

Abstract
Vascular remodeling is an active process that consists in important modifications in the vessel wall. Endothelium-derived nitric oxide (NO) plays a major role in this phenomenon. We assessed wall thickness (WT), total wall area (TWA), lumen diameter, and total nuclei number/cross-section (TN) in cirrhotic rats with ascites and in control rats. A second group of cirrhotic rats received the NO synthesis inhibitor, L-NAME, or vehicle daily for 11 weeks and systemic hemodynamics, arterial compliance, aortic NO synthase 3 (NOS3) protein expression, and vascular morphology were analyzed. Cirrhotic vessels showed a significant reduction in WT, TWA, and TN as compared to control vessels. Long-term inhibition of NOS activity in cirrhotic rats resulted in a significant increase in WT, TWA, and TN as compared to cirrhotic rats receiving vehicle. NOS3 protein abundance was higher in aortic vessels of nontreated cirrhotic animals than in controls. This difference was abolished by chronic treatment with L-NAME. NOS inhibition in cirrhotic rats resulted in higher arterial pressure and peripheral resistance and lower arterial compliance than cirrhotic rats receiving vehicle. Therefore, vascular remodeling in cirrhosis with ascites is a generalized process with significant functional consequences that can be negatively modulated by long-term inhibition of NOS activity.
Many diseases affecting the cardiocirculatory system are associated with structural modifications of the vessel wall. This active process, known as vascular remodeling, occurs in response to long-term modifications in hemodynamic conditions but, subsequently, contributes to the exacerbation of the circulatory dysfunction present in these diseases. 1 After a chronic increase in arterial pressure, the vascular wall undergoes important changes, including augmentation of the muscle mass, rearrangement of cells, augmentation of the wall thickness (WT), and diminution of the vessel lumen. Conversely, a maintained reduction in blood flow usually results in a diminution of the muscular mass of the vessel. 1,2 The cellular signals involved in this process are not fully elucidated, however it is widely recognized that, as the primary sensor of the hemodynamic changes, endothelial cells play a prominent role 3 and that cell mediators such as nitric oxide (NO), growth and vasoactive factors, and cytokines are of major importance in the genesis of the structural changes occurring in the vessel wall. 4 Systemic and pulmonary hypertension, atherosclerosis, arteriovenous fistula, and aneurysm are among the pathological conditions in which vascular remodeling phenomena have been well documented. 1-4
Reduced arterial pressure, high cardiac output (CO), low peripheral vascular resistance, endothelial dysfunction, altered vascular reactivity, increased circulating levels of endogenous vasoactive substances and enhanced aortic mRNA, and protein abundance of NO synthase type 3 (NOS3) are characteristic features in advanced liver disease. 5-7 How the hepatic disease leads to these changes in vascular NOS3 is not fully elucidated. However, it is likely that the augmentation of NOS3 in the vasculature of humans and rats with cirrhosis is a consequence of the profound hemodynamic alterations occurring in advanced liver disease. In fact, collagen deposition resulting from the liver injury produces a marked liver architectural distortion that leads to increased resistance to portal blood inflow and concomitant portal hypertension. This, in turn, results in a marked hyperkinetic circulation with increased vascular shear stress, a well-established mechanism regulating NOS3 expression and activity. In most cases this cardiovascular dysfunction develops throughout a long period of time, thus making the existence of vascular remodeling processes in the circulatory tree of patients with decompensated cirrhosis extremely likely, however this has remained unexplored so far. The existence of several strategies directed toward influencing the remodeling response make it particularly interesting to define whether vascular remodeling occurs in cirrhosis and if so, determine the molecular mechanisms involved in this phenomenon.
In the present study we extensively analyzed the morphology of systemic arteries in cirrhotic rats with ascites to test the hypothesis of whether advanced liver disease is associated with the presence of important structural changes in the vascular wall. Our findings indicate that vascular remodeling in experimental cirrhosis is a generalized process with significant functional consequences and that it can be negatively modulated by long-term inhibition of NOS activity.
Materials and Methods
Animals
The study includes two protocols that were performed in male adult cirrhotic Wistar rats and in control Wistar rats (Charles-River, Saint Aubin les Elseuf, France). Both groups were fed ad libitum with standard chow and distilled water containing phenobarbital. Cirrhosis was induced as described elsewhere. 8 CCl4 was used as hepatotoxin and phenobarbital (0.3 g/L) was administered to shorten the time required to induce cirrhosis. After 1 week of receiving phenobarbital, inhalation of CCl4 was started. Rats were placed in a gas chamber (70 × 25 × 30 cm). Compressed air was passed, via a flowmeter (1 l/min) bubbling through a flask containing CCl4, into the gas chamber. Animals were exposed to the gas atmosphere twice weekly (Monday and Friday) starting with 0.5 minute of bubbling air and 0.5 minute in the gas atmosphere. Afterward, the dosage was increased by 1 minute until 5 minutes of bubbling air and 5 minutes in gas atmosphere was reached.
Experimental Protocols
To assess whether cirrhosis with ascites is associated with the development of structural changes in the systemic vascular tree, six cirrhotic rats with ascites and six control rats were anesthetized with ketamine (100 mg/kg bw) and the thoracic and abdominal aorta and mesenteric and renal arteries harvested and prepared for morphological analysis as described below. Because rats treated with CCl4 and phenobarbital showed ascites within 16 to 18 weeks after starting the cirrhosis induction program, control rats were investigated 16 to 18 weeks after being included in the study.
