

Abstract
The purpose of these experiments was to determine the specific role of reactive oxygen species (ROS) in the blood-retinal barrier (BRB) breakdown that characterizes the early stages of vascular dysfunction in diabetes. Based on our data showing that high glucose increases nitric oxide, superoxide, and nitrotyrosine formation in retinal endothelial cells, we hypothesized that excess formation of ROS causes BRB breakdown in diabetes. Because ROS are known to induce increases in expression of the well-known endothelial mitogen and permeability factor vascular endothelial growth factor (VEGF) we also examined their influence on the expression of VEGF and its downstream target urokinase plasminogen activator receptor (uPAR). After 2 weeks of streptozotocin-induced diabetes, analysis of albumin leakage confirmed a prominent breakdown of the BRB. This permeability defect was correlated with significant increases in the formation of nitric oxide, lipid peroxides, and the peroxynitrite biomarker nitrotyrosine as well as with increases in the expression of VEGF and uPAR. Treatment with a nitric oxide synthase inhibitor (N-ω-nitro-l-arginine methyl ester, 50 mg/kg/day) or peroxynitrite scavenger (uric acid, 160 mg/kg/day) blocked the breakdown in the BRB and prevented the increases in formation of lipid peroxides and tyrosine nitration as well as the increases in expression of VEGF and uPAR. Taken together, these data indicate that early diabetes causes breakdown of the BRB by a mechanism involving the action of reactive nitrogen species in promoting expression of VEGF and uPAR.
Breakdown of the blood-retinal barrier (BRB) is a well-established feature of both clinical and experimental diabetes. 1-5 The permeability defect has been shown to correlate with increases in expression of vascular endothelial growth factor (VEGF). 2-5 Recent studies in streptozotocin (STZ) diabetic rats have shown that the initial BRB breakdown is associated with increases in expression of both the endothelial and neuronal nitric oxide synthase (eNOS and nNOS) as well as with increases in VEGF expression. 4-6 Moreover a VEGF-neutralizing receptor construct has been shown to prevent the diabetes-induced increases in expression of VEGF and eNOS and the BRB breakdown, 3,6 underlining a potential role for VEGF in the early vascular dysfunction. However, the mechanisms by which diabetes increases VEGF expression and causes BRB breakdown are not yet understood.
Multiple interrelated biochemical mechanisms have been postulated to explain the adverse effects of diabetes and hyperglycemia on vascular cells including increased flux of glucose through the polyol pathway, enhanced nonenzymatic glycation and activation of protein kinase C and increased formation of reactive oxygen species (ROS), including nitric oxide (NO), superoxide (O2.−) and their product peroxynitrite (ONOO−). 7-11 Increased formation of ROS and lipid peroxides occurs early in diabetic retinopathy. 12,13 Analyses done with endothelial cells in vitro have shown that high glucose increases the expression of eNOS. 14 High glucose also greatly increases endothelial cell formation of O2.− leading to reduced bioavailability of NO. 15-17 Research demonstrating the presence of nitrotyrosine residues in placental vessels 10 and plasma 11 of diabetic patients supports a role for ONOO− in the development of diabetic complications. Peroxynitrite can contribute to vascular injury by causing lipid peroxidation and nitration of tyrosine residues, inactivating key metabolic enzymes, and reducing cellular antioxidant defenses by oxidation of thiol pools. 18,19 Peroxynitrite may also cause NOS-uncoupled production of O2.− because of oxidation of the NOS co-factor tetrahydrobiopterin (BH4), 20 the l-arginine transporter CAT-1 21 or eNOS itself. 22
Our analyses in vitro support the role of eNOS uncoupling and ONOO− formation in high glucose-induced endothelial dysfunction. We found that retinal endothelial cells maintained in high glucose have significant increases in eNOS expression and activity as well as in formation of O2.− and nitrotyrosine. 23 Each of these alterations was blocked by the NOS inhibitor, L-NAME, or the peroxynitrite scavenger, uric acid, lending further support to the role of eNOS uncoupling and ONOO− formation in high glucose-induced vascular injury. We have also shown that VEGF increases permeability of bovine retinal endothelial cells by activating urokinase plasminogen activator (uPA) and inducing the expression of its receptor urokinase plasminogen activator (uPAR). 24 uPA cleaves tissue plasminogen into the active enzyme plasmin, which in turn activates matrix metalloproteinases. 25 uPAR localizes these events at the cell membrane where cell-cell and cell-substrate attachments are altered. Therefore, we postulated that diabetes causes BRB dysfunction by causing ROS-mediated increases in expression of VEGF and uPAR. The goals of this study are 1) to define the role of NOS activity and formation of reactive nitrogen species in breakdown of the BRB; 2) to determine the correlation between diabetes-induced ROS formation and the expression of VEGF and its down stream target uPAR; and 3) to directly test whether or not reducing NOS activity or peroxynitrite formation protects diabetic rat retinas from diabetes-induced increase in VEGF expression and BRB breakdown.
Materials and Methods
Animal Preparation and Data Analysis
All procedures with animals were done in accordance with the Public Health Service Guide for the Care and Use of Laboratory Animals (DHEW Publication, NIH 80-23). Sprague-Dawley rats were made diabetic by a single intravenous injection of STZ (65 mg/kg) dissolved in 0.1 mol/L of fresh citrate buffer, pH 4.5. Three sets of animals were prepared for a total of 102 rats. In experiment 1, six rats were made diabetic and six rats were controls and in experiment 2, nine rats were made diabetic and nine rats were controls. In experiment 3, to study the effects of inhibitors, 36 rats were made diabetic and 36 rats were control. One group of diabetic animals (12 rats) and one control group (12 rats) received NOS inhibitor N-ω-nitro-l-arginine methyl ester (L-NAME) in their drinking water (50 mg/kg/day). Another group of diabetic animals (12 rats) and controls (12 rats) received uric acid (peroxynitrite scavenger) in their drinking water (160 mg/kg/day). Others were untreated control (n = 12) and untreated diabetic (n = 12) rats. After 2 weeks, the animals were sacrificed by decapitation, blood was collected and their retinas were removed, frozen in liquid nitrogen, and stored at −80°C until further analysis. Both retinas were collected from each rat and analyzed separately in independent experiments. Measurement of nitrite/nitrate, lipid peroxidation, nitrotyrosine, maximum NOS activity, and NOS expression were repeated in three independent experiments. The results are expressed as mean ± SEM. Differences among experimental groups were evaluated by analysis of variance and the significance of differences between groups was assessed by Fisher’s post-hoc least significant difference test when indicated. Significance was defined as P < 0.05.
