

Abstract
Caveolin-1 is the principal protein component of caveolae membrane domains, which are located at the cell surface in most cell types. Evidence has accumulated suggesting that caveolin-1 may function as a suppressor of cell transformation in cultured cells. The human CAV-1 gene is located at a putative tumor suppressor locus (7q31.1/D7S522) and a known fragile site (FRA7G) that is deleted in a variety of epithelial-derived tumors. Mechanistically, caveolin-1 is known to function as a negative regulator of the Ras-p42/44 MAP kinase cascade and as a transcriptional repressor of cyclin D1, possibly explaining its transformation suppressor activity in cultured cells. However, it remains unknown whether caveolin-1functions as a tumor suppressor gene in vivo. Here, we examine the tumor suppressor function of caveolin-1 using Cav-1 (−/−) null mice as a model system. Cav-1 null mice and their wild-type counterparts were subjected to carcinogen-induced skin tumorigenesis, using 7,12-dimethylbenzanthracene (DMBA). Mice were monitored weekly for the development of tumors. We demonstrate that Cav-1 null mice are dramatically more susceptible to carcinogen-induced tumorigenesis, as they develop skin tumors at an increased rate. After 16 weeks of DMBA-treatment, Cav-1 null mice showed a 10-fold increase in tumor incidence, a 15-fold increase in tumor number per mouse (multiplicity), and a 35-fold increase in tumor area per mouse, as compared with wild-type littermate mice. Moreover, before the development of tumors, DMBA-treatment induced severe epidermal hyperplasia in Cav-1 null mice. Both the basal cell layer and the suprabasal cell layers were expanded in treated Cav-1 null mice, as evidenced by immunostaining with cell-type specific differentiation markers (keratin-10 and keratin-14). In addition, cyclin D1 and phospho-ERK1/2 levels were up-regulated during epidermal hyperplasia, suggesting a possible mechanism for the increased susceptibility of Cav-1 null mice to tumorigenesis. However, the skin of untreated Cav-1 null mice appeared normal, without any evidence of epidermal hyperplasia, despite the fact that Cav-1 null keratinocytes failed to express caveolin-1 and showed a complete ablation of caveolae formation. Thus, Cav-1 null mice require an appropriate oncogenic stimulus, such as DMBA treatment, to reveal their increased susceptibility toward epidermal hyperplasia and skin tumor formation. Our results provide the first genetic evidence that caveolin-1 indeed functions as a tumor suppressor gene in vivo.
Caveolae are vesicular invaginations of the plasma membrane that participate in membrane trafficking and signal transduction. They are present in a multitude of cell types including fibroblasts, adipocytes, endothelial cells, and epithelial cells. 1 Caveolin-1 was the first member identified in the caveolin gene family and is the principal structural component of caveolae. Caveolin-1 was initially identified as a major 22-kd v-Src substrate in Rous sarcoma virus (RSV) transformed fibroblasts, implicating caveolin-1 as a target for modification or inactivation by activated oncogenes. 2
Since then, cell culture experiments have contributed a wealth of evidence suggesting that caveolin-1 may function as a transformation or tumor suppressor. Caveolin-1 mRNA and protein levels are strikingly down-regulated in NIH 3T3 fibroblasts transformed with activated oncogenes such as v-Abl, Bcr-Abl, and H-RasG12V, with their ability to grow in soft agar being inversely correlated with caveolin-1 protein levels. 3 Moreover, recombinant expression of caveolin-1 has been shown to inhibit tumor cell proliferation and severely reduces the ability of these cells to proliferate in an anchorage-independent manner. 4-8 In a separate report, ablation of caveolin-1 levels using an anti-sense approach was sufficient to induce NIH 3T3 cell transformation, as evidenced by growth in soft agar and the formation of tumors in nude mice. 9 These findings appear to be mediated in part through hyperactivation of the Ras-p42/44 MAP kinase cascade and are reversible after loss of the Cav-1 anti-sense vector. Conversely, Lee and colleagues 10 have shown that reintroduction of caveolin-1 into a human breast cancer cell line, namely T47D, resulted in an ∼ 50% reduction in proliferation and a remarkable 15-fold decrease in soft agar colony formation. Additionally, other groups have reported that exogenous expression of caveolin-1 dramatically reduces anchorage-independent growth, cell migration, matrix invasion, and lamellipod extension in transformed mammary epithelial cell lines, ie, MCF-7 and MTLn3. 7,8
Aside from data derived from cell culture experiments, there are several lines of clinical and genetic evidence implicating caveolin-1 as a tumor suppressor in vivo. First, caveolin-1 maps to the 7q31.1 region on human chromosome 7, which is frequently deleted in a number of human epithelial-derived cancers, including breast, prostate, ovarian, renal, and colon cancers. 11-18 This region (D7S522) is also a common fragile site known as FRA7G and is suspected to harbor a tumor suppressor locus. 12,19 Second, caveolin-1 levels have been shown to be down-regulated in a number of human-derived cell lines, including mammary adenocarcinomas and in tumors derived from transgenic mouse models of breast cancer. 10,20-22 Finally, Hayashi and colleagues have identified a sporadic P132L mutation in the caveolin-1 gene in up to ∼ 16% of tumors from a human breast cancer cohort. 23 In further support of this clinical finding, we have recently shown that Cav-1 (P132L) behaves in a dominant-negative fashion, causing the intracellular retention of wild-type Cav-1 in the Golgi compartment. 24
Using standard homologous recombination techniques, we and others have reported on the generation of caveolin-1 (−/−) deficient mice. 25,26 While these mice do not experience an increased rate of spontaneous tumor formation, there are several phenotypes that we have recently described which suggest the dysregulation of cellular proliferation. These include hyperproliferation of cultured mouse embryonic fibroblasts (MEFs), an abnormal hypercellular lung phenotype, and mammary epithelial cell hyperplasia in virgin female mice, 24 with the accelerated development of the lobular-alveolar compartment during pregnancy and premature lactation. 27 However, there have been no studies to directly document whether loss of caveolin-1 predisposes mice to tumor formation either by chemically-induced carcinogenesis or by breeding them to genetic tumor-prone mouse models.
Therefore, to more directly examine the role of caveolin-1 in tumor formation, we subjected Cav-1 (−/−) null mice, and their wild-type littermates, to a skin carcinogenesis protocol using 7,12-dimethlybenzanthracene (DMBA). Chemical carcinogenesis induced by DMBA is a well-documented method for generating skin tumors in mice. 28-32 DMBA is a genotoxic agent that forms DNA adducts resulting in DNA damage and genomic instability leading to tumor initiation. Here, we report that loss of caveolin-1 gene expression sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumorigenesis in vivo. These findings clearly provide the first in vivo evidence that caveolin-1 can function either as a tumor suppressor or a tumor susceptibility gene.
In support of these findings, Gumbleton and colleagues examined the expression pattern of caveolin-1 in chronic plaque psoriasis, a well-known hyperproliferative skin disorder. 33 They showed that caveolin-1 expression was greatly reduced or absent in the hyperproliferating basal cell layer of human psoriatic plaques. Based on these data, they hypothesized that down-regulation of caveolin-1 expression may be a critical factor in the pathogenesis or progression of psoriasis. Consistent with this correlative data, we directly show that loss of caveolin-1 gene expression predisposes murine keratinocytes toward epidermal hyperplasia.
Materials and Methods
Materials
Antibodies and their sources were as follows: anti-caveolin-1 IgG (rabbit pAb; Santa Cruz Biotechnology, Inc., Santa Cruz, CA); anti-mouse keratin-10 IgG (rabbit pAb; Covance Research Products, Inc., Princeton, NJ); anti-mouse keratin-14 IgG (rabbit pAb; Covance Research Products, Inc.); anti-phospho-ERK-1/2 IgG (rabbit pAb; Cell Signaling, Inc., Beverly, MA); and anti-cyclin D1 IgG (rabbit pAb; NeoMarkers, Inc., Fremont, CA). A variety of other reagents were purchased commercially and were the highest purity grade available.