To test whether NO synthesis inhibition may modulate the development of structural changes in the arteries of cirrhotic rats, cirrhotic and control animals were randomly assigned to one of the following groups: group A, daily administration of Nω-nitro-l-arginine-methyl-ester (L-NAME, 0.5 mg.kg−1 in drinking water) given for 11 weeks from the seventh week after starting the protocol (11 CCl4-induced cirrhosis rats and 12 control rats). Group B animals (11 CCl4-induced cirrhosis rats and 12 control rats) were identically treated as in group A, with the only exception that they did not receive L-NAME in the drinking water. At the end of the treatment animals were anesthetized and a systemic hemodynamic study was performed to measure mean arterial pressure (MAP), portal pressure (PP), CO, heart rate (HR), stroke volume (SV), total peripheral resistance (TPR), pulse pressure (PuP), and arterial compliance (AC). Afterward a blood sample was obtained to measure electrolytes, osmolality, and standard parameters, or renal and hepatic function. To assess vessel morphology, the vascular tree was perfused and the thoracic and abdominal aorta dissected and included in O.C.T in 14 CCl4-induced cirrhosis rats and in 12 control rats, as described below. In the remaining animals aortic vessels were dissected, placed in a Petri dish containing phosphate-buffered saline (PBS) salt solution (in mmol/L: NaCl, 140; Na2HPO4, 8.5; Na2HPO4.H2O, 1.84; pH 7.4) and cleaned free of surrounding fatty tissue. Samples were immediately frozen in dry ice and stored in liquid nitrogen until further analysis.
Hemodynamic Studies
Cirrhotic and control rats were anesthetized with ketamine (100 mg.kg−1) and prepared with PE-50 polyvinyl catheters in the left femoral artery. A midline abdominal incision (2 cm) was made, and the portal vein was cannulated through an ileocolic vein with a PE-50 catheter to measure PP. After verifying free blood reflux, the catheter was fixed to the mesentery with cyanoacrylate glue and the abdomen closed with silk sutures. The right jugular vein was also isolated and a PE-50 catheter was placed in the right atrium. A thermocouple (Columbus Instruments, Columbus, OH) was advanced to the aortic arch through a left carotid approach to monitor the intra-arterial temperature during CO measurement. The arterial catheter was connected to a highly sensitive transducer (Hewlett Packard, Avondale, PA) that was calibrated before each study. MAP, PuP (the difference between systolic arterial blood pressure and diastolic arterial blood pressure), SV, and HR were determined in a microcomputer system (Cardiomax IIR; Columbus Instruments). MAP, PP, PuP, SV, and HR were recorded in a multichannel system (MX4P and MT4; Lectromed Ltd., Jersey, Channels Islands, UK). CO was measured by thermodilution after the administration of a bolus of 200 μl of Ringer solution (20 to 23°C) into the right atrium. A spring-loaded syringe was used (Hamilton Syringe, model CR-700-200; Hamilton Co., Reno, NV) to ensure a constant injection rate and volume. TPR and AC were obtained using the following formulae: TPR = MAP/CO and AC = SV/PuP. Hemodynamic parameters were allowed to equilibrate for 30 minutes and values of MAP, PP, PuP, HR, CO, SV, TPR, and AC were recorded.
Vascular Perfusion, Harvest of Arteries, and Histology
Anesthetized animals (ketamine, 100 mg.kg−1) were decapitated, the cervical trunk ligated, the chest opened, and the right atrium punctured and cannulated. Then, the vasculature of cirrhotic and control rats was perfused with PBS (pH 7.4, 37°C) containing adenosine (0.1 mmol/L), papaverine (0.3 mmol/L), and heparin sodium (50 U/ml) to relax vascular smooth cells. 3 Venous blood was collected through the atrial incision. The perfusion system was adjusted to achieve a MAP of ≈75-mm Hg and was maintained until obtaining a clear perfusate. Thereafter, and without interruption of flow, perfusion was changed to a PBS fixative (4% paraformaldehyde in PBS, pH 7.4, 37°C) for 5 minutes. Thoracic and abdominal aorta and mesenteric renal and femoral arteries were excised and cleaned in fixative solution. All vessels were postfixed overnight at 4°C and included in O.C.T. Cross sections (4 μm thick) were obtained with a cryostat (1720 Leitz Digital Kryostat; Leitz, Germany) each vessel being oriented perpendicularly to the knife face to maintain symmetry of the WT. Cross sections were taken at similar locations for each vessel and placed on slides pretreated with chromium potassium sulfate to improve section adhesion. Slides were used for hematoxylin and eosin staining and morphometry.
Morphometry and Nuclei Number
Morphometric analysis of arterial vessels was performed using video microscopy at a final magnification of 100 or 200, depending on the size of the vessel. The image was captured and displayed on a computer monitor using the image analysis software, Microimage (Olympus Europe, Hamburg, Germany). After a standard calibration, the perimeter of the vessel lumen was measured on the video image with Microimage and taken as the circumference of a circle. The diameter was calculated as diameter = circumference/π. WT was measured every 45° as the distance between endothelium and adventitia and the average value was calculated for each vessel section. Outer diameter (OD) was calculated as lumen diameter (LD) + 2WT. Total wall area (TWA) was calculated as [(OD/2)2π − (LD/2)2π]. Two consecutive full serial sections were counted and the values averaged. Hematoxylin-positive nuclei including endothelial and smooth muscle cells were counted in three to four full serial cross sections and then averaged.