Measurement of BRB Function
Integerity of the BRB was measured as described by others previously. 1 Rats received tail vein injections of 100 mg/kg of bovine serum albumin (BSA)-Alexa-Fluor 488 conjugate (Molecular Probes, Eugene, OR). After 30 minutes, the animals were sacrificed and the eyes were enucleated, embedded in OCT medium, and snap-frozen in liquid nitrogen. Plasma was assayed for Alexa-Fluor 488 concentration using a Cyto Fluor 4000 spectrofluorometer (Foster City, CA). Frozen retinal sections (10 μm) collected at 60-μm intervals were viewed with a fluorescence microscope fitted with a spot camera. Images were collected from 10 retinal areas (200 μm2) in each section. The average retinal fluorescence intensity was calculated and normalized to a noninjected control retina and to plasma fluorescence intensity for each animal. Through serial sectioning of each eye, this technique allowed quantification of barrier function in each retina.
Measurement of NO Formation in Rat Retinas
NO production was determined by measuring the levels of nitrite and nitrate, the oxidized products of NO, in the supernatant of phosphate-buffered saline (PBS) homogenate of the control or diabetic rat retinas by modified Greiss Reagent assay. Briefly, 210 μl of homogenate were incubated with nitrate reductase enzyme (10 mU) and NADPH (12.5 mmol/L) for 30 minutes at 37°C. Then the total nitrite in each sample was determined by addition of 200 mU of l-glutamate dehydrogenase, 100 mmol/L NH4Cl, and freshly prepared 4 mmol/L of α-ketogluterate. Enzymatic reduction of nitrate to nitrite was monitored by including an NO3− standard. The mixture was incubated at 37°C for 10 minutes followed by addition of 250 μl of Greiss Reagent and incubation for another 5 minutes at 37°C. The absorbance at 543 nm was recorded and concentrations of NO2 were calculated from a standard curve constructed using NaNO2 standards. Protein levels were measured by the Bradford method (Bio-Rad, Hercules, CA) and nitrite/nitrate level was expressed as mmol/mg protein.
Measurement of Retinal NOS Protein Expression
Retinal protein was extracted in RIPA lysis buffer (20 mmol/L Tris, pH 7.4, 2.5 mmol/L EDTA, pH 8, 1% Triton X-100, 1% deoxycholate, 1% sodium dodecyl sulfate, 50 mmol/L sodium fluoride, and 10 mmol/L sodium pyrophosphate) containing 1 mmol/L of phenylmethyl sulfonyl fluoride. Samples equated for protein content were separated on 10% sodium dodecyl sulfate gel and transferred to nitrocellulose membranes. Levels of eNOS, nNOS, and iNOS proteins were analyzed using corresponding monoclonal antibodies (Transduction Laboratories, Lexington, KY) and electrochemiluminescence (ECL; Amersham, Buckinghamshire, UK). The bands were scanned by densitometry and normalized to β-actin as an internal standard.
Determination of Retinal NOS Activity
Maximal NOS activity was quantified using an assay for the conversion of [3H]-l-arginine to [3H]-l-citrulline. 26 Retinas were homogenized on ice in a 250 μl buffer composed of 50 mmol/L Tris-HCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA (pH 7.4), 1 μmol/L pepstatin, 2 μmol/L leupeptin, 1 μmol/L bestatin, and 1 mmol/L phenylmethyl sulfonyl fluoride. The homogenate was centrifuged at 8000 × g for 10 minutes and the supernatant was collected and 100 μl was incubated in the previously described buffer containing 100 nmol/L calmodulin, 1 mmol/L NADPH, 3 μmol/L tetrahydrobiopterin, and 10 μmol/L l-arginine combined with 0.6 μCi l-[2,3-3H]-arginine. The incubation mixture also contained 1 mmol/L l-citrulline to minimize conversion of the formed [3H]-l-citrulline to [3H]-l-arginine. Ice-cold buffer, 1 ml (20 mmol/L Tris-HCl, 0.2 mmol/L EDTA, 0.2 mmol/L EGTA, pH 5.5) was used to stop the reaction. Reaction mixtures were applied to Dowex 50W-8 (Na form) columns and the eluted [3H]-l-citrulline activity was determined by scintillation counting spectroscopy. NOS activity was expressed as pmol/L/30 minutes/mg protein.
Measurement of Retinal Lipid Peroxidation
Lipid peroxide concentration was determined by a method that measures the amount of thiobarbituric acid reactivity by the amount of malondialdehyde formed during acid hydrolysis of the lipid peroxide compound. 27 Retinas were washed with 0.9% NaCl and tissue homogenate was prepared at a ratio of 1 g wet tissue to 9 ml of 1.15% KCl. The reaction mixture contained 0.2 ml of sample, 0.2 ml of 8.1% sodium dodecyl sulfate, 1.5 ml of 20% acetic acid solution (buffered to pH 3.5), and 1.5 ml of 0.8% thiobarbituric acid. The mixture was then incubated at 95°C for 1 hour. After cooling, 1 ml of distilled water and 5 ml of a mixture of n-butanol and pyridine (15:1, v/v) were added and the final mixture was shaken vigorously. After centrifugation at 1500 × g for 10 minutes absorbance of the solvent layer was measured at 532 nm. Tetraethoxypropane was used to establish the standard curve. Lipid peroxide level was expressed in terms of nmol/L malondialdehyde per mg protein.
Measurement of Retinal Nitrotyrosine
Nitrotyrosine immunoreactivity was measured as an indicator for ONOO− formation by slot blot and immunostaining. 28,29 For slot blot analysis of total nitrotyrosine levels, retinas were homogenized in RIPA lysis buffer. Duplicate protein samples were immobilized onto a polyvinylidene difluoride membrane using a slot blot microfiltration unit. After blocking with 5% nonfat milk, the polyvinylidene difluoride membrane was reacted with a monoclonal anti-nitrotyrosine antibody (Cayman Chemical Co., Ann Arbor, MI) followed by peroxidase-labeled goat anti-mouse IgG and enhanced chemiluminescence. Relative levels of nitrotyrosine immunoreactivity were determined by densitometry and comparison with a standard curve generated from ONOO−-modified BSA.