Generation and Husbandry of Caveolin-1 Null Mice
Cav-1 (−/−) knockout mice were generated as previously described, using standard homologous recombination techniques. 26 All animals used for these experiments (Cav-1 null mice and their wild-type control littermates) were in the C57Bl/6 background. Mice were housed and maintained in a barrier facility at the Albert Einstein College of Medicine, Bronx, New York. Mice were kept on 12-hour light/dark cycle and they had ad libitum access to chow (Picolab 20; PMI Nutrition International) and water.
Transmission Electron Microscopy
Skin biopsies from the backs of untreated wild-type and Cav-1 null mice were fixed with 2.5% glutaraldehyde/0.1 mol/L cacodylate, postfixed with OsO4, and stained with uranyl acetate and lead citrate. Samples were then examined under a JEOL 1200EX transmission electron microscope and photographed at a magnification of 16,000×. Caveolae were identified by their characteristic flask-like shape, size (50 to 100 nm), and location proximal to the plasma membrane.
Induction of Tumorigenesis
Skin carcinogenesis was induced essentially as previously described. 30,34 Briefly, a total of 30 mice were used (Cav-1 +/+, n = 15; Cav-1 −/−, n = 15). The backs of 6-day-old mice were shaved and painted once a week with a solution of 0.5% DMBA (7,12-dimethylbenzanthracene; Sigma, St. Louis, MO) dissolved in acetone. Mice were monitored weekly for the appearance of tumors. For calculation of tumor area per mouse, the size of tumors was measured using a caliper and the total area of the tumors was then calculated and divided by the number of mice analyzed.
Collection and Processing of Skin and Tumor Samples
Mice were sacrificed and skin biopsies or tumors were surgically removed and fixed in 10% neutral-buffered formalin for 24 hours, after which the samples were placed in 70% ethanol until processing. Tissue samples were paraffin-embedded, 4- to 5-μm sections were cut, and placed on Super Frost Plus slides (Fisher) for pathological evaluation or immunostaining.
Histopathological Analyses
For tumor evaluation and classification, H&E tissue sections were microscopically examined at low and high magnification by Dr. Steve McClain, an expert dermato-pathologist.
Immunostaining of Tissue Sections
For immunostaining, paraffin-embedded tissue sections were de-paraffinized in xylene (twice, 10 minutes each), dehydrated through a graded series of ethanol washes, and placed in PBS. After use of an Antigen Retrieval Kit (DAKO Corp), tissue sections were blocked with 5% fetal bovine serum in PBS for 30 minutes at room temperature. Primary antibodies were used at the following dilutions: anti-caveolin-1 pAb (1:1000); anti-mouse keratin-10 (1:1000); and anti-mouse keratin-14 (1:10,000), in PBS containing 0.1% BSA. Fluorescently conjugated secondary antibodies (5 μg/ml) were added to the sections for 30 minutes at room temperature. After extensive washing with PBS, the slides were mounted with Slow-Fade anti-fade reagent (Molecular Probes). Slides were observed with an Olympus IX 70 inverted microscope.
Similar experiments were carried out using primary antibodies directed against cyclin D1 and phopho-ERK1/2, and HRP-conjugated secondary antibodies. However, endogenous peroxide activity was quenched by incubating the slides for 10 minutes in 1% H2O2. Bound antibodies were visualized using DAB as the substrate (for 15–20 seconds). Finally, the slides were washed in dH2O to remove excess DAB, counterstained with hematoxylin, dehydrated, and mounted with coverslips.
Statistical Analyses
Results are represented as the mean ± SEM. Statistical significance was determined using Student’s t-test, with P < 0.05 being considered significant.
Results
Cav-1 (−/−) Null Mice Show Normal Skin Morphology but Fail to Express Caveolin-1
To assess the morphology of the epidermis in Cav-1 null mice, skin biopsies were taken and fixed in formalin. After H&E staining, the paraffin-embedded sections were analyzed by light microscopy. Figure 1 ▶ shows the morphology of skin samples derived from the backs of adult Cav-1 null mice and their corresponding wild-type controls (all 4 months old). Note that the epidermal layer appears normal; importantly, no epidermal hyperplasia was evident in Cav-1 null mouse skin.
Figure 1.
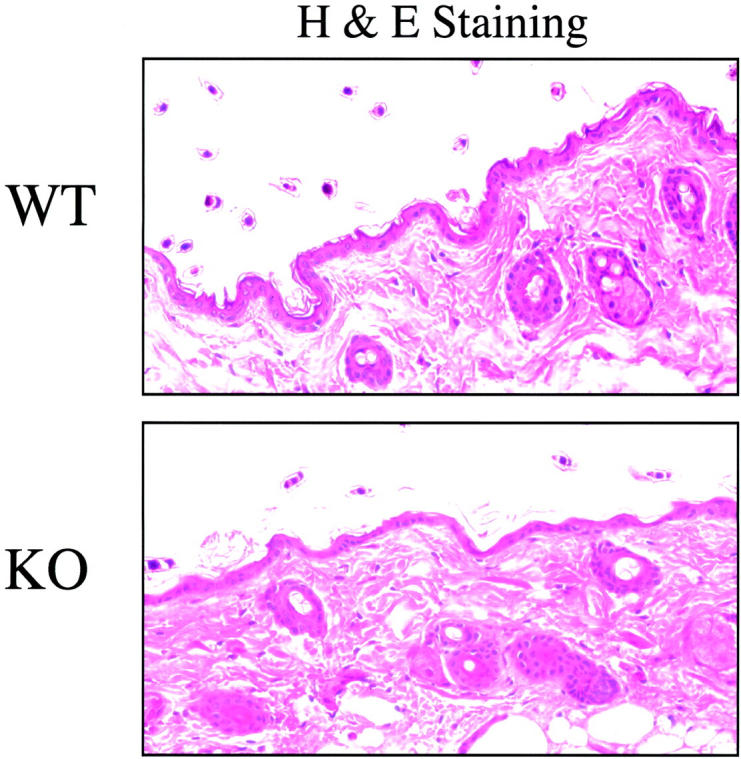
Cav-1 null mice show normal skin morphology. Skin biopsies were taken from the backs of untreated adult wild-type and Cav-1 null mice (4 months old). After excision, samples were formalin-fixed, paraffin-embedded, and stained with H&E. Note that Cav-1 null skin appears normal, as compared with wild-type control mice. Importantly, there is no evidence of epidermal hyperplasia or hyperkeratosis. Images were acquired with a 40× objective. Similar results were obtained with younger mice.
To verify that caveolin-1 is normally expressed in keratinocytes, we immunostained skin tissue sections with anti-caveolin-1 IgG. Figure 2 ▶ (left panel) shows that caveolin-1 is robustly expressed in murine keratinocytes, especially within the basal cell layer (see arrow) and the hair follicles (see asterisks). Similarly, other groups have shown that caveolin-1 is normally expressed at high levels in human keratinocytes, especially within the basal cell layer. 33,35,36 In contrast, skin tissue sections derived from Cav-1 null mice show no caveolin-1 immunostaining, as expected (Figure 2 ▶ , right panel).
Figure 2.

Caveolin-1 immunostaining in wild-type and Cav-1 null skin biopsies. Paraffin-embedded sections of skin biopsies derived from the backs of untreated adult wild-type and Cav-1 null mice were immunostained with antibodies directed against the N-terminal domain of caveolin-1 (rabbit pAb, N-20; Santa Cruz Biotech, Inc.). Note that in wild-type mice, caveolin-1 is highly expressed in keratinocyes, especially within the basal cell layer (arrow) and the hair follicles (asterisks). Images shown were acquired with a 20× objective.