Biochemical Measurements
Vascular concentration of cGMP and aortic NOS3 protein expression were assessed in thoracic aorta of 8 cirrhotic and 12 control rats obtained as described above. Vessels were individually homogenized (PT 10-35; Polytron Kinematica, Kriens-Luzern, Switzerland) in a buffer Tris-HCl, 20 mmol/L, pH 7.4, containing 1% Triton X-100, 0.1% sodium dodecyl sulfate, 50 mmol/L NaCl, 2.5 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L Na4P2O7·10 H2O, 20 mmol/L NaF, 1 mmol/L Na3VO4, 2 mmol/L Pefabloc, and a cocktail of protease inhibitors (Complete Mini, Roche, Basel, Switzerland). Homogenates were rotated at 4°C for 1 hour, centrifuged at 12,000 rpm for 10 minutes at 4°C, and supernatant aliquots were kept at −20°C until further analysis of cGMP concentration. For total NOS3, 40 μg of the denaturated proteins per lane were loaded and separated on a 7.5% sodium dodecyl sulfate-polyacrylamide gel (Mini Protean III; Bio-Rad, Richmond, CA) and transferred to nitrocellulose membranes (Transblot Transfer Medium, Bio Rad), which were stained with Ponceau-S red as a control for protein loading. Subsequently, membranes were blocked with 5% powdered defatted milk in TTBS buffer (50 mmol/L Tris-HCl, pH 8, containing 0.05% Tween 20 and 150 mmol/L NaCl) for 2 hours. Membranes were incubated at room temperature with mouse monoclonal anti-total NOS3 (Transduction Laboratories, Lexington, KY) for 2 hours in a 1:2500 dilution in TTBS buffer containing 5% powdered defatted milk. After incubation with primary antibody the membranes were washed (three times, 10 minutes each) with TTBS buffer and incubated for 60 minutes, at room temperature, with a 1:2000 dilution of horseradish peroxidase-conjugated mouse antibody (Amersham International). For phospho-NOS3 (p-NOS3), 80 μg of the denaturated proteins per lane were loaded and then separated, transferred, and subsequently blocked as described. Membranes were incubated overnight at 4°C with rabbit polyclonal anti-p-NOS3 (Cell Signaling Technologies, Beverly, MA) in a 1:1000 TTBS buffer containing 5% bovine serum albumin. This antibody does not detect nonphosphorylated NOS3. After incubation with primary antibody the membranes were washed (three times, 10 minutes each) with TTBS buffer and incubated for 60 minutes, at room temperature, with a 1:2000 dilution of horseradish peroxidase-conjugated rabbit antibody (Amersham International). After washing with TTBS buffer, the bands for total- and p-NOS3 were visualized by chemiluminescence (ECL Western blotting analysis system, Amersham International). The relative expression of NOS3 proteins in each vessel was quantified by densitometric scanning (Phoretix International Ltd., Newcastle on Tyne, UK).
Tissular cGMP was assessed after acetylation. After tissue homogenization and centrifugation, supernatants were twice extracted with five volumes of water saturated ether. After removing the ether from the aqueous layer, samples were concentrated to dryness (SpeedVac Concentrator; Savant Instruments Inc, Farmingdale, NY), and stored at −20°C for determination of cGMP concentration. The concentration of cGMP was determined by radioimmunoassay (Biomedical Technologies Inc, Stoughton, MA).
Protein concentrations were determined by the method of Lowry and colleagues 9 with bovine serum albumin as the standard. Serum and urinary osmolality were determined from osmometric depression of the freezing point (Advanced Instruments Osmometer 3MO; Advanced Instruments, Needham Heights, MA) and sodium and potassium concentration by flame photometry (IL 943; Instrumentation Laboratory, Lexington, MA). Serum total bilirubin, albumin, total proteins, and alanine aminotransferase were measured by the Ektachem Clinical Chemistry Slide method (Johnson & Johnson Clinical Diagnostic Inc., Rochester, NY).
Statistical Analysis
Statistical analysis of results was performed by using unpaired Student’s t-test. Data are expressed as mean ± SEM and were considered significant at a P level of 0.05 or less. The study was performed according to the criteria of the Investigation and Ethics Committee of the Hospital Clínic Universitari.
Results
The liver histology of all of the animals treated with CCl4 included in the study had a finely granulated surface and histological examination showed marked architectural distortion leading to fully developed cirrhosis. 8 All cirrhotic rats included in the first protocol had ascites. The volume of ascites ranged from 14 to 65 ml. Control rats showed no appreciable alterations in liver histology.
Vascular Remodeling in Cirrhotic Rats
Macroscopically, arterial vessels of cirrhotic rats with ascites displayed slackened consistency and histological examination of aorta of these animals showed marked abnormalities in comparison to that of control rats. The thoracic and abdominal aortas were examined separately. Microscopically, the thoracic aorta of control rats consisted in a single layer of endothelial cells, 10 to 11 layers of media composed of vascular smooth muscle cells and elastic lamellas and loose connective adventitial tissue. In contrast, the media of thoracic aorta of cirrhotic rats with ascites was markedly thinner than that of control animals (Figure 1) ▶ . No apparent differences were observed in the elastic lamellas.
Figure 1.
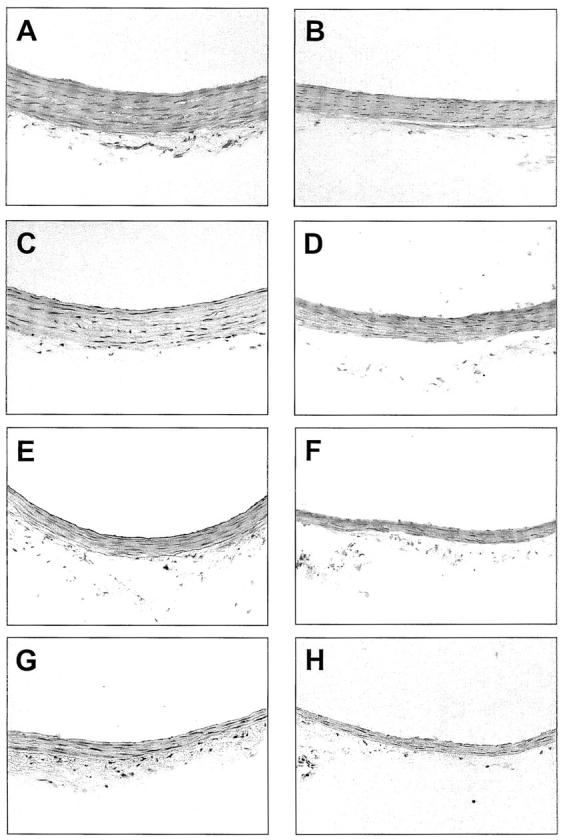
Photomicrographs of representative cross sections of thoracic and abdominal aorta, and mesenteric and renal arteries from a control rat (A, C, E, and G, respectively) and a cirrhotic rat with ascites (B, D, F, and H, respectively). Note the marked reduction in WT and the diminution in the number of nuclei in the cirrhotic vessel (H&E staining; original magnifications, ×200).