The distribution of nitrotyrosine in retinal sections was analyzed using immunolocalization techniques. Retinal sections were fixed with 4% paraformaldehyde then reacted with a polyclonal rabbit anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY) followed by Oregon green-conjugated goat anti-rabbit antibody (Molecular Probes). Data were analyzed using fluorescence microscopy and Ultra View morphometric software to quantify intensity of immunostaining.
Control experiments to demonstrate specificity of the nitrotyrosine antibody were done by processing the sections in the absence of primary antibody and by neutralizing the primary antibody. 30 Briefly, the antibody was neutralized by incubation with 10 mmol/L of 3-nitrotyrosine in PBS solution (Cayman Chemical Co.). The specificity of measurement of nitrotyrosine as indicator for peroxynitrite and the efficacy of uric acid as a scavenger of peroxynitrite were confirmed by in vitro techniques. Cultured bovine retinal endothelial cells were treated with 100 μmol/L of peroxynitrite (Upstate Biotechnology) in the presence or absence of 1 mmol/L of uric acid (Sigma Chemical Co., St. Louis, MO) for 1 hour and analyzed for nitrotyrosine formation as described above.
Real-Time Quantitative Polymerase Chain Reaction (PCR) Analysis of VEGF and uPAR RNA
SYBR Green provides a convenient way to detect and quantify reverse transcriptase (RT)-PCR product. The quantification is based on the amounts of SYBR Green-bound DNA measured at the end of each cycle of PCR. In brief, reverse transcription of the retinal RNA was performed by conventional methods using AMV RT and random primers. The use of random primers allowed the simultaneous amplification of the 18-S rRNA as reference or internal marker for each sample. A pair of rat-specific uPAR primers was synthesized based on the cDNA sequence 31 accession no. x71899 and was used to amplify a 200-bp DNA fragment (corresponding to nucleotides 764 to 963) by conventional PCR. For VEGF analysis, primers designed for bovine VEGF based on the cDNA sequence 32 accession no. M32976 were used to amplify a 200-bp amplicon (corresponding to nucleotides 853 to 1053). The uPAR and VEGF amplicons prepared by conventional PCR were used as standard templates in the subsequent analysis. Quantitative PCR was performed using a Light Cycler apparatus (Roche Diagnostics Ltd., Lewes, UK) using a kit provided by the same vendor. The specificity of the assay was determined at the completion of cycling by plotting the melting profile of each PCR product. In our experiments, this melting profile always overlapped with the melting profile of the standard samples in which pure template was used. Two RT reactions were performed and two light cycler reactions were performed for both uPAR and VEGF for each RNA sample. For internal controls, the RT samples were diluted 50- to 100-fold and were primed with 18S rRNA primers (Universal 18S Primer Pair; Ambion, Austin, TX).
Results
Inhibitor Treatment Does Not Alter Body Weight or Blood Glucose
The body weights of the diabetic rats decreased significantly (19%) whereas those of the controls increased (18%, Table 1 ▶ ). Blood glucose levels were significantly increased (fourfold) in the STZ-induced diabetic rats compared with the controls. Administration of L-NAME and uric acid did not affect body weight or blood glucose levels of either control or diabetic animals.
Table 1.
Effects of STZ-Induced Diabetes on Body Weight and Blood Glucose Levels in Control and Diabetic Rats
| Animals | n | % Weight change | Blood glucose level |
|---|---|---|---|
| Control | 21 | 19.6 ± 0.9 | 118.6 ± 12.12 |
| Diabetic | 21 | −18.8 ± 1.6 | 411.7 ± 15.2* |
| Control+ L-NAME | 12 | 15.6 ± 1.35 | 100.6 ± 9.5 |
| Diabetic+ L-NAME | 12 | −20.6 ± 0.8 | 446.4 ± 21.9* |
| Control+ uric acid | 12 | 15.9 ± 1.2 | 96.5 ± 8.3 |
| Diabetic+ uric acid | 12 | −19.4 ± 1.4 | 468.5 ± 15.47* |
Data are mean ± SE. Animals were made diabetic by a single STZ injection (65 mg/kg) in freshly prepared 1 mmol/L sodium citrate buffer, pH 4.5. Average starting weight of untreated control and diabetic rats was 236 ± 5.95. Average starting weight of L-NAME and uric acid-treated rats was 275 ± 2.1 and 264 ± 9.1, respectively.
*, P < 0.05 compared to corresponding control.
Inhibiting NOS or Scavenging Peroxynitrite Prevents the Diabetes-Induced Breakdown of the BRB
Assessment of BRB function using BSA-Alexa-Fluor 488 showed significant permeability increases within the diabetic retina. Microscopic images showed prominent accumulation of the tracer within retinal layers containing blood vessels—the nerve fiber layer and the inner and outer plexiform layers (Figure 1A) ▶ . Quantitative analysis showed a twofold increase (P < 0.01) in the fluorescence intensity in the diabetic retinas (Figure 1B) ▶ . Treatment with the NOS inhibitor, L-NAME (50 mg/kg/day) or the ONOO− scavenger, uric acid (160 mg/kg/day), completely blocked the permeability breakdown in the diabetic rats. The control rat retinas were not affected by either treatment.
Figure 1.

Permeability as determined by Alexa-Fluor 488-BSA in STZ-induced diabetic and control rat retina. Diabetic and control rats were injected intravenously with Alexa-Fluor 488-BSA (100 mg/kg each). A: Representative images showing the fluorescence distribution in different retinal layers; the nerve fiber layer (NFL), inner plexiform layer (IPL), outer plexiform layer (OPL), and the inner segment layer (ISL). B: Morphometric analysis of fluorescence intensity in serial sections of rat eyes shows that diabetic rats had a twofold increase in fluorescence compared with controls. Treatment of diabetic rats with L-NAME (50 mg/kg/day) or with uric acid (160 mg/kg/day) blocked the permeability increase. Data shown is the mean ± SEM of six animals in each group. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic. Original magnification, ×200 (A).
Diabetes Increases Oxidative Stress and Formation of Reactive Nitrogen Species
Our results showed that diabetes increased the formation of reactive nitrogen species as indicated by nitrite/nitrate, nitrotyrosine, maximum NOS activity, and NOS expression and increased oxidative stress as indicated by lipid peroxidation. Biochemical measures of oxidative stress and reactive nitrogen species formation were repeated and confirmed in three independent experiments. The data shown represent results from experiments 2 and 3.
