

Abstract
α-Synucleinopathies, including Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy, are neurodegenerative disorders in which abnormal inclusions containing α-synuclein accumulate in selectively vulnerable neurons and glia. In this report, immunohistochemistry demonstrates ubiquitin in subsets of α-synuclein inclusions in dementia with Lewy bodies and multiple system atrophy. Biochemistry demonstrates that α-synuclein in the sodium dodecyl sulfate-soluble fractions of diseased brains is ubiquitinated, with mono- and di-ubiquitinated species predominating over polyubiquitinated forms. Similar immunohistochemical and biochemical characteristics were observed in an A53T mutant human α-synuclein transgenic mouse model of neurodegenerative α-synucleinopathies. Furthermore, in vitro ubiquitination of α-synuclein fibrils recapitulated the pattern of α-synuclein ubiquitination observed in human disease and the A53T α-synuclein mouse model. These results suggest that ubiquitination of α-synuclein is not required for inclusion formation and follows the fibrillization of α-synuclein.
Many neurodegenerative disorders are characterized by the presence of abnormal protein aggregates in neurons and/or glia of the central nervous system. One set of these disorders, characterized by pathological lesions in which α-synuclein (α-syn) is a major component, has been termed α-synucleinopathies. 1-3 A genetic link between α-syn and these neurodegenerative disorders came with the seminal discovery of point mutations in the α-synuclein gene in families afflicted with autosomal-dominant inherited Parkinson’s disease (PD). 4,5 This stimulated studies showing that the hallmark lesions of α-synucleinopathies, such as Lewy bodies (LBs) and Lewy neurites (LNs) in PD and dementia with Lewy bodies (DLB), as well as glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA), are comprised of α-syn 6-8 in the form of abnormal filaments. 9-11 Furthermore, in vitro studies demonstrated that recombinant α-syn forms fibrils and that α-syn mutants associated with familial PD have a greater propensity to fibrillize. 12-14 Recent studies have shown that the formation of α-syn inclusions in transgenic mice and flies overexpressing α-syn causes neurodegeneration associated with motor impairments. 15-20
Mounting evidence has implicated the ubiquitin-proteasomal pathway in the pathogenesis of PD. A point mutation in the ubiquitin C-terminal hydrolase (UCH)-L1 gene has been linked to autosomal-dominant inherited parkinsonism in one family, 21 whereas autosomal-recessive juvenile parkinsonism is caused by various mutations and deletions in the parkin gene. 22,23 UCH-L1 is thought to be critical in the recycling of free ubiquitin and regulating the extent of ubiquitin-protein conjugation, which is an important marker for targeting proteins to proteasomes for degradation. 24 Parkin is associated with E3 ubiquitin-ligase activity, and several putative substrates have been reported. 25-30 These findings suggest that defects in the ubiquitin-proteasomal pathway, resulting in the accumulation of toxic protein species, might contribute to the pathogenesis of sporadic PD. Indeed, proteasomal activity in the substantia nigra of patients with sporadic PD has been reported to be decreased compared to controls. 31 Pathological inclusions comprised of α-syn are also immunoreactive to antibodies to ubiquitin, 11,32,33 although it is unclear if this reflects sequestered free ubiquitin or ubiquitinated proteins in the inclusions. Cell culture models, however, have pointed to a role for proteasomal dysfunction in the formation of intracellular α-syn-containing aggregates, which are sometimes also ubiquitin-immunoreactive. 34,35 The aggregates formed in these in vitro paradigms, therefore, recapitulate certain properties of the aggregates found in human α-synucleinopathies.
Nevertheless, the exact process by which ubiquitin-positive inclusions form in α-synucleinopathies and the temporal relationship between inclusion formation and ubiquitination remains unclear. In this report, immunohistochemical analysis of filamentous inclusions in DLB, MSA, and an A53T α-synucleinopathy mouse model indicate that not all α-syn inclusions are immunoreactive for ubiquitin. Biochemical analysis reveals that ubiquitinated α-syn is a component of LBs and that in vitro ubiquitination of filamentous α-syn more closely recapitulates the pattern of ubiquitination observed in human disease than that of monomeric α-syn. Together, these data suggest that ubiquitination of α-syn is not required for inclusion formation and that the assembly of α-syn into fibrillar aggregates may precede their ubiquitination in α-synucleinopathies.