As shown in Figure 2 ▶ , WT reduction in cirrhotic rats was a generalized feature, because similar changes were observed in the abdominal aorta and mesenteric and renal arteries. Arterial wall thinning of cirrhotic rats was not associated with any difference in LD and, consequently, the TWA and the WT/LD ratio were significantly lower in the thoracic and abdominal aorta and mesenteric and renal arteries of cirrhotic rats with ascites than in controls (Figure 2) ▶ .
Figure 2.

WT, LD, TWA, and WT/LD ratio in control rats and in cirrhotic rats with ascites. THORAC, thoracic aorta; ABDOM, abdominal aorta; MES, mesenteric artery; REN, renal artery. Data are mean ± SE; n = 6 rats per group.
A marked reduction was also observed in the number of nuclei per cross-section of cirrhotic rats in comparison with control animals (Table 1) ▶ . This was because of a diminution in the amount of nuclei in the media, because no significant differences were observed on analyzing the values of endothelial nuclei. Therefore, the lesser thickening of systemic arterial vessels in cirrhotic rats was secondary to a reduction in the amount of vascular smooth muscle cells in the media.
Table 1.
Number of Nuclei per Cross Section in the Endothelium and the Media of Control Rats and Cirrhotic Rats with Ascites
| Control rats (n = 6) | Cirrhotic rats (n = 6) | |||
|---|---|---|---|---|
| Endothelium | Media | Endothelium | Media | |
| Thoracic aorta | 182 ± 9 | 1778 ± 129 | 178 ± 9 | 1302 ± 126* |
| Abdominal aorta | 123 ± 15 | 1107 ± 83 | 134 ± 11 | 864 ± 39* |
| Mesenteric artery | 91 ± 3 | 450 ± 20 | 89 ± 4 | 364 ± 28* |
| Renal artery | 81 ± 3 | 273 ± 32 | 72 ± 3 | 189 ± 20* |
*P < 0.05 versus number of nuclei in the media of control rats.
Effect of Chronic NO Synthase Inhibition on Vascular Remodeling and Systemic Hemodynamics in Cirrhotic Rats
After completing the protocol, 16 CCl4-treated rats had ascites, 7 of which had received L-NAME and the remaining 9 received vehicle. Ascites volume ranged from 5 to 40 ml in the former group of animals and from 10 to 40 ml in the latter. Cirrhotic rats were investigated after they had developed marked abnormalities in liver function tests. As anticipated, rats submitted to the cirrhosis induction protocol, treated or not treated with L-NAME, showed decreased serum proteins (40.0 ± 3.6 g/L, P < 0.001, and 41.5 ± 2.4 g/L, P < 0.001) and albumin (21.0 ± 2.0 g/L, P < 0.001, and 20.0 ± 2.0 g/L, P < 0.001), increased activity of alanine aminotransferase (40 ± 9 U/L, P < 0.05, and 31 ± 5 U/L, P < 0.01) and enhanced serum concentration of bilirubin (0.41 ± 0.07 mg/dl, P < 0.05, and 0.46 ± 0.14 mg/dl, P < 0.05) than the corresponding control rats (58.0 ± 1.9 g/L and 57.8 ± 1.1 g/L, 34.0 ± 1.0 g/L and 37.0 ± 0.4 g/L, 18 ± 2 U/L and 17 ± 2 U/L, respectively). Cirrhotic animals showed similar values of serum electrolytes and osmolality to those of control rats. In addition, CCl4-treated rats had a tendency toward presenting decreased body weight, although statistical differences were only achieved on comparing the values of cirrhotic and control rats receiving L-NAME (468 ± 17 g versus 552 ± 12 g, P < 0.001). No significant differences were found in any of these parameters between cirrhotic rats chronically treated with L-NAME or vehicle. Neither were differences observed in control rats.
Daily treatment with the NOS inhibitor for 11 weeks promoted important modifications in the structure of systemic arterial vessels of cirrhotic rats. In fact, cirrhotic vessels treated with L-NAME showed greater WT and TWA than those receiving vehicle (Figure 3) ▶ . Consequently, the vessel ability to contract markedly improved in all of the assessed arteries of cirrhotic rats treated with the NOS inhibitor, as indicated by the significantly higher WT/LD ratio of cirrhotic vessels chronically receiving L-NAME as compared to vessels of nontreated cirrhotic rats (Figure 3) ▶ . As previously reported, 10 L-NAME treatment also increased WT and TWA in thoracic (78.75 ± 3.64 μm versus 67.01 ± 1.70 μm, P < 0.01 and 499 ± 35 × 103 μm2 versus 409 ± 18 × 103 μm2, P < 0.05, respectively) and abdominal aorta (66.87 ± 2.16 μm versus 57.75 ± 1.26 μm, P < 0.001 and 388 ± 13 × 103 μm2 versus 326 ± 11 × 103 μm2, P < 0.001, respectively) of control rats. Of note was, however, that no differences were recorded between the mesenteric and renal arteries on analyzing the WT/LD ratios of treated and nontreated control animals.