In addition, we evaluated the status of caveolae formation in Cav-1 null keratinocytes. Figure 3 ▶ shows by transmission electron microscopy that Cav-1 null keratinocytes lack morphologically identifiable caveolae membranes, while wild-type keratinocytes have abundant plasmalemmal caveolae.
Figure 3.

Cav-1 null keratinocytes lack morphologically identifiable caveolae. Skin biopsies taken from untreated wild-type and Cav-1 null mice were processed for transmission electron microscopy. Note that in wild-type animals, caveolae organelles (50- to 100-nm vesicles and invaginations; arrows) are particularly abundant in keratinocytes within the basal cell layer (A) and the hair follicles (B). Interestingly, the distribution of caveolae in keratinocytes is restricted to the basal membrane, but they are absent from the lateral and apical regions of the plasma membrane. However, in Cav-1 null keratinocytes, note that caveolae formation is completely ablated. The cytoplasm and extracellular collagen fibrils are as indicated. N, nucleus. Scale bar, 250 nm.
Interestingly, in wild-type animals, both caveolin-1 immunostaining and caveolae organelles were preferentially localized to the basal plasma membrane of keratinocytes, ie, in a polarized distribution (Figures 2 and 3) ▶ , and were virtually absent from the lateral and apical surfaces. Consistent with these findings, a similar polarized distribution of caveolin-1 and caveolae has been noted in both renal and intestinal epithelial cells. 37-39
Cav-1 Null Mice Are Dramatically More Susceptible to DMBA-Induced Skin Carcinogenesis
To assess the tumor suppressor role of caveolin-1 in a whole animal model, we treated the skin of Cav-1 null mice and their wild-type counterparts with a known carcinogen, DMBA (7,12-dimethylbenzanthracene), using a standard protocol. DMBA treatments were administered weekly for a period of up to 16 weeks and tumor appearance was monitored visually.
Figure 4A ▶ shows that the tumor incidence in Cav-1 null mice is greatly increased. At 8 weeks of treatment, only 10% of wild-type mice developed tumors and this remained constant for up to 16 weeks. In striking contrast, at 8 weeks of treatment, > 80% of Cav-1 null mice developed tumors, reaching 100% at 12 weeks. Thus, at 16 weeks of treatment, Cav-1 null mice show a 10-fold increase in tumor incidence. Figure 4B ▶ shows that tumor multiplicity is also greatly increased in Cav-1 null mice. For example, at 8 and 12 weeks of treatment Cav-1 null mice show an ∼10-fold increase in tumor number per mouse. In addition, at 16 weeks of treatment, Cav-1 null mice show an ∼15-fold increase in tumor multiplicity.
Figure 4.
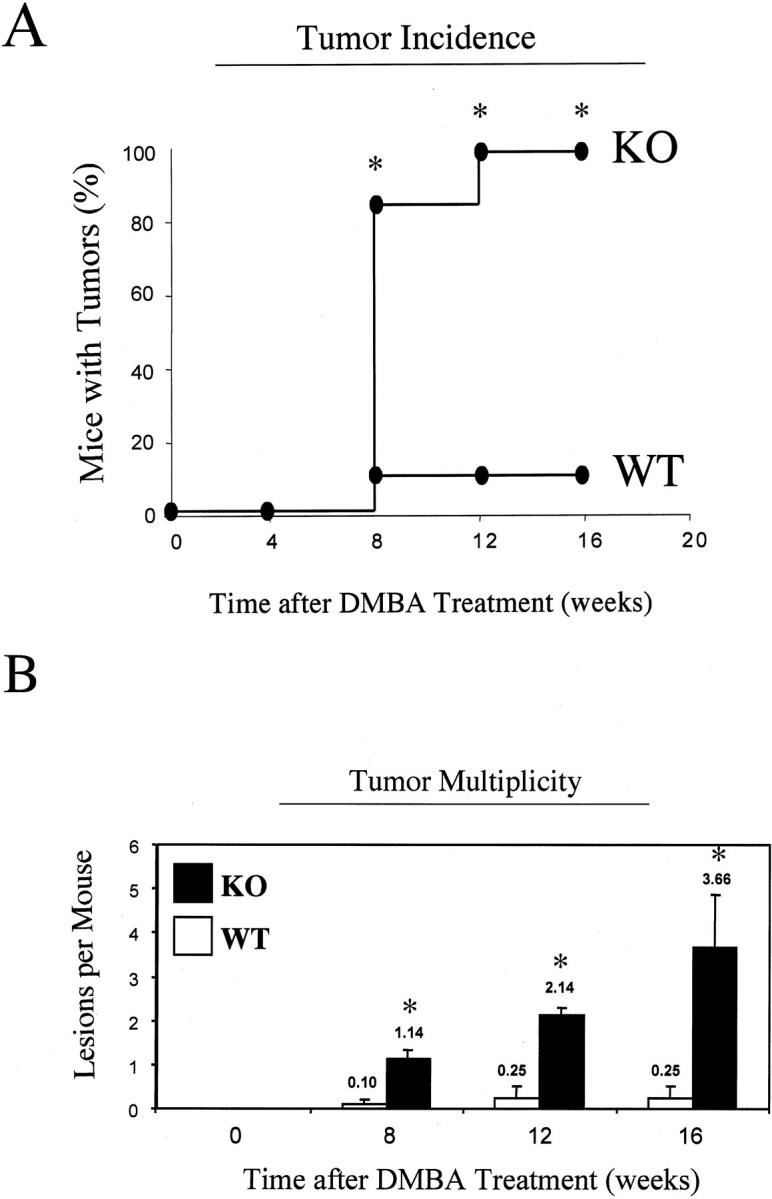
Cav-1 null mice are dramatically more susceptible to DMBA-induced skin carcinogenesis. The backs of 6-day-old wild-type and Cav-1 null mice were shaved and painted once a week with a solution of 0.5% DMBA. Mice were then monitored weekly for the appearance of tumors. A: Tumor incidence. Note that tumor incidence in Cav-1 null mice is greatly increased. At 8 weeks of treatment, only 10% of wild-type mice developed tumors and this remained constant for up to 16 weeks. In striking contrast, at 8 weeks of treatment, >80% of Cav-1 null mice developed tumors, reaching a maximum of 100% at 12 weeks. Thus, at 16 weeks of treatment, Cav-1 null mice show a 10-fold increase in tumor incidence. B: Tumor multiplicity. Note also that tumor multiplicity is greatly increased in Cav-1 null mice. At 8 and 12 weeks of treatment, Cav-1 null mice show an ∼10-fold increase in tumor number per mouse. In addition, at 16 weeks of treatment, Cav-1 null mice show an ∼15-fold increase in tumor multiplicity. In A and B, an asterisk denotes statistical significance; P < 0.05.
At 16 weeks of treatment, we assessed tumor growth by measuring the total area occupied by tumors in a given mouse. Figure 5 ▶ shows an ∼35-fold increase in tumor area per Cav-1 null mouse.
Figure 5.

Skin tumor growth is accelerated in Cav-1 null mice. The backs of 6-day-old wild-type and Cav-1 null mice were shaved and painted once a week with a solution of 0.5% DMBA. Total tumor area per mouse was then quantitated after 16 weeks of treatment. Note that Cav-1 null mice showan ∼35-fold increase in tumor area per mouse. An asterisk denotes statistical significance; P < 0.05.
Tumors were also characterized morphologically. Both wild-type and Cav-1 null mice showed skin lesions that grossly appeared as papillomas (Figures 6A, B and Figure 7) ▶ . After 16 weeks of treatment, tumors and their adjacent normal tissue were excised and subjected to histopathological analysis. Figure 8 ▶ illustrates three such representative lesions that were all classified as noninvasive papillomas, with significant epidermal hyperplasia. One wild-type tumor and two Cav-1 null tumors are shown for comparison. However, tumor grade was not increased in Cav-1 null mice, but the incidence, the multiplicity, and the growth of tumors were increased.