Materials and Methods
Antibodies
LB509, Syn208, and Syn211 are mouse monoclonal antibodies (mAbs) specific for human α-syn, whereas Syn102 is a mouse mAb that detects both α- and β-syn. 36 Syn303 is a mouse mAb that specifically detects pathological α-syn inclusions. 37 SNL-4 is a rabbit antibody raised to a synthetic peptide corresponding to amino acid residues 2 to 12 in α-syn. 36 A mouse mAb (1510) to ubiquitin was purchased from Chemicon International, Inc. (Temecula, CA), whereas rabbit anti-ubiquitin antibody Conj8, generated as described, 38 was kindly provided by Dr. C. Pickart (Johns Hopkins University, Baltimore, MD).
Immunohistochemical Staining and Quantification of Pathological Inclusions
The harvesting, fixation, and further processing of the tissue specimens used in this study were conducted as previously described. 39,40 Briefly, tissue blocks of cingulate cortex from DLB or cerebellum from MSA brains were fixed with 70% ethanol with 150 mmol/L of NaCl and infiltrated with paraffin. The diagnostic assessment of all DLB and MSA cases was performed in accordance with published guidelines. 41,42
Immunohistochemistry was performed using the avidin-biotin complex detection system (Vector Laboratories, Burlingame, CA) and 3,3′-diaminobenzidine (DAB) as described. 40 Briefly, sections were deparaffinized and rehydrated, endogenous peroxidases were quenched with 5% H2O2 in methanol for 30 minutes and sections were blocked in 0.1 mol/L of Tris with 2% donor horse serum for 5 minutes. Primary antibodies were incubated overnight at 4°C. After washing, sections were sequentially incubated with biotinylated secondary antibodies for 1 hour and avidin-biotin complex for 1 hour. Bound antibody complexes were visualized by incubating sections in a solution containing 100 mmol/L Tris, pH 7.6, 0.1% Triton X-100, 1.4 mmol/L 3,3′-diaminobenzidine, 10 mmol/L imidazole, and 8.8 mmol/L H2O2. Sections were then lightly counterstained with hematoxylin.
The percentage of cortical LBs or GCIs that are ubiquitin-positive was determined using published methods. 43 Consecutive 6-μm sections from DLB (cingulate cortex) or MSA (cerebellum) brains were immunostained with either anti-syn antibody Syn303 or anti-ubiquitin mAb 1510. Three adjacent photomicrographs were taken from the DLB cingulate cortex or MSA cerebellar white matter tissue sections and immunoreactive inclusions were counted. The percentage of LBs or GCIs labeled with anti-ubiquitin was determined as a ratio of ubiquitin inclusion counts over α-syn inclusion counts.
Double-labeling immunofluorescence analyses were performed as previously described 44 using Alexa Fluor 488- and 594-conjugated secondary antibodies (Molecular Probes, Eugene, OR) and coverslipped with Vectashield-DAPI mounting medium (Vector Laboratories, Burlingame, CA).
Sequential Biochemical Fractionation
Gray matter from cingulate cortex (DLB, NL) was dissected and weighed. Tissue was homogenized in 10 ml/g of low-salt (LS) buffer [10 mmol/L Tris, pH 7.5, 5 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L dithiothreitol, 10% sucrose, and a cocktail of protease inhibitors] and sedimented at 25,000 × g for 30 minutes at 4°C. Supernatants were saved as the LS fraction and pellets were washed by re-extraction in LS buffer. Resulting pellets were subjected to two sequential extractions in 10 ml/g of Triton-X (TX) buffer (LS + 1% Triton X-100 + 0.5 mol/L NaCl) and sedimented at 180,000 × g for 30 minutes at 4°C. Supernatants from the first of these TX buffer extractions were saved as the TX fraction. Pellets were then homogenized in 10 ml/g of sarkosyl buffer (LS + 1% N-lauroyl-sarcosine + 0.5 mol/L NaCl) and incubated at 22°C on a shaker for 1 hour before sedimentation at 180,000 × g for 30 minutes at 22°C. Supernatants were saved as the sarkosyl-soluble fraction. Remaining pellets were extracted in 2.5 ml/g of sodium dodecyl sulfate (SDS) buffer (2% SDS, 50 mmol/L Tris, pH 7.6, and a cocktail of protease inhibitors) before centrifugation at 25,000 × g for 30 minutes at 22°C. Supernatants were saved as the SDS-soluble fraction. Fresh mouse brain and spinal cord were sequentially extracted in a similar manner, except 3 ml/g of LS buffer, 3 ml/g of TX buffer, 2 ml/g of sarkosyl buffer, and 1 ml/g of SDS buffer were used. For the mouse spinal cord, an additional step with homogenization in TX buffer containing 30% sucrose followed by centrifugation was inserted after the TX buffer extractions to float and remove myelin. SDS sample buffer (10 mmol/L Tris, pH 6.8, 1 mmol/L EDTA, 40 mmol/L dithiothreitol, 1% SDS, 10% sucrose) was added to samples of LS, TX, and sarkosyl-soluble fractions, and sample buffer without SDS (10 mmol/L Tris, pH 6.8, 1 mmol/L EDTA, 40 mmol/L dithiothreitol, 10% sucrose) was added to SDS-soluble samples, followed by heating to 100°C for 5 minutes.