Figure 3.

WT, LD, TWA, and WT/LD ratio in cirrhotic rats receiving vehicle or chronically treated with L-NAME (0.5 mg/kg/day). THORAC, thoracic aorta; ABDOM, abdominal aorta; MES, mesenteric artery; REN, renal artery. Data are mean ± SE; n = 7 rats per group.
As shown in Table 2 ▶ , chronic treatment with the NOS inhibitor also resulted in substantial effects on the number of nuclei in cirrhotic vessels. In fact, the total number of nuclei per cross-section was significantly higher in all of the studied vessels of cirrhotic animals receiving L-NAME in comparison to those treated with vehicle. The augmentation in the number of nuclei occurred in the medial layer of vessel (data not shown). In control vessels, the number of nuclei tended toward to increase in rats treated with L-NAME as well. However, differences only reached statistical significance in the abdominal aorta.
Table 2.
Total Number of Nuclei per Cross Section in Control and Cirrhotic Rats Receiving Vehicle or Chronically Treated with L-NAME (0.5 mg.kg−1)
| Control rats | Cirrhotic rats | |||
|---|---|---|---|---|
| Vehicle (n = 6) | L-NAME (n = 6) | Vehicle (n = 7) | L-NAME (n = 7) | |
| Thoracic aorta | 2193 ± 171 | 2528 ± 167 | 1506 ± 85 | 2210 ± 99‡ |
| Abdominal aorta | 1483 ± 72 | 1954 ± 103§ | 1180 ± 75 | 1522 ± 100* |
| Mesenteric artery | 579 ± 25 | 613 ± 34 | 469 ± 27 | 652 ± 36‡ |
| Renal artery | 333 ± 37 | 438 ± 40 | 321 ± 21 | 453 ± 29† |
*, P < 0.05;
†, P < 0.01;
‡, P < 0.001 compared with cirrhotic rats receiving vehicle.
§, P < 0.001 compared with control rats receiving vehicle.
To ascertain the effectiveness of L-NAME in inhibiting the vascular NO-dependent metabolic pathway, we assessed the tissular concentration of cGMP in aorta of cirrhotic and control rats (Figure 4) ▶ . As previously shown, 11 cirrhotic rats with ascites had increased tissular concentration of cGMP than control rats. This difference between cirrhotic and control rats was no longer present in those animals chronically receiving the NOS inhibitor. Furthermore, vessels of nontreated cirrhotic animals displayed enhanced abundance of both total-NOS3 and p-NOS3 protein. Indeed, this difference between cirrhotic and control vessels was abolished by chronic treatment with L-NAME (Figure 5) ▶ .
Figure 4.

Aortic cGMP concentration in control and cirrhotic rats receiving vehicle or chronically treated with L-NAME (0.5 mg/kg/day). Thoracic aortas were individually homogenized and the concentration of cGMP was measured in the acetylated extracts by radioimmunoassay. Eight cirrhotic and 12 control rats were studied, half of the animals in each group being treated with L-NAME. Data are mean ± SE.
Figure 5.
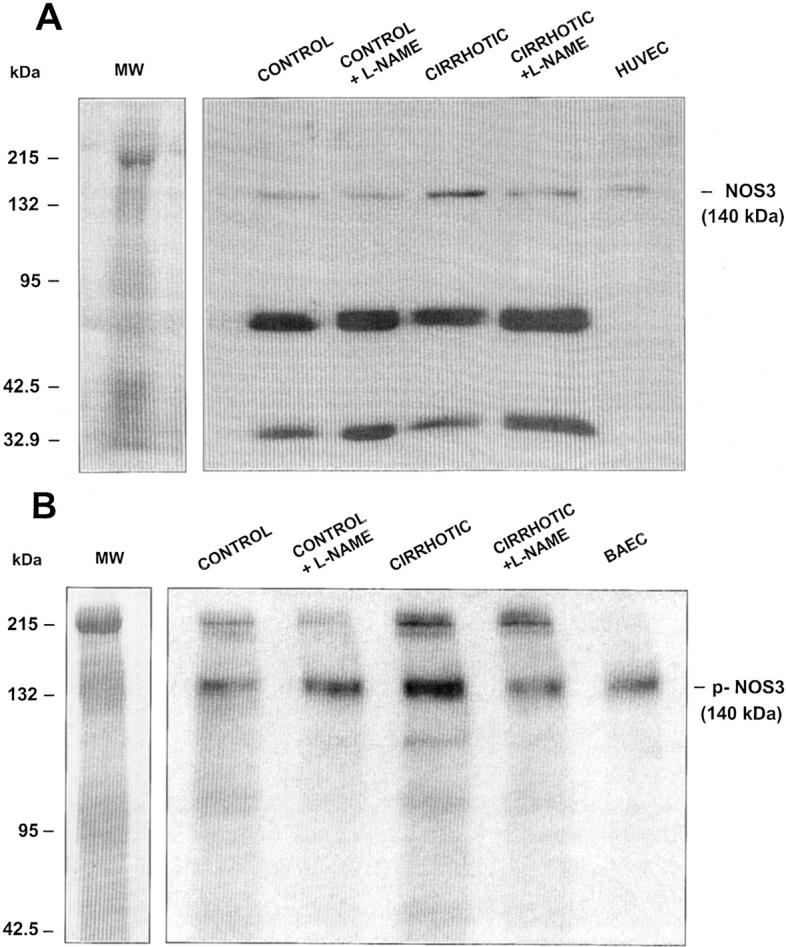
Representative Western blot of total NOS3 (A) and phospho-NOS3 (p-NOS3) (B) protein in thoracic aorta of control and cirrhotic rats receiving vehicle or chronically treated with L-NAME (0.5 mg/kg/day). Protein extracts were prepared as described in Material and Methods and 40 μg (total NOS3) or 80 μg (p-NOS3) of protein were loaded per lane. Ten μg of human vein endothelial cell (HUVEC) lysate or stimulated bovine aortic endothelial cells (BAEC) were used as a positive control.
