VEGF-A164, and it seems more likely that VEGF-A164 acted directly on lymphatic endothelial cells through VEGFR-2. 51 Our findings predict that malignant tumors that express large amounts of VEGF-A164/5 would also induce the formation of abnormal lymphatics. Tumors have not been thought to generate new lymphatics 52 but this issue needs to be reinvestigated in light of our findings.
In summary, our work has demonstrated that a single cytokine, VEGF-A164, induces remarkably diverse vascular responses and can account for at least three of the distinct types of abnormal blood vessels found in tumors. We do not yet know the secondary factors that are responsible for generating each of these vessel types and the larger number of such factors that must be needed to form the diverse types of mature blood vessels that are found in normal adult tissues. Discovering these secondary factors and elucidating their mode of action is an important future goal that will have important implications for understanding normal vascular development and for developing both pro- and anti-angiogenic therapies that have clinical utility.
Footnotes
Address reprint requests to Harold F. Dvorak, M.D., Department of Pathology, Beth Israel Deaconess Medical Center, 330 Brookline Avenue, Boston, MA 02215. E-mail: hdvorak@bidmc.harvard.edu.
Supported in part by U.S. Public Health Service grants CA-50453, HL-59316, and P01 CA92644 and by a contract from the National Foundation for Cancer Research.
The Rous-Whipple Award was established by the American Society for Investigative Pathology to recognize a career of outstanding scientific contribution. Harold F. Dvorak, the 2002 recipient of the Rous-Whipple Award, delivered a lecture entitled, “How Tumors Make Bad Blood Vessels and Stroma” after accepting the award at the 2002 annual meeting of the American Society for Investigative Pathology in New Orleans, Louisiana.
References
- 1.Dvorak HF: Tumors: wounds that do not heal: similarities between tumor stroma generation and wound healing. N Engl J Med 1986, 315:1650-1659 [DOI] [PubMed] [Google Scholar]
- 2.Dvorak H, Nagy J, Feng D, Dvorak A: Tumor architecture and targeted delivery. Abrams P Fritzberg A eds. Radioimmunotherapy of Cancer. 2000:pp 107-135 Marcel Dekker, Inc., New York
- 3.Folkman J: Tumor angiogenesis. Adv Cancer Res 1985, 43:175-203 [DOI] [PubMed] [Google Scholar]
- 4.Brown LF, Detmar M, Claffey K, Nagy JA, Feng D, Dvorak AM, Dvorak HF: Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. Exs 1997, 79:233-269 [DOI] [PubMed] [Google Scholar]
- 5.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol 1999, 237:97-132 [DOI] [PubMed] [Google Scholar]
- 6.Warren BA: The vascular morphology of tumors. Peterson H-I eds. Tumor Blood Circulation: Angiogenesis, Vascular Morphology and Blood Flow of Experimental and Human Tumors. 1979:pp 1-47 CRC Press, Inc., Boca Raton, FL
- 7.Dvorak HF, Nagy JA, Dvorak AM: Structure of solid tumors and their vasculature: implications for therapy with monoclonal antibodies. Cancer Cells 1991, 3:77-85 [PubMed] [Google Scholar]
- 8.Beck L, Jr, D’Amore PA: Vascular development: cellular and molecular regulation. EMBO J 1997, 11:365-373 [PubMed] [Google Scholar]
- 9.Carmeliet P: Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000, 6:389-395 [DOI] [PubMed] [Google Scholar]
- 10.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J: Vascular-specific growth factors and blood vessel formation. Nature 2000, 407:242-248 [DOI] [PubMed] [Google Scholar]
- 11.Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L, Churchill WH: Fibrin gel investment associated with line 1 and line 10 solid tumor growth, angiogenesis, and fibroplasia in guinea pigs: role of cellular immunity, myofibroblasts, microvascular damage, and infarction in line 1 tumor regression. J Natl Cancer Inst 1979, 62:1459-1472 [PubMed] [Google Scholar]