Western Blot Analysis
Proteins were separated by 15% SDS-polyacrylamide gel electrophoresis and subsequently transferred electrophoretically onto nitrocellulose membrane (Schleicher & Schuell, Keene, NH), in buffer containing 25 mmol/L Tris, 190 mmol/L glycine, and 10% methanol. Membranes were blocked with a 5% solution of powdered skimmed milk dissolved in Tris-buffered saline (50 mmol/L Tris, pH 7.6, 150 mmol/L NaCl), incubated with primary antibodies, followed with either goat anti-mouse or goat anti-rabbit antibody conjugated to horseradish peroxidase, developed with Renaissance Enhanced Luminol Reagents (NEN Life Science Product, Inc., Boston, MA), and exposed onto X-Omat Blue XB-1 films (Kodak, Rochester, NY).
Immunoprecipitation
SDS-soluble fraction from the cingulate cortex of DLB brain was diluted 20-fold in immunoprecipitation (IP) buffer (50 mmol/L Tris, pH 7.5, 100 mmol/L NaCl, 2 mmol/L EGTA, 50 mmol/L NaF, 1% Triton X-100, and a cocktail of protease inhibitors) and immunoprecipitated with anti-α-syn antibodies Syn h310, Syn h312, and Syn h313 covalently linked to Carbolink beads (Pierce, Rockford, IL) according to the manufacturer’s instructions. Immunocomplexes were washed three times with IP buffer, protein was eluted from the beads by boiling in SDS sample buffer, and samples were analyzed by Western blotting.
In Vitro Deubiquitination with UCH-L1
SDS-soluble fractions from the cingulate cortex of neuropathologically normal (NL) or DLB brains were diluted 40-fold in UCH buffer (50 mmol/L HEPES, pH 7.8, 0.5 mmol/L EDTA, 1 mmol/L dithiothreitol) and then concentrated 40-fold using a MicroconYM-10 (Millipore Corp., Bedford, MA) to remove SDS. UCH-L1 (Boston Biochem, Cambridge, MA) was preincubated in activation buffer (50 mmol/L HEPES, pH 7.8, 0.5 mmol/L EDTA, 10 mmol/L dithiothreitol) for 15 minutes at 25°C. UCH-L1 (or UCH buffer of equal volume without UCH-L1) was then added to samples to a final enzyme concentration of 5 nmol/L and incubated for 1 hour at 25°C. Reactions were stopped by adding SDS sample buffer and heating to 100°C for 5 minutes. Samples were analyzed by Western blotting.
In Vitro Ubiquitination of Unassembled and Fibrillar α-Syn
Bacterial expressed recombinant human α-syn was purified as previously described 13 and assembled in vitro by shaking at 37°C for 48 hours in 100 mmol/L of sodium acetate, pH 7.0. 45 Fibrillized α-syn was isolated by centrifugation at 100,000 × g for 20 minutes 45 and resuspended into ubiquitination reaction buffer (50 mmol/L Tris, pH 7.5, 2.5 mmol/L MgCl2, 2 mmol/L ATP, 1 mmol/L dithiothreitol). Monomeric (0.5 mg/ml) or fibrillar α-syn (0.5 mg/ml) was ubiquitinated in vitro by incubation with ubiquitin conjugation fractions A and B (derived from mammalian cell lysate, each at 280 μg/ml; Boston Biochem), purified bovine ubiquitin (480 μg/ml, Sigma Chemical Co. St. Louis, MO), energy-regenerating system (1×, Boston Biochem), and ubiquitin-aldehyde (2.5 μg/ml, Boston Biochem) for 2 hours at 37°C. Reactions were stopped by the addition of SDS sample buffer and heating to 100°C for 5 minutes. Samples were then analyzed by Western blotting.