Figure 6.
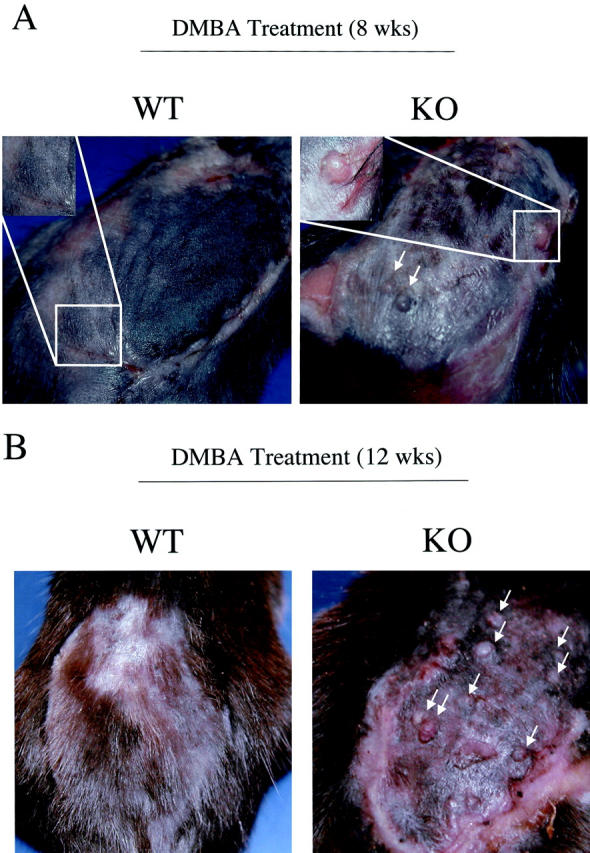
Gross morphology of DMBA-induced skin tumors at 8 and 12 weeks of treatment. The backs of 6-day-old wild-type and Cav-1 null mice were shaved and painted once a week with a solution of 0.5% DMBA. The appearance of tumors was documented with the use of a digital camera. The gross morphology of DMBA-induced tumors at 8 weeks (A) and 12 weeks (B) of treatment is shown. One representative example is included for each genotype. In Cav-1 null mice, note that the lesions grossly appeared as papillomas (arrows). Very few, if any, lesions were detected in wild-type mice at the same time points.
Figure 8.
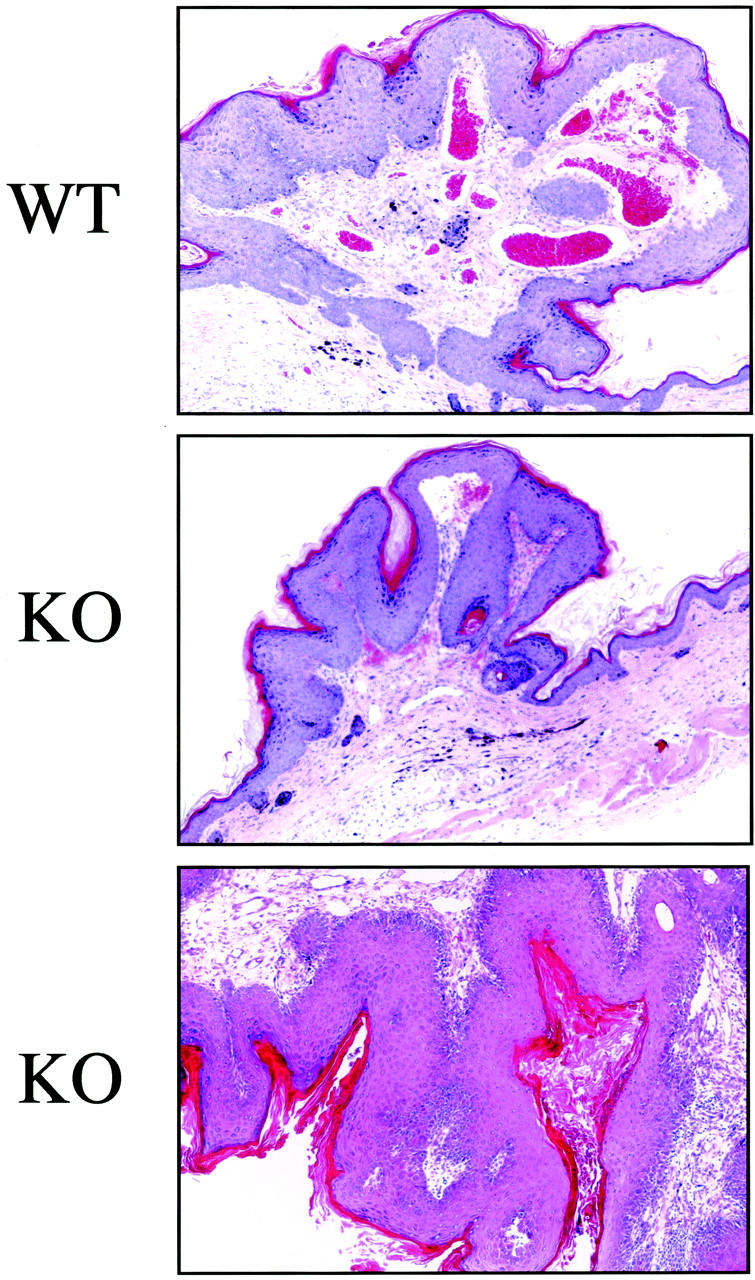
Histopathological appearance of DMBA-induced skin tumors from wild-type and Cav-1 null mice. After 16 weeks of treatment, tumors and their adjacent normal tissue were excised and subjected to histopathological analysis. Three such lesions that were classified as noninvasive papillomas, with significant epidermal hyperplasia are shown. However, tumor grade was not increased in Cav-1 null mice, but rather only the incidence, the multiplicity, and the growth of tumors were increased. Images were acquired with a 10× objective.
Before Tumor Formation, Cav-1 Null Mice Develop Extensive Epidermal Hyperplasia in Response to DMBA Treatment
Because the tumor grade was not increased in Cav-1 null mice, we next examined the morphology of the skin at 7 weeks of DMBA-treatment, just before the development of tumors. Skin tissue samples were excised, formalin-fixed, paraffin-embedded, and stained with H&E.
Figure 9 ▶ shows medium- and high-power views of treated wild-type and Cav-1 null skin. Note that in wild-type animals, there is only a modest increase in the thickness of the epidermal layer, as compared with non-treated animals (see Figure 1 ▶ for comparison). However, in Cav-1 null mice, there is a dramatic increase in the thickness of the epidermal layer, ie, severe epidermal hyperplasia. The change in epidermal thickness is best appreciated at high power (B), while the extent of these changes is best visualized at medium power (A). Note also that all of the layers of the epidermis, especially the basal, granular, and cornified layers are hyperplastic or thickened. Thus, it appears that Cav-1 null mice are more susceptible to DMBA-induced epidermal hyperplasia, possibly explaining their accelerated rate of tumorigenesis.
Figure 9.