Diabetes Increases Retinal Nitrite/Nitrate Levels
Direct measurement of NO formation in vivo is difficult because of its short half-life. Therefore, the evaluation of the stable NO end products, nitrite and nitrate, has been widely used to estimate NO production. As shown in Figure 2 ▶ , the formation of nitrate/nitrite in diabetic retinas was approximately twofold higher than in the controls (P < 0.01). Administration of either L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day) blocked this increase, but did not alter basal values in controls.
Figure 2.

Levels of nitrate/nitrite in STZ-induced diabetic and control rat retinas as determined by the Greiss reaction assay. Diabetic retinas had significant increases in formation of nitrate/nitrite. Treatment of diabetic rats with L-NAME (50 mg/kg/day) or with uric acid (160 mg/kg/day) blocked the increased nitrate/nitrite formation. Data shown is the mean ± SEM of six animals in the treated groups and nine animals in untreated groups. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic.
Diabetes Increases Retinal Lipid Peroxidation
The neural retina has a high content of polyunsaturated fatty acids and hence, is extremely susceptible to oxidative insult by ROS. Levels of lipid peroxides were measured by the thiobarbituric acid test and expressed as nmol/L of malondialdehyde. Malondialdehyde levels were increased ∼2.5-fold in the diabetic retinas in comparison with the controls (Figure 3) ▶ . Administration of either L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day) completely blocked the increased formation of lipid peroxides in diabetic retina. Neither agent altered lipid peroxidation in the control retina.
Figure 3.

Levels of lipid peroxides in STZ-induced diabetic and control rat retinas as determined by the amount of thiobarbituric acid reactivity with malondialdehyde formed during acid hydrolysis of the lipid peroxide. Diabetic retinas had significant increases in formation of lipid peroxides. Treatment of diabetic rats with L-NAME (50 mg/kg/day) or with uric acid (160 mg/kg/day) blocked the increased lipid peroxidation. Data shown is the mean ± SEM of six animals in the treated groups and nine animals in untreated groups. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic.
Diabetes Increases Retinal Nitrotyrosine Formation
ONOO− is a short-lived molecule at physiological pH, but it has been shown to nitrate protein tyrosine residues. Therefore, determination of nitrotyrosine can be used as an indicator of ONOO− formation. 29 The specificity of the nitrotyrosine antibody was confirmed by complete blocking of the signal in the absence of the primary antibody or when the primary antibody was neutralized using 3-nitrotyrosine (Figure 4A) ▶ . The specificity of detecting nitrotyrosine as indicator of peroxynitrite was further confirmed by our in vitro studies in bovine endothelial cells exposed to 100 μm of ONOO−. These experiments showed significant increases in tyrosine nitration in ONOO−-treated cells. Further, this effect was totally blocked by uric acid (1 mmol/L) demonstrating the efficiency of this agent as a scavenger for ONOO− (Figure 4B) ▶ .
Figure 4.

Measures of nitrotyrosine formation in peroxynitrite-treated endothelial cells and STZ-induced diabetic and control rats. A: Control experiments demonstrating the specificity of the nitrotyrosine antibody. The signal was absent in the absence of the primary antibody or in the presence of neutralized primary antibody. B: Immunocytochemistry of nitrotyrosine in cultured bovine endothelial cells. Peroxynitrite (100 μmol/L) increases nitrotyrosine formation and uric acid (1 mmol/L) blocked the increased nitrotyrosine formation. C: Slot-blot analysis of nitrotyrosine immunoreactivity. Window shows a representative slot blot. Diabetes increases nitrotyrosine formation by 1.8-fold. Treatment of diabetic rats with L-NAME (50 mg/kg/day) or with uric acid (160 mg/kg/day) blocked the increased nitrotyrosine formation. Data shown is the mean ± SEM of six animals in the treated groups and nine animals in untreated groups. D: Immunohistochemistry of nitrotyrosine in diabetic and control rats, note strong immunoreactivity in the vascularized layers [nerve fiber layer (NFL), inner plexiform layer (IPL), and outer plexiform layer (OPL)]. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic. Original magnification, ×200 (D).
Immunoblot analysis showed that formation of nitrotyrosine in the diabetic retina was increased approximately twofold in comparison to controls (P < 0.01, Figure 4C ▶ ). Treatment of the diabetic rats with either L-NAME or uric acid completely blocked this effect, whereas control levels were unchanged. Parallel studies also examined nitrotyrosine immunolocalization in retinal sections. In the control animals, nitrotyrosine residues were distributed within the inner segment, outer plexiform, inner plexiform, and nerve fiber/ganglion cell layers of the retina. In the diabetic retina, the intensity of the immunoreactivity of nitrotyrosine residues increased significantly in these layers, particularly within the areas where blood vessels are found—the nerve fiber/ganglion cell layer and the inner and outer plexiform layers (Figure 4D) ▶ . Treatment of diabetic animals with L-NAME or uric acid inhibited the increase in nitrotyrosine immunostaining.
Diabetes Increases Constitutive NOS Expression
Maximal NOS activity was determined in retinal tissue homogenates using an assay for conversion of [3H]-l-arginine to [3H]-l-citrulline. This assay serves to indicate changes in total levels of NOS protein because the enzyme is fully activated when tissue homogenate is reacted with both substrate and co-factors. NOS activity was increased 1.5-fold in the diabetic retina in comparison to the control retina (P < 0.01, Figure 5A ▶ ). Treatment of diabetic rats with L-NAME or uric acid blocked the effect of diabetes increasing maximum NOS activity, but did not alter the NOS activity in the control retinas. Addition of L-NAME to the tissue homogenates almost totally blocked formation of [3H]-l-citrulline (95% inhibition), demonstrating specificity of the reaction.
Figure 5.