- 12.Dvorak HF, Orenstein NS, Carvalho AC, Churchill WH, Dvorak AM, Galli SJ, Feder J, Bitzer AM, Rypysc J, Giovinco P: Induction of a fibrin-gel investment: an early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J Immunol 1979, 122:166-174 [PubMed] [Google Scholar]
- 13.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219:983-985 [DOI] [PubMed] [Google Scholar]
- 14.Senger DR, Perruzzi CA, Feder J, Dvorak HF: A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 1986, 46:5629-5632 [PubMed] [Google Scholar]
- 15.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT: Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246:1309-1312 [DOI] [PubMed] [Google Scholar]
- 16.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J: Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest 1989, 84:1470-1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung DW, Cachianes G, Kuang W-J, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 18.Ferrara N, Keyt B: Vascular endothelial growth factor: basic biology and clinical implications. Exs 1997, 79:209-232 [DOI] [PubMed] [Google Scholar]
- 19.Veikkola T, Alitalo K: VEGFs, receptors and angiogenesis. Semin Cancer Biol 1999, 9:211-220 [DOI] [PubMed] [Google Scholar]
- 20.Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Manseau EJ, Dvorak AM, Dvorak HF: VEGF-A induces angiogenesis, arteriogenesis, lymphangiogenesis and vascular malformations. Cold Spring Harbor Symp Quant Biol 2002, 67:227-237 [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A: Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380:435-439 [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard JC, Dewerchin M, Flameng W, Nagy A, Lupu F, Moons L, Collen D, D’Amore PA, Shima DT: Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med 1999, 5:495-502 [DOI] [PubMed] [Google Scholar]
- 23.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E: Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999, 103:159-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe Y, Lee SW, Detmar M, Ajioka I, Dvorak HF: Vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) delays and induces escape from senescence in human dermal microvascular endothelial cells. Oncogene 1997, 14:2025-2032 [DOI] [PubMed] [Google Scholar]
- 25.Miao HQ, Lee P, Lin H, Soker S, Klagsbrun M: Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. EMBO J 2000, 14:2532-2539 [DOI] [PubMed] [Google Scholar]
- 26.Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF: Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest 2000, 80:99-115 [DOI] [PubMed] [Google Scholar]
- 27.Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, Bitzer AM, Carvalho AC: Tumor shedding and coagulation. Science 1981, 212:923-924 [DOI] [PubMed] [Google Scholar]
- 28.Dvorak HF, Senger DS, Dvorak AM, Harvey VS, McDonagh J: Regulation of extravascular coagulation by microvascular permeability. Science 1985, 227:1059-1061 [DOI] [PubMed] [Google Scholar]
- 29.Dvorak HF, Harvey VS, Estrella P, Brown LF, McDonagh J, Dvorak AM: Fibrin-containing gels induce angiogenesis: implications for tumor stroma generation and wound healing. Lab Invest 1987, 57:673-686 [PubMed] [Google Scholar]
- 30.Dvorak HF, Harvey VS, McDonagh J: Quantitation of fibrinogen influx and fibrin deposition and turnover in line 1 and line 10 guinea pig carcinomas. Cancer Res 1984, 44:3348-3354 [PubMed] [Google Scholar]
- 31.Kohn S, Nagy JA, Dvorak HF, Dvorak AM: Pathways of macromolecular tracer transport across venules and small veins: structural basis for the hyperpermeability of tumor blood vessels. Lab Invest 1992, 67:596-607 [PubMed] [Google Scholar]
- 32.Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM: Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med 1996, 183:1981-1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM: Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation 1999, 6:23-44 [PubMed] [Google Scholar]