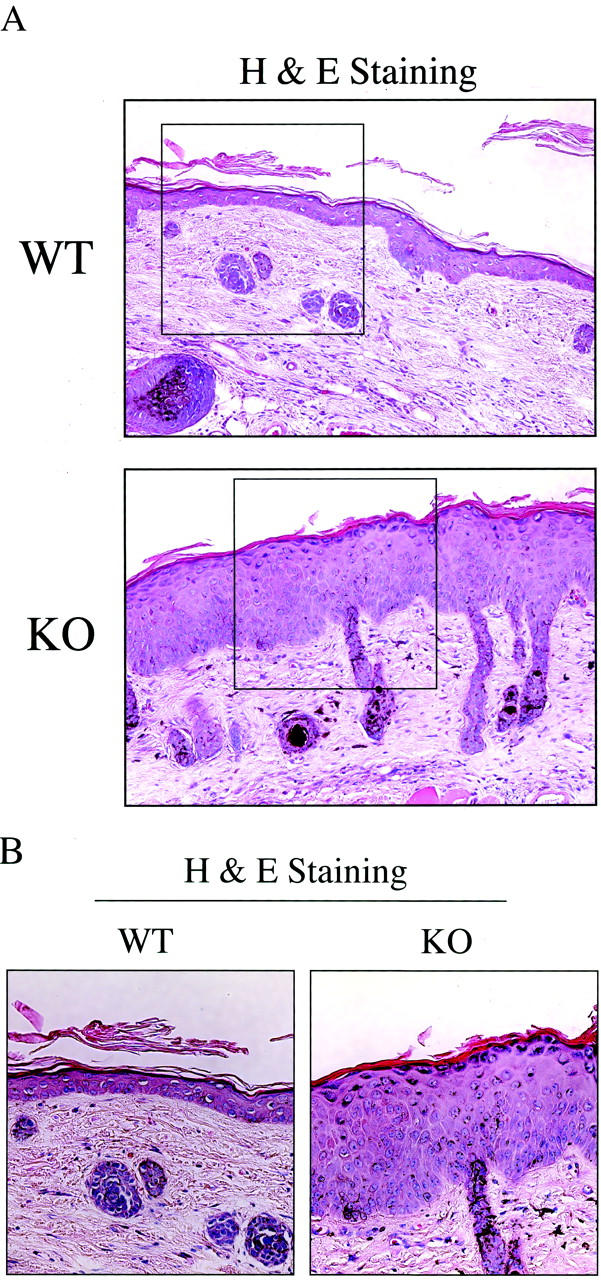
Before tumor formation, Cav-1 null mice develop extensive epidermal hyperplasia in response to DMBA treatment. We also examined the morphology of the skin at 7 weeks of DMBA treatment, just before the development of tumors. Skin tissue samples were excised, formalin-fixed, paraffin-embedded and stained with H&E. Note that in wild-type animals, there is only a modest increase in thickness of the epidermal layer, as compared with non-treated animals (Figure 1) ▶ . In contrast, in Cav-1 null mice there is a dramatic increase in the thickness of the epidermal layer, ie, severe epidermal hyperplasia. Medium- and high-power views are shown in A and B, respectively. Images were acquired with a 20× objective (A). Boxed areas in A are shown further magnified in B. Note that in Cav-1 null mice all of the layers of the epidermis, especially the basal, granular, and cornified layers are hyperplastic or thickened.
To further evaluate the epidermal hyperplasia phenotype of Cav-1 null mice, we next performed immunostaining with keratin-specific differentiation markers. Keratin-10 is most highly expressed in cells of the suprabasal layer (spinous/granular), while keratin-14 is confined to the basal cell layer. 40 Figure 10 ▶ clearly shows that in Cav-1 null mice both the suprabasal cell layer (keratin-10 immunostaining, panel A) and the basal cell layer (keratin-14 immunostaining, panel B) are increased in thickness and appear hypercellular. These results directly support the idea that the skin of Cav-1 null mice undergoes significant epidermal hyperplasia in response to DMBA treatment.
Figure 10.
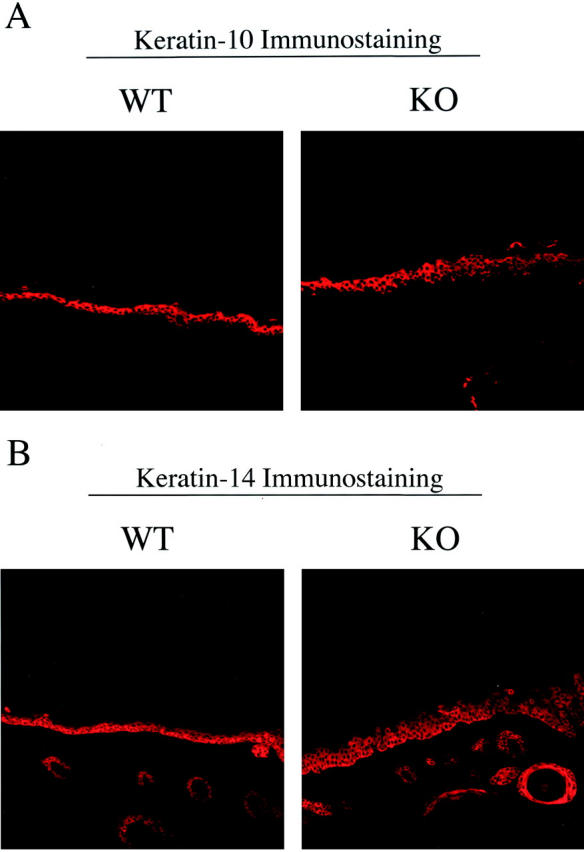
Immunostaining with cell-type specific markers (keratin-10 and keratin-14) reveals that both the basal cell layer and the suprabasal cell layers are expanded in treated Cav-1 null mouse skin. To further evaluate the DMBA-induced epidermal hyperplasia in Cav-1 null mice, we next performed immunostaining with keratin markers. Note that in Cav-1 null mice both the suprabasal cell layer (keratin-10 immunostaining, A) and the basal cell layer (keratin-14 immunostaining, B) are increased in thickness and appear hypercellular.
Up-Regulation of Cyclin D1 and Phospho-ERK1/2 in Cav-1 Null Keratinocytes During DMBA-Induced Epidermal Hyperplasia
Cav-1 is known to function as a negative regulator of the Ras-p42/44 MAP kinase cascade and as a transcriptional repressor of cyclin D1, possibly explaining its transformation suppressor activity in cultured cells. 4,9,41,42 Thus, we next examined the levels of cyclin D1 and activated ERK1/2 during DMBA-induced epidermal hyperplasia. At 7 weeks of DMBA-treatment, just before the development of tumors, skin tissue samples were excised, formalin-fixed, paraffin-embedded and immunostained with antibodies to cyclin D1 and activated ERK1/2.
Figure 11A ▶ shows that in wild-type animals, mainly the proliferative basal cell layer is immunostained for cyclin D1; in contrast, in Cav-1 null animals, both the basal cell layer and the suprabasal layers are heavily immunostained. Similarly, phospho-ERK1/2 immunostaining was clearly elevated in Cav-1 null keratinocytes and notably absent in wild-type keratinocytes (Figure 11B) ▶ . Thus, cyclin D1 and phospho-ERK1/2 levels are up-regulated in Cav-1 null keratinocytes during DMBA-induced epidermal hyperplasia. These findings provide a possible mechanism to explain why Cav-1 null mice are more susceptible to DMBA-induced skin carcinogenesis.
Figure 11.