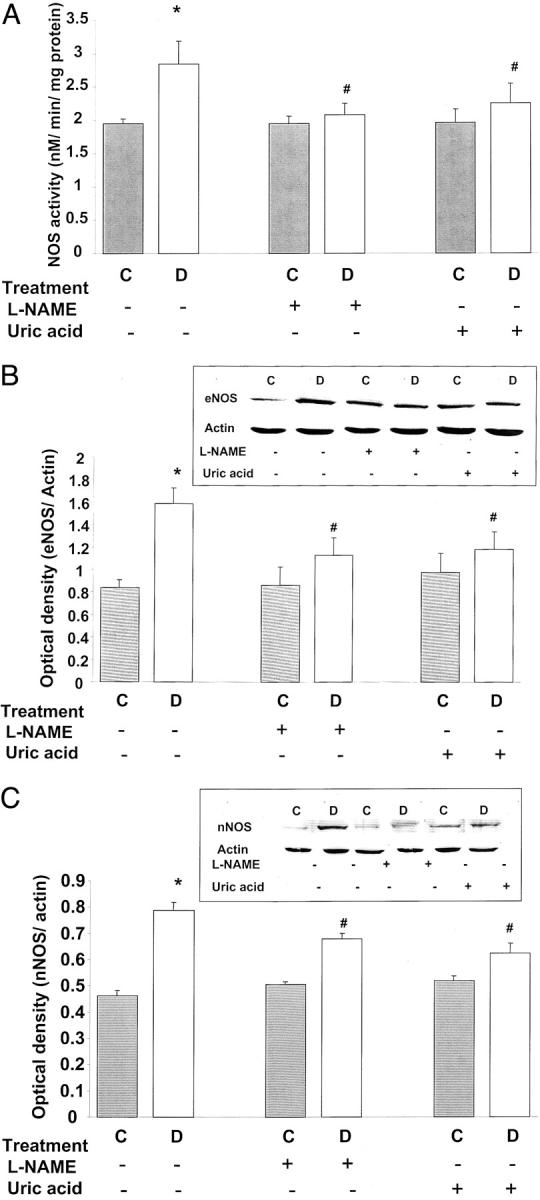
Analysis of NOS protein expression and activity in STZ-induced diabetic and control rat retinas. Data shown is the mean ± SEM of six animals in the treated groups and nine animals in untreated groups. A: Analysis of maximal NOS activity by conversion of [3H]-l-arginine to [3H]-l-citrulline in tissue homogenates shows significant increases in NOS activity in diabetic retinas. Treatment of diabetic rats with L-NAME (50 mg/kg/day) or with uric acid (160 mg/kg/day) blocked the increased NOS activity. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic. B: Western blot analysis shows a significant increase in eNOS expression in diabetic retinas, which is blunted by treatment with L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day). C: Western blot analysis shows a significant increase in nNOS expression in diabetic retinas, which is blocked by treatment with L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day).
Western blot analysis of eNOS and nNOS protein expression showed that both isoforms of constitutive NOS were significantly increased (2- and 1.8-fold, respectively) in the diabetic retinas in comparison to the control group (Figure 5, B and C) ▶ . Treatment with L-NAME or uric acid inhibited the increases in eNOS and nNOS expression in diabetic retina, but did not alter NOS expression in the control rat retinas. No difference in iNOS expression was found between diabetic and normal retinas (data not shown).
Diabetes Induces Increased Expression of VEGF and uPAR
Quantitative analysis of VEGF mRNA expression using real-time PCR showed a significant increase (P < 0.01) in the diabetic retina as compared to the control retina (Figure 6) ▶ . This was positively correlated with the increases in ROS formation and the BRB breakdown shown by the above results. Treatment with L-NAME or uric acid prevented the increase in VEGF expression in diabetic animals.
Figure 6.

Expression of VEGF mRNA in diabetic and control rat retina as determined by real-time PCR. STZ-induced diabetic retinas had significant increases in VEGF mRNA expression. This increase was totally blocked by treatment with L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day). Data shown is the mean ± SEM of three animals in each group. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic.
Our previous experiments showed that VEGF-induced permeability increases in retinal endothelial cells are associated with activation of uPA and expression of its receptor uPAR. To see if the diabetes-induced breakdown of the BRB could be related to VEGF activation of uPA/uPAR, we measured the expression of uPAR mRNA and protein levels in retinal extract. Real-time PCR analysis showed a significant increase in uPAR in the diabetic retina as compared to control retina (P < 0.01) (Figure 7A) ▶ . The increase in uPAR mRNA expression was associated with a significant increase in uPAR protein expression levels in diabetic retina compared to control retinas (Figure 7B) ▶ . Treatment with L-NAME or uric acid inhibited the increases in uPAR in diabetic retinas.
Figure 7.

Expression of uPAR mRNA and protein levels in STZ-induced diabetic and control rat retina as determined by real-time PCR and Western blot analysis. A: Diabetic retinas had significant increases in uPAR mRNA expression, which was inhibited by treatment with L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day). Data shown is the mean ± SEM of three animals in each group. B: Diabetic retinas had significant increases in uPAR protein expression, which was inhibited by treatment with L-NAME (50 mg/kg/day) or uric acid (160 mg/kg/day). Data shown is the mean ± SEM of six animals in each group. C, control; D, diabetic; *, P < 0.01 versus untreated control; #, P < 0.05 versus untreated diabetic.
Discussion
The major findings of this study are that diabetes-induced breakdown of the BRB is correlated with increases in the formation of ROS and increased expression of VEGF and uPAR and that treatment with a NOS inhibitor or a peroxynitrite scavenger reduced ROS formation, blocked the increases in expression of VEGF and uPAR, and prevented the BRB breakdown. Taken together, these data suggest that oxidative stress plays an important role in the initial breakdown of the BRB during diabetes and that this process could be mediated by VEGF-induced increases in expression of uPAR.
The source of the increase in ROS in the diabetic retina is likely to include NOS. Our data showed a significant increase in nitrate/nitrite levels in the diabetic retinas indicating an increase in NO formation. Measurement of maximal NOS activity confirmed significant increases in levels of NOS protein within the diabetic retina. Significant increases in eNOS and nNOS protein were also observed in the diabetic retina, whereas iNOS expression was unchanged from the control levels. A previous study observed similar increases in constitutive NOS protein expression in diabetic rats. 5 However, another group attributed increases in NOS activity to iNOS, based on their findings in diabetic and control retinas comparing a selective iNOS inhibitor, aminoguanidine, with a selective constitutive NOS inhibitor, L-NAME. 33 Because our Western blotting analyses failed to show an increase in iNOS expression, we determined diabetes’ effects on the expression of iNOS mRNA. Our RT-PCR analyses also showed no difference in iNOS expression between the diabetic and control retinas (data not shown). Because the increases we observed in eNOS and nNOS protein expression (2- and 1.8-fold, respectively) were similar in magnitude to the increases in maximum NOS activity and nitrite formation (1.5- and 2-fold, respectively), we conclude that the increase in nitrite formation reflects an overall increase in expression of the constitutive NOS isoforms.