Results
Pathological Inclusions in α-Synucleinopathy Brains Are Ubiquitinated to Varying Degrees
The accumulation of ubiquitin in α-syn pathological inclusions was studied by immunohistochemical analysis. Cortical LBs in the cingulate cortex of patients with DLB (Figure 1A) ▶ and GCIs in the cerebellum of patients with MSA (Figure 1C) ▶ were labeled with the anti-α-syn antibody Syn303. Cortical LBs have the typical more regular round shape, whereas the morphology of GCIs is less regular, but often showing a crescent shape. Sections stained with the anti-ubiquitin antibody mAb 1510 demonstrated that a significant number of LBs and GCIs also contain ubiquitin (Figure 1, B and D) ▶ . The abundance of ubiquitin-positive inclusions was further demonstrated by double-labeling immunofluorescence microscopy (Figure 1; E to J) ▶ . A large proportion of cortical LBs were stained with anti-ubiquitin antibody, and the staining pattern for α-syn and ubiquitin overlapped (Figure 1; E to G) ▶ . Many GCIs were stained with anti-ubiquitin antibodies, but a large subset of GCIs demonstrated little or no ubiquitin immunoreactivity (Figure 1; H to J) ▶ . Moreover, in many GCIs, anti-ubiquitin labeling displayed spatial variations in intensity so that within individual inclusions, some regions were intensely stained, while other regions remained unstained. Quantification of the percentage of α-syn inclusions that contain ubiquitin using adjacent 3,3′-diaminobenzidine-labeled sections indicated case-to-case variability (Table 1) ▶ . The majority (79 to 96%) of cortical LBs in the five DLB cases examined was ubiquitin-positive, whereas the percentage of GCIs that were ubiquitinated in the nine MSA cases analyzed was more variable (19 to 96%).
Figure 1.
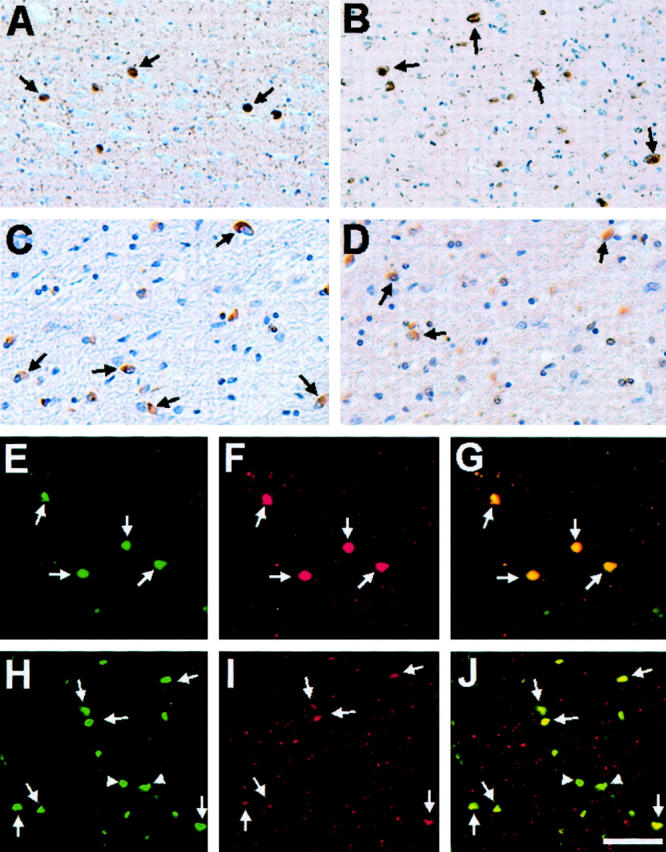
Ubiquitin immunostaining of α-syn pathological inclusions in DLB and MSA. Immunohistochemistry of cingulate cortex from a DLB patient (DLB-2) (A and B) and cerebellum from a MSA patient (MSA-6) (C and D) stained using monoclonal anti-α-syn antibodies Syn303 (A and C) and monoclonal anti-ubiquitin antibody mAb 1510 (B and D). In A and B, arrows highlight immunoreactive cortical LBs, whereas in C and D arrows indicate stained GCIs. Double-label immunofluorescence of cortical LBs in the cingulate cortex of a patient with DLB (DLB-5) (E–G) and GCIs in the cerebellum of a patient with MSA (MSA-8) (H–J) with rabbit anti-α-syn antibody SNL-4 (green, E and H) and murine anti-ubiquitin antibody mAb 1510 (red, F and I). The overlays of staining with both antibodies are shown in G and J. In H to J, arrows indicate inclusions stained with antibodies to both α-syn and ubiquitin, whereas arrowheads depict inclusions stained only with antibodies to α-syn. Scale bars: 80 μm (A, B, E–J), 40 μm (C, D).