- 34.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF: The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol 1996, 59:100-115 [PubMed] [Google Scholar]
- 35.Feng D, Nagy JA, Dvorak AM, Dvorak HF: Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res 2000, 59:24-37 [DOI] [PubMed] [Google Scholar]
- 36.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, van de Water L: Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med 1992, 176:1375-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swayne GT, Smaje LH: Dynamic compliance of single perfused frog mesenteric capillaries and rat venules: a filtration coefficient correction. Int J Microcirc Clin Exp 1989, 8:43-52 [PubMed] [Google Scholar]
- 38.Rafii S, Meeus S, Dias S, Hattori K, Heissig B, Shmelkov S, Rafii D, Lyden D: Contribution of marrow-derived progenitors to vascular and cardiac regeneration. Semin Cell Dev Biol 2002, 13:61-67 [DOI] [PubMed] [Google Scholar]
- 39.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM: Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275:964-967 [DOI] [PubMed] [Google Scholar]
- 40.Nagy JA, Morgan ES, Herzberg KT, Manseau EJ, Dvorak AM, Dvorak HF: Pathogenesis of ascites tumor growth: angiogenesis, vascular remodeling, and stroma formation in the peritoneal lining. Cancer Res 1995, 55:376-385 [PubMed] [Google Scholar]
- 41.Lantos P, VandenBerg S, Kleihues P: Tumors of the nervous system. Graham D Lantos P eds. Greenfield’s Neuropathology. 1997:pp 583-879 Arnold, London
- 42.Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF: Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am J Pathol 2001, 158:1145-1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKee P: Pathology of the skin with clinical correlations. 1996:pp 16.57-16.66 Mosby International, London
- 44.Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, Lawitts JA, Benjamin L, Tan X, Manseau EJ, Dvorak AM, Dvorak HF: Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med 2002, 196:1497-1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leak LV, Schannahan A, Scully H, Daggett WM: Lymphatic vessels of the mammalian heart. Anat Rec 1978, 191:183-201 [DOI] [PubMed] [Google Scholar]
- 46.Feng D, Nagy J, Dvorak H, Dvorak A: Ultrastructural studies define soluble macromolecular, particulate, and cellular transendothelial cell pathways in venules, lymphatic vessels, and tumor-associated microvessels in man and animals. Microsc Res Tech 2002, 57:289-326 [DOI] [PubMed] [Google Scholar]
- 47.Brown LF, Lanir N, McDonagh J, Tognazzi K, Dvorak AM, Dvorak HF: Fibroblast migration in fibrin gel matrices. Am J Pathol 1993, 142:273-283 [PMC free article] [PubMed] [Google Scholar]
- 48.Dvorak HF, Form DM, Manseau EJ, Smith BD: Pathogenesis of desmoplasia: I: immunofluorescence identification and localization of some structural proteins of line 1 and line 10 guinea pig tumors and of healing wounds. J Natl Cancer Inst 1984, 73:1195-1205 [PubMed] [Google Scholar]
- 49.Losordo DW, Vale PR, Isner JM: Gene therapy for myocardial angiogenesis. Am Heart J 1999, 138:132-141 [DOI] [PubMed] [Google Scholar]
- 50.Oh SJ, Jeltsch MM, Birkenhager R, McCarthy JE, Weich HA, Christ B, Alitalo K, Wilting J: vEGF and VEGF-C: specific induction of angiogenesis and lymphangiogenesis in the differentiated avian chorioallantoic membrane. Dev Biol 1997, 188:96-109 [DOI] [PubMed] [Google Scholar]
- 51.Feng D, Nagy JA, Brekken RA, Pettersson A, Manseau EJ, Pyne K, Mulligan R, Thorpe PE, Dvorak HF, Dvorak AM: Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors. J Histochem Cytochem 2000, 48:545-556 [DOI] [PubMed] [Google Scholar]
- 52.Jain RK, Fenton BT: Intratumoral lymphatic vessels: a case of mistaken identity or malfunction? J Natl Cancer Inst 2002, 94:417-421 [DOI] [PubMed] [Google Scholar]