Increases in the formation of NO and O2.− have been described previously in early diabetic retinopathy and in bovine aortic endothelial cells cultured in high glucose conditions. 14-17 However, our study is the first we know of to show that diabetic retinas also have significant increases in formation of reactive nitrogen species as indicated by increases in protein tyrosine nitration. Although several pathways of tyrosine nitration have been suggested in addition to peroxynitrite formation, nitrotyrosine is considered to be a likely indicator for peroxynitrite, particularly under conditions of simultaneous production of NO and superoxide. 34-36
In diabetic rat retinas, the increases in formation of NO, lipid peroxidation, and tyrosine nitration were correlated with the BRB breakdown. This observation is in agreement with previous studies showing that S-nitroso-N-acetylpenicillamine (SNAP) and 3-morpholinosydnonimine (SIN-1) can cause increases in permeability of epithelial cells in vitro 37 and of rat brain endothelial cells in vivo. 38 SIN-1 releases O2.− and NO simultaneously, whereas SNAP releases only NO. However, SNAP appears to induce intracellular production of O2.− under some conditions. 37,38 These effects of SNAP and SIN-1 on increasing permeability were blocked by scavengers of ONOO− or O2.−, suggesting that the permeability enhancing actions involve increased ONOO− formation.
Further support for the role of ONOO− in causing the BRB breakdown comes from our finding that the diabetes-induced increases in ROS formation and the BRB breakdown could be blocked by inhibiting NOS and by scavenging ONOO−. Uric acid is a naturally occurring product of purine metabolism that has a selective effect in inhibiting tyrosine nitration. It’s action as a peroxynitrite scavenger has been demonstrated by its capacity to bind peroxynitrite but not nitric oxide 39 and by its ability to inhibit the action of peroxynitrite in causing tyrosine nitration 40 (see Figure 4B ▶ ). Interestingly, the diabetic rats in our study had significantly lower levels of uric acid compared to the control rats. In support of this observation, patients with type I diabetes also show significantly lower serum uric acid levels in comparison to healthy controls or to type II diabetics. 41
It is important to note that treatments that inhibit NOS can reduce ONOO− formation by blocking the action of eNOS in forming O2.− as well as preventing the formation of NO. 42 Our tissue culture analyses in retinal endothelial cells maintained in high glucose conditions indicate that eNOS becomes uncoupled and generates O2.− as well as NO, leading to ONOO− formation. 23 These data support a relationship between ONOO− and diabetes-induced vascular dysfunction. Observation of increased nitrotyrosine formation in the ganglion cell layer suggest that ONOO− may also contribute to neuronal injury that has been described previously in diabetic rats and humans. 43
Diabetic retinopathy is clinically divided into an early stage of nonproliferative retinopathy and a late stage of proliferative neovascular disease. Breakdown of the BRB and progressive vascular occlusion are hallmarks of early diabetic retinopathy that continue with progression of the disease. 44,45 Under diabetic conditions, BRB breakdown is thought to occur because of diabetes-induced increases in the formation of vasoactive factors such as VEGF/vascular permeability factor. 1-5 Although diabetes-induced vascular dysfunction in the rodent model does not progress to proliferative retinopathy, increases in retinal vascular permeability are well established. Our data confirmed a breakdown of the BRB at 2 weeks after the onset of diabetes, which is accompanied by a substantial increase in VEGF expression.
Our finding that retinal VEGF mRNA expression is increased in the early stages of diabetes is consistent with previous data showing that ischemia, hyperglycemia, and oxidative stress can increase growth factor expression. 46-50 Induction of VEGF expression by hypoxia provides a pathogenic link between ischemic retinal diseases and the accompanying neovascularization. 51 Experiments in animal models of hypoxia-induced ocular neovascularization have shown that VEGF is up-regulated several fold before the onset of neovascularization. 50,52-54
Another potential inducer of VEGF gene expression in diabetic tissue is the increased generation of ROS. 55 Our data showing a significant increase of lipid peroxidation in diabetic retinas are in agreement with several reports correlating formation of lipid peroxides with oxidative stress in early experimental diabetes. 12,13 The mechanism by which ROS increase VEGF gene expression has been suggested to involve an increase in VEGF mRNA stability. 54 It has also been demonstrated that ROS activate intracellular signaling events leading to the stabilization of HIF-1α 55 and the subsequent VEGF mRNA expression during hypoxia. 56,57
The effects of VEGF in increasing vascular permeability are well established, yet the mechanisms of this action remain poorly understood. Our studies in cultured retinal endothelial cells have shown that VEGF-induced permeability increases are mediated by activation of urokinase plasminogen activator (uPA) and expression of its receptor (uPAR). 24 uPA is involved in numerous biological processes requiring cell movement and tissue remodeling. It has been suggested that uPAR enhances uPA function by localizing plasmin formation at the cell membrane. Hence, increases in uPAR expression may be involved in the breakdown of cell junctions and permeability barrier function. This concept was supported by our analyses of uPAR mRNA showing significant increases in both uPAR mRNA and protein expression in the diabetic retina. The fact that the increase in uPAR expression was inhibited in by L-NAME or uric acid strongly supports the role of ROS-induced VEGF expression in the diabetic retina. This is also consistent with the above findings showing that treatment with the NOS inhibitor or peroxynitrite scavenger could prevent the diabetes-induced increases in VEGF expression and maintain the integrity of the BRB. These observations also indicate a critical role of uPA and uPAR in the diabetes-induced permeability breakdown. Taken together, these data strongly indicate that ONOO− plays an important role in the pathogenesis of early diabetic vascular dysfunction as indicated by the BRB breakdown and that this process could be mediated by VEGF-induced increases in expression of uPAR. Although the influence of ROS on increasing VEGF expression 54,55 and the involvement of VEGF in diabetes-induced BRB breakdown has been reported, 3,50-52 the increased formation of reactive nitrogen species and potential involvement of uPAR in BRB breakdown have not been shown previously. Further investigation is required to define the role of uPAR in the diabetes-induced breakdown in retinal endothelial cell permeability barrier function and to elucidate the relationship between uPA/uPAR activation and previously identified mediators of the BRB breakdown during early diabetes, such as increased expression of intercellular adhesion molecule (ICAM-1) and leukeostasis. 6,58
Footnotes
Address reprint requests to R. B. Caldwell, Ph.D., Vascular Biology Center, Medical College of GA, Augusta, GA, 30909. E-mail: rcaldwel@mail.mcg.edu.