Table 1.
Summary of Study Participants
| Case | Age (years) | Sex | PMI (hour) | % Inclusions Ub+/Syn+ |
|---|---|---|---|---|
| DLB-1 | 66 | M | 11 | 96 |
| DLB-2 | 75 | F | 6 | 91 |
| DLB-3 | 79 | M | 20.5 | 79 |
| DLB-4 | 69 | F | 12 | 87 |
| DLB-5 | 71 | M | 9 | 89 |
| DLB-6 | 90 | F | 5 | * |
| MSA-1 | 55 | M | 7 | 53 |
| MSA-2 | 67 | F | 5 | 85 |
| MSA-3 | 60 | F | 10.5 | 96 |
| MSA-4 | 43 | M | 20 | 36 |
| MSA-5 | 79 | M | 16 | 19 |
| MSA-6 | 65 | M | 43 | 49 |
| MSA-7 | 73 | F | 20.5 | 53 |
| MSA-8 | 73 | M | 8 | 72 |
| MSA-9 | 57 | F | 8 | 35 |
| NL-1 | 69 | M | 11 | n/a |
| NL-2 | 74 | F | 3.5 | n/a |
DLB, MSA, and neuropathologically normal (NL) patients used in this study are numbered in the left-most column. The right-most column indicates the percentage of LBs and GCIs that were positive for ubiquitin staining in the cingulate cortex of DLB brains or cerebellum of MSA brains, respectively. Abbreviations: PMI, post-mortem interval; n/a, not applicable; M, male; F, female.
*This brain was used for biochemical studies only and not for the quantitative studies here.
α-Syn Is Ubiquitinated in Pathological Inclusions of Diseased Brains
The high percentage of ubiquitin-immunoreactive α-syn inclusions in most DLB and some MSA brains suggests that α-syn may be ubiquitinated in these inclusions, but it does not exclude the possibility that other ubiquitinated proteins accumulate in these lesions. To determine whether α-syn within these inclusions is ubiquitinated, sequential biochemical extractions using buffers with increasing protein solubilization strengths were performed on samples from diseased brains (see Materials and Methods). Western blot analysis of biochemical fractions from DLB cingulate cortex revealed the presence of higher molecular mass (Mr) α-syn-immunoreactive species in the SDS-soluble fraction, but not in the more soluble fractions (Figure 2A) ▶ . This finding was confirmed with the anti-α-syn antibodies LB509 and Syn208, and bands similar to these higher Mr species of α-syn also were labeled by the anti-ubiquitin mAb 1510 (Figure 2A) ▶ . The presence of these higher Mr α-syn species was observed with antibodies to α-syn and ubiquitin in the SDS-soluble fraction of multiple DLB brains, but not in the SDS-soluble fraction of NL cingulate cortex (Figure 2B) ▶ . To further confirm that these higher Mr species were α-syn, the SDS-soluble fraction of a DLB brain was analyzed with additional anti-α-syn antibodies (Figure 2C) ▶ . Monomeric α-syn as well as α-syn-positive bands running at apparent Mr of ∼24 kd, ∼32 kd, and ∼40 kd were relatively more abundant compared to α-syn species of higher Mr. Protein bands with similar gel mobility as the higher Mr α-syn species, but not monomeric α-syn, were detected with mAb 1510 as well as another anti-ubiquitin antibody Conj8 (Figure 2C) ▶ , suggesting that these species represent ubiquitinated forms of α-syn. Additionally, the presence of monomeric α-syn in the SDS-soluble fraction indicates that not all α-syn in inclusions is ubiquitinated, consistent with the immunohistochemical properties of α-syn inclusions described above (Figure 1) ▶ .