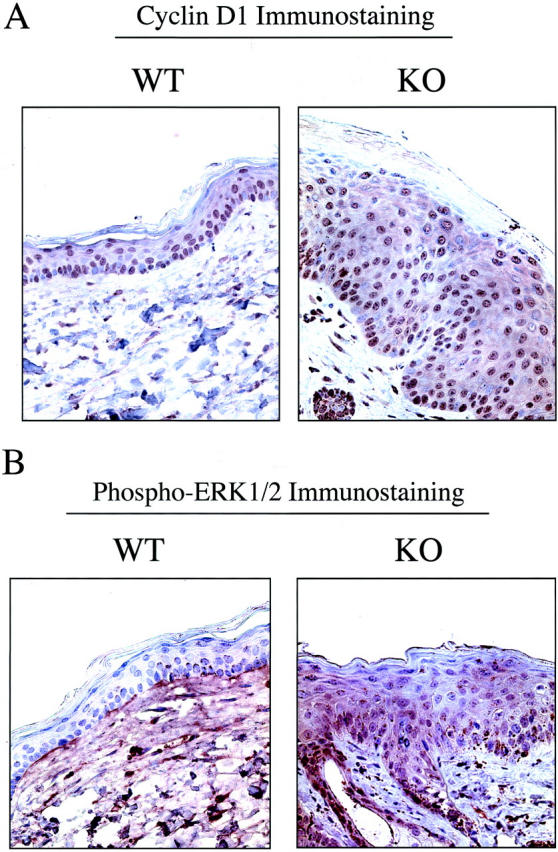
Up-regulation of cyclin D1 and phospho-ERK1/2 in Cav-1 null keratinocytes during DMBA-induced epidermal hyperplasia. Skin tissue samples (at 7 weeks of DMBA treatment) were excised, formalin-fixed, and paraffin-embedded. A: Cyclin D1 immunostaining. In wild-type animals, mainly the proliferative basal cell layer is immunostained for cyclin D1; in contrast, in Cav-1 null animals, both the basal cell layer and the suprabasal layers are heavily immunostained. B: Phospho-ERK1/2 immunostaining. Phospho-ERK1/2 immunostaining was clearly elevated in Cav-1 null keratinocytes and notably absent in wild-type keratinocytes (even on overexposure to the DAB substrate, as shown for the wild-type image). In this case, the wild-type section was intentionally exposed twice as long to DAB to illustrate that the wild-type epidermis is truly negative; however, background staining of the underlying dermis is evident as a consequence.
Discussion
In this report, we have directly assessed the proposed tumor suppressor role of the caveolin-1 gene by using genetically-engineered mice that harbor a targeted disruption of the Cav-1 locus. We subjected these Cav-1 null mice (and their wild-type littermates) to a carcinogen-induced skin tumorigenesis protocol over a 16-week period using DMBA. We showed that Cav-1 null mice have much higher rates of tumor incidence, tumor multiplicity, and tumor growth. More specifically, at 16 weeks of treatment, Cav-1 null mice demonstrated a 10-fold increase in tumor incidence, a 15-fold increase in tumor multiplicity, and a 35-fold increase in tumor area per mouse. In addition, at 7 weeks of treatment, DMBA induced severe epidermal hyperplasia in Cav-1 null mice before the development of tumors. Both suprabasal and basal cell layers appeared hyperplastic. This was confirmed by immunostaining with cell-type specific differentiation markers (keratins 10 and 14). In contrast, untreated Cav-1 null mice did not show any signs of epidermal hyperplasia or hyperkeratosis. Thus, Cav-1 null mice clearly show increased susceptibility toward epidermal hyperplasia and skin tumor formation.
What is the role of Cav-1 in skin tumorigenesis? We and others have demonstrated that skin tissue exhibits significant amounts of caveolin-1 expression, including the epidermis, but there is not much known regarding what role caveolin-1 plays in different skin cell types. For the two main caveolar functions that have garnered the most attention, membrane trafficking and signal transduction, a large body of evidence has accumulated supporting a role for caveolin-1 as a negative regulator of cell transformation and tumorigenesis. Indeed, caveolae microdomains are enriched in a number of signaling molecules including H-Ras, Src-family tyrosine kinases, hetero-trimeric G-protein subunits, receptor tyrosine kinases (RTKs; EGF-R, c-Neu, PDGF-R, and insulin receptor), and endothelial nitric-oxide synthase (eNOS), and many of these molecules have been shown to interact directly with caveolin-1 via the caveolin-scaffolding domain (CSD) (reviewed in 1 ). These results suggest that caveolins are important signaling modulators within the context of caveolae membranes.
Functionally, it appears that caveolin binding serves to inhibit downstream signaling events. For example, Cav-1 has been shown to interact with and suppress the kinase activity of the EGF receptor and several members of the Ras-p42/44 MAP kinase cascade, including MEK and ERK. 41,43 Conversely, down-regulation of Cav-1 by an anti-sense methodology results in hyperactivation of the p42/44 MAP kinase cascade and cellular transformation. 9 Moreover, down-regulation of Cav-1 expression by RNAi in Caenorhabditis elegans results in hyperactivation of the meiotic cell cycle, which is controlled by the Ras-MAP kinase pathway. 44 In accordance with these findings, we show here that phospho-ERK1/2 levels are up-regulated in Cav-1 null keratinocytes during DMBA-induced epidermal hyperplasia in vivo.
There is also an emerging role for caveolin-1 in cell cycle control. Indeed, the balance between cellular differentiation and proliferation is extremely relevant to the process of tumorigenesis. Besides controlling tumor growth rates, this balance can also determine tumor grade since the most invasive tumors are generally among the most poorly-differentiated morphologically. Cav-1 is highly expressed in well-differentiated and slowly proliferating cell types, such as adipocytes and endothelial cells. Primary mouse embryonic fibroblasts (MEFs) derived from Cav-1 (−/−) null mice proliferate faster, show increased DNA-synthesis rates (thymidine incorporation), and demonstrate an increased percentage of cells in S-phase, as compared to their Cav-1 expressing wild-type counterparts. 26
In addition to repression of the Ras-p42/44 MAP kinase pathway, Cav-1 has been found to transcriptionally repress cyclin D1 expression. 42 Cyclin D1 is a well-characterized protein that forms complexes with the cyclin-dependent kinases, cdk4 and cdk6, thereby activating them and allowing entry into S-phase. In addition, the cyclin D1 gene is amplified or over-expressed in a number of epidermal-derived tumors, such as human squamous cell carcinomas or mouse epidermal papillomas harboring activated ras genes. 45,46 Interestingly, DMBA-treatment of transgenic C57Bl/6 mice with keratinocyte-specific over-expression of cyclin D1 resulted in the appearance of papillomas in 100% of mice after 40 weeks; in contrast, wild-type control mice showed a tumor incidence of only 9.5% under identical conditions. 34 Additionally, primary cultured keratinocytes from these cyclin D1 transgenic mice were refractory to calcium-induced terminal differentiation and continued to divide in culture. Thus, in Cav-1 null mice, an absence of transcriptional repression by caveolin-1 could result in overexpression of cyclin D1, providing a possible mechanism for the accelerated appearance of skin papillomas. In support of this prediction, we show here that cyclin D1 levels are up-regulated in Cav-1 null keratinocytes during DMBA-induced epidermal hyperplasia.
There are a number of oncogenes, including H-Ras, v-Abl, middle T antigen, Bcr-Abl, v-Src, Neu T (ErbB2), and c-Myc which target caveolin-1 for transcriptional repression. 1 Indeed, one of the first experiments implicating Cav-1 as a target for cell transformation showed that stable expression of activated H-Ras (G12V mutant) in NIH 3T3 cells causes down-regulation of Cav-1 mRNA and protein levels. 3 Importantly, down-regulation of Cav-1 was completely reversed by treatment with an inhibitor of the Ras-p42/44 MAP kinase pathway, namely PD 98059. 4 Furthermore, recombinant expression of Cav-1 in H-RasG12V-transformed NIH 3T3 cells resulted in abrogation of the transformed phenotype, with dramatic inhibition of the Ras-p42/44 MAP kinase cascade. 4 Interestingly, DMBA-induced carcinogenesis often results in papillomas with activating mutations in the H-Ras proto-oncogene (especially at codon 61). 47,48 Others have shown that activated Ras can then induce over-expression of cyclin D1. 49 Taken together, these data provide an additional mechanism by which loss of caveolin-1 could enhance the initiation of papilloma formation.