Baseline MAP, PP, CO, TPR, HR, SV, PuP, and AC in control and cirrhotic rats receiving or not receiving the NOS inhibitor are shown in Table 3 ▶ . Compared with untreated control rats, cirrhotic animals receiving vehicle showed arterial hypotension and portal hypertension; reduced TPR; and increased CO, SV, and AC. No significant differences in HR and PuP were observed between cirrhotic and control animals. This characteristic hyperkinetic circulatory syndrome of cirrhotic animals was significantly attenuated by the chronic inhibition of NOS activity because cirrhotic rats treated with L-NAME showed significantly higher MAP and TPR than nontreated cirrhotic rats. In addition, cirrhotic animals receiving L-NAME also displayed an ∼50% decrease in AC in comparison to nontreated cirrhotic rats. This occurred in the setting of a moderate diminution in SV and without noticeable changes in PuP. In control rats, L-NAME promoted arterial hypertension and increased TPR with a compensatory reduction in CO. No differences were observed in any other of the remaining hemodynamic parameters.
Table 3.
Mean Arterial Pressure (MAP), Portal Pressure (PP), Cardiac Output (CO), Total Peripheral Resistance (TPR), Heart Rate (HR), Stroke Volume (SV), Pulsatile Pressure (PuP), and Arterial Compliance (AC) in Control and Cirrhotic Rats Receiving Vehicle or Chronically Treated with L-NAME (0.5 mg.kg−1)
| Control rats | Cirrhotic rats | |||
|---|---|---|---|---|
| Vehicle (n = 12) | L-NAME (n = 12) | Vehicle (n = 11) | L-NAME (n = 11) | |
| MAP (mm Hg) | 121 ± 1 | 132 ± 3† | 94 ± 3¶ | 106 ± 2† |
| PP (mm Hg) | 5.6 ± 0.2 | 6.2 ± 0.2 | 12.6 ± 1.0¶ | 12.7 ± 1.3§ |
| CO (ml/min) | 199 ± 8 | 167 ± 11* | 311 ± 13¶ | 306 ± 20¶ |
| TPR (mm Hg · min · ml−1) | 0.63 ± 0.03 | 0.78 ± 0.06* | 0.30 ± 0.01¶ | 0.37 ± 0.02*¶ |
| HR (beat/min) | 355 ± 15 | 348 ± 22 | 315 ± 21 | 362 ± 14 |
| SV (ml/beat) | 0.59 ± 0.03 | 0.71 ± 0.13 | 1.17 ± 0.12¶ | 0.86 ± 0.05* |
| PuP (mm Hg) | 13.8 ± 0.8 | 12.1 ± 1.2 | 10.9 ± 1.3 | 13.5 ± 1.1 |
| AC (ml/mm Hg) | 0.046 ± 0.004 | 0.060 ± 0.010 | 0.140 ± 0.027§ | 0.070 ± 0.010* |
*, P < 0.05;
†, P < 0.01; compared with vehicle rats.
§, P < 0.01;
¶, P < 0.001 compared with control rats receiving the same treatment.
Discussion
The results of the present investigation indicate that the conductive vessels of cirrhotic rats with ascites undergo an intense process of vascular remodeling. The most remarkable features of this phenomenon are a decrease in the thickness and the total area of the vascular wall and a reduction in the WT/LD ratio, which is an estimation of vessel ability to contract. This impairment in vessel structure is likely secondary to a decrease in the amount of vascular smooth muscle cells because we also observed a diminution in the number of nuclei per cross-section in the medial layer of the vessels. Although this, to the best of our knowledge, is the first experimental evidence documenting major architectural changes in the large cirrhotic arteries, the existence of vascular remodeling could be anticipated considering the profound circulatory dysfunction developed by patients and experimental animals with advanced liver disease.
Actually, it is well established that chronic modifications in blood flow cause adaptive changes in the structure of the vessels. Vascular endothelial cells play a major role orchestrating these changes by producing and secreting several vasoactive molecules and growth factors. 12,13 Among them, we focused on NO because it may inhibit migration and proliferation of vascular smooth muscle cells, 14 neointima formation, and extracellular matrix turnover, 15 all of which are necessary events for vascular remodeling. In addition, advanced liver disease is associated with a marked endothelial dysfunction characterized by impaired responsiveness to endogenous vasoconstrictors, 16 increased NO-dependent vasorelaxation, 17 and a higher production of endothelium-derived NO. 18 These features occur in the setting of increased vascular expression of NOS3 mRNA and protein. 7
The results of the present study are in line with these findings because aortas of cirrhotic rats not receiving the NOS inhibitor have increased total NOS3 protein abundance and higher vascular content of cGMP than the corresponding control rats. Moreover, by analyzing p-NOS3 expression, this study further confirms a major involvement of NOS3 in the pathogenesis of the endothelial dysfunction in cirrhosis. NOS3 phosphorylation increases the activation state of the enzyme at any given level of free Ca2+, thereby enhancing NO release from cells. 19,20 Aortas of cirrhotic rats presented a clear signal corresponding to p-NOS3, which was not observed in the vessels of control rats, thus demonstrating NOS3 phosphorylation in conductive vessels of cirrhotic animals.