Supported by grants from the National Institutes of Health (EY04618 and EY11766), postdoctoral fellowships from Association for Research in Vision and Ophthalmology/Ciba Vision Inc., and the American Heart Association.
References
- 1.Antonetti DA, Barber AJ, Khin S, Lieth E, Tarbell JM, Gardner TW: Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content. Diabetes 1998, 47:1953-1959 [DOI] [PubMed] [Google Scholar]
- 2.Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, Yeo TK, Yeo KT: Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 1994, 118:445-450 [DOI] [PubMed] [Google Scholar]
- 3.Qaum T, Xu Q, Houssen A, Clemens MW, Qin W, Miyamoto K, Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP: VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci 2001, 42:2408-2413 [PubMed] [Google Scholar]
- 4.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE: Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994, 331:1480-1487 [DOI] [PubMed] [Google Scholar]
- 5.Takeda M, Mori F, Yoshida A, Takamiya A, Nakagomi S, Sato E, Kiyama H: Constitutive nitric oxide synthase is associated with retinal vascular permeability in early diabetic rats. Diabetologia 2001, 44:1043-1050 [DOI] [PubMed] [Google Scholar]
- 6.Joussen AM, Poulaki V, Tsujikawa A, Qin W, Qaum T, Xu Q, Moromizato Y, Bursell SE, Wiegand SJ, Rudge J, Ioffe E, Yancopoulos GD, Adamis AP: Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol 2002, 160:1683-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M: Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404:787-790 [DOI] [PubMed] [Google Scholar]
- 8.Koya D, King GL: Protein kinase C activation and the development of diabetic complications. Diabetes 1998, 47:859-866 [DOI] [PubMed] [Google Scholar]
- 9.Nakamura N, Obayashi H, Fujii M: Induction of aldose reductase in cultured human microvascular endothelial cells by advanced glycation end products. Free Radic Biol Med 2000, 29:17-25 [DOI] [PubMed] [Google Scholar]
- 10.Kossenjans W, Eis A, Sahay R, Brockman D, Myatt L: Role of peroxynitrite in altered fetal-placental vascular reactivity in diabetes or preeclampsia. Am J Physiol 2000, 278:H1311-H1319 [DOI] [PubMed] [Google Scholar]
- 11.Ceriello A, Mercuri F, Quagliaro L, Assaloni R, Motz E, Tonutti L, Taboga C: Detection of nitrotyrosine in the diabetic plasma: evidence of oxidative stress. Diabetologia 2001, 44:834-838 [DOI] [PubMed] [Google Scholar]
- 12.Armstrong D, Al-Awadi F: Lipid peroxidation and retinopathy in streptozotocin-induced diabetes. Free Radic Biol Med 1991, 11:433-436 [DOI] [PubMed] [Google Scholar]
- 13.Fathallah L, Obrosova IG: Increased retinal lipid peroxidation in early diabetes is not associated with ascorbate depletion or changes in ascorbate redox state. Exp Eye Res 2001, 72:719-723 [DOI] [PubMed] [Google Scholar]
- 14.Cosentino F, Hishikawa K, Katusic SZ, Luscher TF: High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 1997, 96:25-28 [DOI] [PubMed] [Google Scholar]
- 15.Graier WF, Simecek S, Kukovetz WR, Kostner GM: High D-glucose-induced changes in endothelial Ca2+/EDRF signaling are due to generation of superoxide anions. Diabetes 1996, 45:1386-1395 [DOI] [PubMed] [Google Scholar]
- 16.Graier WF, Posch K, Fleischhacker E, Wascher TC, Kostner GM: Increased superoxide anion formation in endothelial cells during hyperglycemia: an adaptive response or initial step of vascular dysfunction? Diabetes Res Clin Pract 1999, 2-3:153-160 [DOI] [PubMed] [Google Scholar]
- 17.Pieper GM, Langenstroer P, Siebeneich W: Diabetic-induced endothelial dysfunction in rat aorta: role of hydroxyl radicals. Cardiovasc Res 1997, 34:145-156 [DOI] [PubMed] [Google Scholar]
- 18.Salgo G, Bermudez E, Squadrito L, Pryor A: Peroxynitrite causes DNA damage and oxidation of thiols in rat thymocytes. Arch Biochem Biophys 1995, 322:500-505 [DOI] [PubMed] [Google Scholar]
- 19.Salvemini D, Wang Z, Stern M, Currie M, Misko T: Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci USA 1998, 95:2695-2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milstien S, Katusic Z: Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun 1999, 263:681-684 [DOI] [PubMed] [Google Scholar]
- 21.Patel JM, Abeles AJ, Block ER: Nitric oxide exposure and sulfhydryl modulation alter L-arginine transport in cultured pulmonary artery endothelial cells. Free Radic Biol Med 1996, 20:629-637 [DOI] [PubMed] [Google Scholar]
- 22.Zou MH, Shi C, Cohen RA: Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 2002, 109:817-826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB: High glucose increases tyrosine nitration and superoxide anion formation in endothelial cells. Invest Ophthalmol Vis Sci (In press) [DOI] [PubMed]
- 24.Behzadian MA, Windsor JL, Ghaly N, Liou GI, Tsai N, Caldwell RB: VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEBJ. 2003, 17:752-754 [DOI] [PubMed] [Google Scholar]
- 25.Mandriota SJ, Seghezzi G, Vassalli JD, Ferrara N, Wasi S, Mazzieri R, Mignatti P, Pepper MS: Vascular endothelial growth factor increases urokinase receptor expression in vascular endothelial cells. J Biol Chem 1995, 270:9709-9716 [DOI] [PubMed] [Google Scholar]
- 26.Bredt DS, Snyder SH: Isolation of nitric oxide synthase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 1990, 87:682-689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohkawa H, Ohishi N, Yagi K: Assay for lipid peroxides in animal tissue by thiobarbituric acid reaction. Anal Biochem 1979, 95:351-358 [DOI] [PubMed] [Google Scholar]
- 28.Brooks SE, Gu X, Samuel S, Marcus DM, Bartoli M, Huang PL, Caldwell RB: Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Invest Ophthalmol Vis Sci 2001, 42:222-228 [PubMed] [Google Scholar]
- 29.Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D: Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem 1998, 273:15646-15653 [DOI] [PubMed] [Google Scholar]
- 30.Viera L, Ye Y, Estevez AG, Beckman JS: Immunohistochemical methods to detect nitrotyrosine. Methods Enzymol 1999, 301:373-381 [DOI] [PubMed] [Google Scholar]