Further support for a pro-differentiation or anti-proliferative function for caveolin-1 in skin comes from an in vitro model of keratinocyte differentiation, where others have shown that caveolin-1 protein expression is significantly up-regulated during the differentiation of primary human keratinocytes cultured on a collagen-based matrix. 50 Interestingly, caveolin-1 expression was up-regulated by ∼8-fold in parallel with certain well-known keratinocyte differentiation markers, such as involucrin and fillagrin. 50 Thus, caveolin-1 may play a role in suppressing the proliferation of keratinocytes, perhaps facilitating their ability to undergo differentiation. Furthermore, using PCR and subtractive hybridization techniques, caveolin-1 was independently identified as a KGF (keratinocyte growth factor)-regulated gene. 35 For example, after KGF treatment of keratinocytes in culture, both caveolin-1 mRNA and protein levels increased dramatically. Similarly, caveolin-2 was also induced in response to KGF treatment. This is in contrast to the finding that treatment of NIH 3T3 fibroblasts with other growth factors (such as PDGF and FGF) leads to the p42/44 MAP kinase-induced suppression of caveolin-1 gene expression. 35
In accordance with our current findings, Gumbleton and colleagues 33 have recently examined the expression pattern of caveolin-1 in a well-known hyperproliferative skin disorder in humans, chronic plaque psoriasis. In 20 of the 22 patients examined, caveolin-1 expression in the hyperproliferating basal cell layer of psoriatic plaques was greatly reduced or absent. Based on these findings, they postulated that down-regulation of caveolin-1 expression may be a critical factor in the pathogenesis or progression of psoriasis. Consistent with this correlative data, we directly demonstrate that loss of caveolin-1 gene expression predisposes murine keratinocytes toward epidermal hyperplasia if they are provided with the appropriate stimulus.
In conclusion, our results argue that caveolin-1 may indeed play an important role in the suppression of tumor formation. Given a carcinogenic-stimulus, we have shown that loss of caveolin-1 expression by targeted gene-deletion in mice predisposes these animals toward epidermal hyperplasia and accentuates skin tumor initiation, multiplicity, and growth. As such, this study represents the first clear genetic evidence that caveolin-1 can function either as a tumor suppressor or tumor susceptibility gene in an in vivo animal model.
Interestingly, we have recently observed that Cav-1 (−/−) null mice show endothelial-based defects in angiogenesis in vivo (51). Thus, the dramatic increases in DMBA-induced skin tumorigenesis we observe here in Cav-1 (−/−) null mice may actually be an underestimate of the tumor suppressor capacity of caveolin-1.
Figure 7.

Gross morphology of DMBA-induced skin tumors at 16 weeks of treatment. The gross morphology of DMBA-induced tumors at 16 weeks of treatment is shown. Two representative wild-type examples are shown (left and center) along with one representative Cav-1 null mouse (right). Note that in wild-type and Cav-1 null mice the lesions grossly appeared as papillomas (arrows). However, in Cav-1 null mice these lesions were dramatically more numerous. See Figures 4 and 5 ▶ for quantitation of tumor incidence, multiplicity, and area.
Acknowledgments
We thank Dr. David Neufeld for help with image acquisition, Dr. Radma Mahmood for her expertise with tissue processing and sectioning, Dr. Dolores Di Vizio for her advice regarding immunohistochemistry, Dr. Udayan Guha for many helpful suggestions, and Dr. Richard G. Pestell for insightful discussions.
Footnotes
Address reprint requests to Michael P. Lisanti, M.D., Ph.D., Albert Einstein College of Medicine, Department of Molecular Pharmacology, Golding Building, Room 202, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: lisanti@aecom.yu.edu.
Supported by grants from the National Institutes of Health, the Muscular Dystrophy Association, the American Heart Association, and the Breast Cancer Alliance, as well as a Hirschl/Weil-Caulier Career Scientist Award (all to M.P.L.). T.M.W. was supported by a National Institutes of Health Medical Scientist Training Grant (T32-GM07288).
References
- 1.Razani B, Woodman SE, Lisanti MP: Caveolae: from cell biology to animal physiology. Pharmacol Rev 2002, 54:431-467 [DOI] [PubMed] [Google Scholar]
- 2.Glenney JR, Jr: Tyrosine phosphorylation of a 22-kDa protein is correlated with transformation by Rous sarcoma virus. J Biol Chem 1989, 264:20163-20166 [PubMed] [Google Scholar]
- 3.Koleske AJ, Baltimore D, Lisanti MP: Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA 1995, 92:1381-1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelman JA, Wycoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP: Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem 1997, 272:16374-16381 [DOI] [PubMed] [Google Scholar]
- 5.Park DS, Razani B, Lasorella A, Schreiber-Agus N, Pestell RG, Iavarone A, Lisanti MP: Evidence that Myc isoforms transcriptionally repress caveolin-1 gene expression via an INR-dependent mechanism. Biochemistry 2001, 40:3354-3362 [DOI] [PubMed] [Google Scholar]
- 6.Razani B, Altschuler Y, Zhu L, Pestell RG, Mostov KE, Lisanti MP: Caveolin-1 expression is down-regulated in cells transformed by the human papilloma virus in a p53-dependent manner. Replacement of caveolin-1 expression suppresses HPV-mediated cell transformation. Biochemistry 2000, 39:13916-13924 [DOI] [PubMed] [Google Scholar]
- 7.Fiucci G, Ravid D, Reich R, Liscovitch M: Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 2002, 21:2365-2375 [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Razani B, Altschuler Y, Bouzahzah B, Mostov KE, Pestell RG, Lisanti MP: Caveolin-1 inhibits epidermal growth factor-stimulated lamellipod extension and cell migration in metastatic mammary adenocarcinoma cells (MTLn3): transformation suppressor effects of adenovirus-mediated gene delivery of caveolin-1. J Biol Chem 2000, 275:20717-20725 [DOI] [PubMed] [Google Scholar]
- 9.Galbiati F, Volonté D, Engelman JA, Watanabe G, Burk R, Pestell R, Lisanti MP: Targeted down-regulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J 1998, 17:6633-6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE: Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998, 16:1391-1397 [DOI] [PubMed] [Google Scholar]
- 11.Engelman JA, Zhang XL, Galbiati F, Lisanti MP: Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3): Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett 1998, 429:330-336 [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Zhang XL, Lisanti MP: Genes encoding human caveolin-1 and -2 are co-localized to the D7S522 locus (7q31.1), a known fragile site (FRA7G) that is frequently deleted in human cancers. FEBS Lett 1998, 436:403-410 [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA, Zhang XL, Galbiati F, Volonte D, Sotgia F, Pestell RG, Minetti C, Scherer PE, Okamoto T, Lisanti MP: Molecular genetics of the caveolin gene family: implications for human cancers, Diabetes, Alzheimer’s disease, and muscular dystrophy. Am J Hum Genetics 1998, 63:1578-1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins RB, Qian J, Lee HK, Huang H, Hirasawa K, Bostwick DG, Proffitt J, Wilber K, Lieber MM, Liu W, Smith DI: A molecular cytogenetic analysis of 7q31 in prostate cancer. Cancer Res 1998, 58:759-766 [PubMed] [Google Scholar]
- 15.Kerr J, Leary JA, Hurst T, Shih YC, Antalis TM, Friedlander M, Crawford E, Khoo SK, Ward B, Chenevix-Trench G: Allelic loss on chromosome 7q in ovarian adenocarcinomas: two critical regions and a rearrangement of the PLANH1 locus. Oncogene 1996, 13:1815-1818 [PubMed] [Google Scholar]