Daily administration of L-NAME for 11 weeks was effective to inhibit the overactivity of NOS3 in cirrhotic animals because all of the assessed molecular manifestations of increased activity of this enzyme reverted with treatment. In fact, cirrhotic rats receiving the NOS activity inhibitor showed similar vascular content of cGMP and total- and p-NOS3 protein abundance to that of control rats. Therefore, we assessed whether the normalization in the vascular production of NO in cirrhotic rats is associated with any improvement in the architectural distortions observed in the arterial vessels of these animals.
Rats with CCl4-induced cirrhosis presented similar body weight, hepatic dysfunction, and when present, ascites volume, regardless of whether they were treated or not with the NOS activity inhibitor. However, the morphological analysis of arterial vessels revealed marked differences between the two groups of cirrhotic rats. Conductance arteries of cirrhotic rats receiving L-NAME showed significantly higher WT and number of nuclei than cirrhotic rats not receiving L-NAME. An important observation was that WT and nuclei number per cross-section in all of the arteries examined were similar in cirrhotic rats treated with L-NAME compared to untreated control rats, indicating that long-term NOS activity inhibition is associated with vascular structure normalization in cirrhotic rats. Collectively these results indicate that, in addition to its role as vasodilator, endothelium-derived NO has an important, and so far, not considered role in the control of vascular morphology in experimental cirrhosis. In concordance with previous studies, 10 chronic treatment with L-NAME also produced a significant effect on WT of major conductance vessels of control rats. The action of the NOS inhibitor on the vascular wall of the animals is probably related with the well-known effect of NO as an endogenous inhibitor of smooth muscle cells. In this regard, there is evidence indicating that NO inhibits smooth muscle cell proliferation and migration via distinct cell-cycle arrests in phases G1 and S. 21,22
Identification of an abnormal reorganization of the vessel wall in cirrhotic rats as well as the central role played by endothelium-dependent NO in the pathogenesis of this phenomenon may have relevant implications in terms of circulatory performance. Endothelial dysfunction is a potentially reversible condition, as previously shown in clinical trials using angiotensin-converting enzyme inhibitors or cholesterol-lowering drugs. 23,24 Because these substances exert their beneficial effects, at least in part, by acting on the endothelial NO-dependent metabolic pathway, we were next interested in analyzing the effect on the cardiovascular function resulting from the correction of the abnormal structure of large vessels in cirrhotic rats.
In addition to its well-characterized hyperdynamic circulatory syndrome, cirrhotic rats not receiving L-NAME also had more than a threefold greater increase in AC than control rats treated with vehicle. AC is a measure of the elasticity of the arterial system and, therefore, this parameter is closely dependent on the structure of the arterial wall. 25 Along with the well-known increase in MAP and TPR, 11 chronic inhibition of NOS activity in cirrhotic rats, also resulted in a 50% reduction in AC as compared to cirrhotic animals not treated with L-NAME. This effect was not observed in control rats in which no changes in AC were recorded. Cirrhotic rats treated with L-NAME showed similar AC to control animals, indicating a normalization of AC after long-term inhibition of NOS activity. Because L-NAME treatment resulted in a similar hypertensive effect in control and cirrhotic animals, the marked improvement of AC in cirrhotic rats receiving the NOS activity inhibitor is an indication that the elevation in arterial pressure does not account for the reversal of the abnormal vascular remodeling occurring in these animals. Moreover, previous studies in chronically L-NAME-treated rats receiving hydralazine or captopril also indicated that, rather than the elevation in arterial pressure, NO per se is responsible for the structural changes in aorta and coronary vessels of hypertensive rats. 26,27
There was a clear dissociation between the effect of NOS inhibition on vascular structure and AC and the effect on cardiocirculatory function. Whereas L-NAME treatment normalized vascular architecture and AC, it only partially improved systemic hemodynamics. Cirrhotic rats under long-term inhibition of NO still showed arterial hypotension, high CO, low TPR, and ascites. These data indicate that, although NO is a major player in vascular remodeling, it only plays a contributory role in the pathogenesis of the circulatory dysfunction in experimental cirrhosis. This is not surprising, because in this condition the alteration in the vascular structure is a generalized phenomenon while systemic circulatory dysfunction is because of arterial vasodilation in the splanchnic circulation.
We proposed that, likely through a shear stress-dependent mechanism, the hyperdynamic circulation in cirrhosis results in a chronic increase in endothelium-derived NO in the large conductive vessels that promotes an important architectural modification in the structure of these vessels and an aggravation in the circulatory dysfunction. This unwanted structural modification may be modulated by acting on the endothelium-dependent metabolic pathway.
Footnotes
Address reprint requests to Dr. Wladimiro Jiménez, Laboratorio Hormonal, Hospital Clinic Universitari, Villarroel 170, Barcelona 08036, Spain. E-mail: wjimenez@medicina.ub.es.
Supported by the Dirección General de Investigación Científica y Técnica (grants SAF99-0016 to W. J. and SAF 01-2585 to M. M.), the Fondo de Investigación Sanitaria (FIS00/0398 to J. R. and FIS01/1514 to W. J.), Institut d’Investígacíons Biomèdìques August pí í Sunyer (to P. C.-M.), and Díreccíon General de Investígacíon Cíentífíca y Tecníca (SAF99-0016 to G. F.-V.).