- 31.Rabbani SA, Rajwans N, Achbarou A, Murthy KK, Goltzman D: Isolation and characterization of multiple isoforms of the rat urokinase receptor in osteoblasts. FEBS Lett 1994, 338:69-74 [DOI] [PubMed] [Google Scholar]
- 32.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 33.Carmo A, Lopes C, Santos M, Proenca R, Cunha-Vaz J, Carvalho AP: Nitric oxide synthase activity and L-arginine metabolism in the retinas from streptozotocin-induced diabetic rats. Gen Pharmacol 1998, 30:319-324 [DOI] [PubMed] [Google Scholar]
- 34.Ischiropoulos H: Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys 1998, 356:1-11 [DOI] [PubMed] [Google Scholar]
- 35.Sawa T, Akaike T, Maeda H: Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J Biol Chem 2000, 275:32467-32474 [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, Joseph J, Feix J, Hogg N, Kalyanaraman B: Nitration and oxidation of a hydrophobic tyrosine probe by peroxynitrite in membranes: comparison with nitration and oxidation of tyrosine by peroxynitrite in aqueous solution. Biochemistry 2001, 40:7675-7686 [DOI] [PubMed] [Google Scholar]
- 37.Menconi MJ, Unno N, Smith M, Aguirre DE, Fink MP: Nitric oxide donor-induced hyperpermeability of cultured intestinal epithelial monolayers: role of superoxide radical, hydroxyl radical and peroxynitrite. Biochem Biophys Acta 1998, 1425:189-203 [DOI] [PubMed] [Google Scholar]
- 38.Mayhan WG: Nitric oxide donor-induced increase in permeability of the blood-brain barrier. Brain Res 2000, 866:101-108 [DOI] [PubMed] [Google Scholar]
- 39.Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, Koprowski H: Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA 1998, 95:675-680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiteman M, Halliwell B: Protection against peroxynitrite-dependent tyrosine nitration and alpha 1-antiproteinase inactivation by ascorbic acid. A comparison with other biological antioxidants. Free Radic Res 1996, 25:275-283 [DOI] [PubMed] [Google Scholar]
- 41.Zazgornik J, Biesenbach G, Janko O, Grafinger P, Kaiser W, Stuby U, Hubmann R: Serum uric acid level in type 1 and type 2 diabetic patients. Wien Med Wochenschr 1996, 146:102-144 [PubMed] [Google Scholar]
- 42.Kaesemeyer WH, Ogonowski AA, Jin L, Caldwell RB, Caldwell RW: Endothelial nitric oxide synthase is a site of superoxide synthesis in endothelial cells treated with glyceryl trinitrate. Br J Pharmacol 2000, 131:1019-1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW: Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 1998, 102:783-791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enea NA, Hollis TM, Kern JA, Gardner TW: Histamine H1 receptors mediate increased blood-retinal barrier permeability in experimental diabetes. Arch Ophthalmol 1989, 107:270-274 [DOI] [PubMed] [Google Scholar]
- 45.Engler C, Krosgaa B, Lund-Anderson H: Blood-retinal barrier permeability and its relation to the progression of diabetic retinopathy in type I diabetes: a 8-year follow-up study. Graefe’s Arch Clin Exp Ophthalmol 1991, 229:442-446 [DOI] [PubMed] [Google Scholar]
- 46.Gilbert RE, Vranes D, Berka JL, Kelly DJ, Cox A, Wu LL, Stacker SA, Cooper ME: Vascular endothelial growth factor and its receptors in control and diabetic rat eyes. Lab Invest 1998, 78:1017-1027 [PubMed] [Google Scholar]
- 47.Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA: Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol 1995, 113:1538-1544 [DOI] [PubMed] [Google Scholar]
- 48.Duh E, Aiello LP: Vascular endothelial growth factor and diabetes. The agonist versus antagonist paradox. Diabetes 1999, 48:1899-1906 [DOI] [PubMed] [Google Scholar]
- 49.Tilton RG, Kawamura T, Chang KC, Ido Y, Bjercke RJ, Stephan CC, Brock TA, Williamson JR: Vascular dysfunction induced by elevated glucose levels in rats is mediated by vascular endothelial growth factor. J Clin Invest 1997, 99:2192-2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammes HP, Lin J, Bretzel RG, Brownlee M, Breier G: Upregulation of the vascular endothelial growth factor/vascular endothelial growth factor receptor system in experimental background diabetic retinopathy of the rat. Diabetes 1998, 47:401-406 [DOI] [PubMed] [Google Scholar]
- 51.Peer J, Shweiki D, Itin A, Hemo I, Gnessin H, Keshet E: Hypoxia-induced expression of vascular endothelial growth factor by retinal cells is a common factor in neovascularizing ocular diseases. Lab Invest 1995, 72:638-645 [PubMed] [Google Scholar]
- 52.Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LEH: Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA 1995, 92:905-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith LEH, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA: Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 1994, 35:101-111 [PubMed] [Google Scholar]
- 54.Kuroki M, Voest EE, Amano S, Beerepoot LV, Takashima S, Tolentino M, Kim RY, Rohan RM, Colby KA, Yeo KT, Adamis AP: Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J Clin Invest 1996, 98:1675-1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT: Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 during hypoxia. A mechanism of O2 sensing. J Biol Chem 2000, 275:25130-25138 [DOI] [PubMed] [Google Scholar]
- 56.Duyndam MC, Hulscher, Fontijn D, Pinedo HM, Boven E: Induction of vascular endothelial growth factor expression and hypoxia-inducible factor 1 α protein by the oxidative stressor arsenite. J Biol Chem 2001, 276:48066-48076 [DOI] [PubMed] [Google Scholar]
- 57.Gorlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, Kietzmann T, Busse R: Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22phox-containing NADPH oxidase. Circ Res 2001, 89:47-54 [DOI] [PubMed] [Google Scholar]
- 58.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP: Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA 1999, 96:10836-10841 [DOI] [PMC free article] [PubMed] [Google Scholar]