- 16.Shridhar V, Sun QC, Miller OJ, Kalemkerian GP, Petros J, Smith DI: Loss of heterozygosity on the long arm of human chromosome 7 in sporadic renal cell carcinomas. Oncogene 1997, 15:2727-2733 [DOI] [PubMed] [Google Scholar]
- 17.Zenklusen JC, Thompson JC, Troncoso P, Kagan J, Conti CJ: Loss of heterozygosity in human primary prostate carcinomas: a possible tumor suppressor gene at 7q31.1. Cancer Res 1994, 54:6370-6373 [PubMed] [Google Scholar]
- 18.Zenklusen JC, Bieche I, Lidereau R, Conti CJ: (C-A)n microsatellite repeat D7S522 is the most commonly deleted region in human primary breast cancer. Proc Natl Acad Sci USA 1994, 91:12155-12158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H, Qian C, Jenkins RB, Smith DI: Fish mapping of YAC clones at human chromosomal band 7q31.2: identification of YACS spanning FRA7G within the common region of LOH in breast and prostate cancer. Genes Chromosomes Cancer 1998, 21:152-159 [DOI] [PubMed] [Google Scholar]
- 20.Sager R, Sheng S, Anisowicz A, Sotiropoulou G, Zou Z, Stenman G, Swisshelm K, Chen Z, Hendrix MJC, Pemberton P, Rafidi K, Ryan K: RNA genetics of breast cancer: maspin as a paradigm. Cold Spring Harbor Sym. Quant. Biol. 1994, LIX:537-546 [DOI] [PubMed] [Google Scholar]
- 21.Suzuki T, Suzuki Y, Hanada K, Hashimoto A, Redpath JL, Stanbridge EJ, Nishijima M, Kitagawa T: Reduction of caveolin-1 expression in tumorigenic human cell hybrids. J Biochem 1998, 124:383-388 [DOI] [PubMed] [Google Scholar]
- 22.Engelman JA, Lee RJ, Karnezis A, Bearss DJ, Webster M, Siegel P, Muller WJ, Windle JJ, Pestell RG, Lisanti MP: Reciprocal regulation of Neu tyrosine kinase activity and caveolin-1 protein expression in vitro and in vivo. Implications for mammary tumorigenesis. J Biol Chem 1998, 273:20448-20455 [DOI] [PubMed] [Google Scholar]
- 23.Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y, Hayakawa T, Hamaguchi M: Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res 2001, 61:2361-2364 [PubMed] [Google Scholar]
- 24.Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP: Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (−/−) null mice show mammary epithelial cell hyperplasia. Am J Pathol 2002, 161:1357-1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV: Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293:2449-2452 [DOI] [PubMed] [Google Scholar]
- 26.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP: Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 2001, 276:38121-38138 [DOI] [PubMed] [Google Scholar]
- 27.Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF, Russell RG, Hulit J, Pestell RG, Lisanti MP: Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell 2002, 13:3416-3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA: Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85:27-37 [DOI] [PubMed] [Google Scholar]
- 29.Kupferman ME, Fini ME, Muller WJ, Weber R, Cheng Y, Muschel RJ: Matrix metalloproteinase 9 promoter activity is induced coincident with invasion during tumor progression. Am J Pathol 2000, 157:1777-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA: Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413:86-91 [DOI] [PubMed] [Google Scholar]
- 31.Weinberg WC, Fernandez-Salas E, Morgan DL, Shalizi A, Mirosh E, Stanulis E, Deng C, Hennings H, Yuspa SH: Genetic deletion of p21WAF1 enhances papilloma formation but not malignant conversion in experimental mouse skin carcinogenesis. Cancer Res 1999, 59:2050-2054 [PubMed] [Google Scholar]
- 32.Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA: Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 2000, 26:114-117 [DOI] [PubMed] [Google Scholar]
- 33.Campbell L, Laidler P, Watson RE, Kirby B, Griffiths CE, Gumbleton M: Downregulation and altered spatial pattern of caveolin-1 in chronic plaque psoriasis. Br J Dermatol 2002, 147:701-709 [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto H, Ochiya T, Takeshita F, Toriyama-Baba H, Hirai K, Sasaki H, Sakamoto H, Yoshida T, Saito I, Terada M: Enhanced skin carcinogenesis in cyclin D1-conditional transgenic mice: cyclin D1 alters keratinocyte response to calcium-induced terminal differentiation. Cancer Res 2002, 62:1641-1647 [PubMed] [Google Scholar]
- 35.Gassmann MG, Werner S: Caveolin-1 and -2 expression is differentially regulated in cultured keratinocytes and within the regenerating epidermis of cutaneous wounds. Exp Cell Res 2000, 258:23-32 [DOI] [PubMed] [Google Scholar]
- 36.Campbell L, Gumbleton M: Aberrant caveolin-1 expression in psoriasis: a signalling hypothesis. IUBMB Life 2000, 50:361-364 [DOI] [PubMed] [Google Scholar]
- 37.Breton S, Lisanti MP, Tyszkowski R, McLaughlin M, Brown D: Basolateral distribution of caveolin-1 in the kidney: absence from H+-ATPase-coated endocytic vesicles in intercalated cells. J Histochem Cytochem 1998, 46:205-214 [DOI] [PubMed] [Google Scholar]
- 38.Mora R, Bonilha VL, Marmorstein A, Scherer PE, Brown D, Lisanti MP, Rodriguez-Boulan E: Caveolin-2 localizes to the Golgi complex but redistributes to plasma membrane, caveolae, and rafts when co-expressed with caveolin-1. J Biol Chem 1999, 274:25708-25717 [DOI] [PubMed] [Google Scholar]
- 39.Vogel U, Sandvig K, van Deurs B: Expression of caveolin-1 and polarized formation of invaginated caveolae in Caco-2 and MDCK II cells. J Cell Sci 1998, 111:825-832 [DOI] [PubMed] [Google Scholar]
- 40.Fuchs E: Keratins and the skin. Annu Rev Cell Dev Biol 1995, 11:123-153 [DOI] [PubMed] [Google Scholar]
- 41.Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP: Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo: a role for the caveolin-scaffolding domain. FEBS Lett 1998, 428:205-211 [DOI] [PubMed] [Google Scholar]
- 42.Hulit J, Bash T, Fu M, Galbiati F, Albanese C, Sage DR, Schlegel A, Zhurinsky J, Shtutman M, Ben-Ze’ev A, Lisanti MP, Pestell RG: The cyclin D1 gene is transcriptionally repressed by caveolin-1. J Biol Chem 2000, 275:21203-21209 [DOI] [PubMed] [Google Scholar]
- 43.Couet J, Sargiacomo M, Lisanti MP: Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem 1997, 272:30429-30438 [DOI] [PubMed] [Google Scholar]
- 44.Scheel J, Srinivasan J, Honnert U, Henske A, Kurzchalia TV: Involvement of caveolin-1 in meiotic cell-cycle progression in Caenorhabditis elegans. Nat Cell Biol 1999, 1:127-129 [DOI] [PubMed] [Google Scholar]
- 45.Lammie GA, Peters G: Chromosome 11q13 abnormalities in human cancer. Cancer Cells 1991, 3:413-420 [PubMed] [Google Scholar]
- 46.Bianchi AB, Fischer SM, Robles AI, Rinchik EM, Conti CJ: Overexpression of cyclin D1 in mouse skin carcinogenesis. Oncogene 1993, 8:1127-1133 [PubMed] [Google Scholar]
- 47.Bizub D, Wood AW, Skalka AM: Mutagenesis of the Ha-ras oncogene in mouse skin tumors induced by polycyclic aromatic hydrocarbons. Proc Natl Acad Sci USA 1986, 83:6048-6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quintanilla M, Brown K, Ramsden M, Balmain A: Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322:78-80 [DOI] [PubMed] [Google Scholar]
- 49.Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti C: Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9:3627-3633 [PubMed] [Google Scholar]
- 50.Li WP, Liu P, Pilcher BK, Anderson RG: Cell-specific targeting of caveolin-1 to caveolae, secretory vesicles, cytoplasm or mitochondria. J Cell Sci 2001, 114:1397-1408 [DOI] [PubMed] [Google Scholar]
- 51.Woodman SE, Ashton AW, Schubert W, Lee H, Williams TM, Medina FA, Wyckoff JB, Combs TP, Lisanti MP: Caveolin-1 knock-out mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol 2003, 162:2059-2068 [DOI] [PMC free article] [PubMed] [Google Scholar]