References
- 1.Gibbons GH, Dzau VJ: The emerging concept of vascular remodeling. N Engl J Med 1994, 330:1431-1438 [DOI] [PubMed] [Google Scholar]
- 2.Mulvany MJ: Vascular remodeling of resistance vessels: can we define this? Cardiovasc Res 1999, 41:9-13 [DOI] [PubMed] [Google Scholar]
- 3.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC: Direct evidence of the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 1998, 101:731-736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dzau VJ, Horiuchi M: Vascular remodeling: the emerging paradigm of programmed cell death (apoptosis). Chest 1998, 114:91S-99S [DOI] [PubMed] [Google Scholar]
- 5.Arroyo V, Jiménez W: Renal and circulatory dysfunction in cirrhosis. Lights and shadows in an important clinical problem. J Hepatol 2000, 32(Suppl 1):157-170 [DOI] [PubMed] [Google Scholar]
- 6.Arroyo V, Ginès P, Jiménez W, Rodés J: Renal dysfunction in cirrhosis. Bircher J Benhamou JP McIntyre N Rizzetto M Rodés J eds. Oxford Textbook of Clinical Hepatology, ed 2 1999:pp 733-761 Oxford Medical Publications, Oxford
- 7.Morales M, Jiménez W, Pérez-Sala D, Ros J, Leivas A, Lamas S, Rivera F, Arroyo V: Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology 1996, 24:1481-1486 [DOI] [PubMed] [Google Scholar]
- 8.Clària J, Jiménez W: Renal dysfunction and ascites in carbon-tetrachloride-induced cirrhosis in rats. Ascites and renal dysfunction in liver disease. Arroyo V Ginès P Rodés J Schrier RW eds. Pathogenesis Diagnosis and Treatment. 1999:pp 379-396 Blackwell Science Inc., Malden, MA
- 9.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with folin phenol reagent. J Biol Chem 1951, 193:265-275 [PubMed] [Google Scholar]
- 10.Zhao H, Shimokaw H, Uragami-Harasawa L, Igarashi H, Takeshita A: Long-term vascular effects of Nω-nitro-l-arginine methyl ester are not solely mediated by inhibition of endothelial nitric oxide synthesis in the rat mesenteric artery. J Cardiovasc Pharmacol 1999, 33:554-566 [DOI] [PubMed] [Google Scholar]
- 11.Niederberger M, Martin PY, Ginés P, Morris K, Tsai P, Xu DL, McMurtry I, Schrier RW: Normalization of nitric oxide production corrects arterial vasodilation and hyperdynamic circulation in cirrhotic rats. Gastroenterology 1995, 109:1624-1630 [DOI] [PubMed] [Google Scholar]
- 12.Cornwell TL, Arnold E, Boerth NJ, Lincoln TM: Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol 1994, 267:C1405-C1413 [DOI] [PubMed] [Google Scholar]
- 13.von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, Dzau VJ: Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci USA 1995, 92:1137-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC: Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res 1996, 78:225-230 [DOI] [PubMed] [Google Scholar]
- 15.Murrell GA, Jang D, Williams RJ: Nitric oxide activates metalloprotease enzymes in articular cartilage. Biochem Biophys Res Commun 1995, 206:15-21 [DOI] [PubMed] [Google Scholar]
- 16.Castro A, Jiménez W, Clària J, Ros J, Martínez JM, Bosch M, Arroyo V, Piulats J, Rivera F, Rodés J: Impaired responsiveness to angiotensin II in experimental cirrhosis: role of nitric oxide. Hepatology 1993, 18:367-372 [PubMed] [Google Scholar]
- 17.Clària J, Jiménez W, Ros J, Rigol M, Angeli P, Arroyo V, Rivera F, Rodés J: Increased nitric oxide dependent vasorelaxation in aortic rings of cirrhotic rats with ascites. Hepatology 1994, 20:1615-1621 [DOI] [PubMed] [Google Scholar]
- 18.Ros J, Jiménez W, Lamas S, Clària J, Arroyo V, Rivera F, Rodés J: Nitric oxide production in arterial vessels of cirrhotic rats. Hepatology 1995, 21:554-560 [PubMed] [Google Scholar]
- 19.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC: Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399:597-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCabe TJ, Fulton D, Roman LT, Sessa WC: Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J Biol Chem 2000, 275:6123-6128 [DOI] [PubMed] [Google Scholar]
- 21.Sarkar R, Gordon D, Stanley JC, Webb RC: Cell cycle effect of nitric oxide on vascular smooth muscle cells. Am J Physiol 1997, 272:H1810-H1818 [DOI] [PubMed] [Google Scholar]
- 22.Guo K, Andrés V, Walsh K: Nitric oxide-induced down regulation of Cdk2 activity and cyclin A gene transcription in vascular smooth muscle cells. Circulation 1998, 97:2066-2072 [DOI] [PubMed] [Google Scholar]
- 23.Mancini GB, Henry GC, Macaya C, O′Neill BJ, Pucillo AL, Carere RG, Wargovich TJ, Mudra H, Luscher TF, Klibaner MI, Haber HE, Uprichard AC, Pepine CJ, Pitt B: Angiotensin converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. The TREND (Trial on Reversing Endothelial Dysfunction). Circulation 1996, 94:258-265 [DOI] [PubMed] [Google Scholar]
- 24.Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, Zhang J, Boccuzzi SJ, Cedarholm JC, Alexander RW: Beneficial effects of cholesterol lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med 1995, 332:481-487 [DOI] [PubMed] [Google Scholar]
- 25.Safar M, Simon A, Leverson J: The arterial wall: relationships between haemodynamic and structural aspects in man. Camilleri JP Berry CL Fiessinger JN Bariety J eds. Diseases of the Arterial Wall. 1989:pp 269-278 Springer-Verlag, Berlin
- 26.Numaguchi K, Egashira K, Takemoto M, Kadokami T, Shimokawa H, Sueishi K, Takeshita A: Chronic inhibition of nitric oxide synthesis causes coronary microvascular remodeling in rats. Hypertension 1995, 26:957-962 [DOI] [PubMed] [Google Scholar]
- 27.Rossi MA, Colombini-Netto M: Chronic inhibition of NO synthesis per se promotes structural intimal remodeling of the rat aorta. J Hypertens 2001, 19:1567-1579 [DOI] [PubMed] [Google Scholar]