Figure 2.

Insoluble α-syn in diseased brain is ubiquitinated. A: Western blot analysis of biochemically fractionated cingulate cortex from a patient with DLB (DLB-3). Immunoblots were developed with anti-α-syn antibodies LB509 and Syn208 as well as anti-ubiquitin antibody mAb 1510. Twenty μl of LS fraction (lane 1), TX fraction (lane 2), sarkosyl-soluble fraction (lane 3), and SDS-soluble fraction (lane 4) were loaded in separate lanes of 15% SDS-polyacrylamide gels. Note that the SDS-soluble fraction is four times as concentrated as each of the other fractions in that 2.5 ml/g of SDS buffer was used for tissue extraction versus 10 ml/g for each of the other fractions. Arrowhead indicates α-syn monomer (αS) and bracket indicates ubiquitin monomer (Ub). Arrows depict mono-, di-, and tri-ubiquitinated forms of α-syn. B: Western blot analysis (with antibodies Syn208 and mAb 1510) of the SDS-soluble fraction from the cingulate cortex of normal brains (NL-1, NL-2) and DLB brains (DLB-1, DLB-2, and DLB-3). Twenty μl of SDS-soluble fraction was loaded in each lane of a 15% gel. One hundred ng of recombinant human α-syn was loaded in the indicated lanes. C: Western blot analysis of the SDS-soluble fraction from the cingulate cortex of case DLB-1 using various anti-α-syn (LB509, Syn102, Syn211) and anti-ubiquitin (mAb 1510, Conj8) antibodies. Arrows depict mono-, di-, and tri-ubiquitinated forms of α-syn. D: Immunoprecipitation followed by Western blot analysis. α-Syn in the SDS-soluble fraction of the cingulate cortex of DLB-1 was isolated by immunoprecipitation with anti-α-syn antibodies. The sample was analyzed by Western blot analysis using anti-α-syn antibody LB509 and anti-ubiquitin antibody mAb 1510 (arrows, mono- and di-ubiquitinated forms of α-syn; * and **, possible ubiquitinated forms of α-syn in which ubiquitin moieties may be masking the LB509 epitope; ***, modified form of α-syn, possibly dimerized α-syn). E: Ubiquitinated α-syn from DLB brain can be deubiquitinated by UCH-L1 in vitro. SDS-soluble fraction from the cingulate cortex of NL-1 or DLB-1 was untreated or reacted with 50 nmol/L of UCH-L1. The samples were analyzed by Western blot analysis using LB509. R represents a lane loaded with 100 ng of recombinant human α-syn. The mobility of molecular mass markers (kd) is depicted on the left of each panel.
To directly determine whether these α-syn immunoreactive bands correspond to ubiquitinated α-syn, α-syn was isolated from the SDS-soluble fraction of DLB brain by immunoprecipitation. Monomeric as well as higher Mr species were detected by Western blotting with LB509 after immunopurification of α-syn. Several of the higher Mr bands, but not the α-syn monomer, were detected by mAb 1510, demonstrating that aggregated α-syn is ubiquitinated (Figure 2D) ▶ . Some protein bands, with apparent Mr of ∼24 kd and ∼32 kd (Figure 2D ▶ , arrows), that were detected with LB509 were also labeled by mAb 1510, consistent with the idea that these are mono- and di-ubiquitinated forms of monomeric α-syn. Bands detected by mAb 1510 that failed to react with LB509, such as the ∼29-kd band (Figure 2D ▶ , *) and ∼40-kd band (Figure 2D ▶ , **), may be ubiquitinated forms of α-syn wherein ubiquitination or some additional posttranslational modifications have masked the epitope recognized by anti-α-syn antibodies. On the other hand, bands recognized by LB509, but not mAb 1510 (Figure 2D ▶ , ***), may represent α-syn modified in a manner other than ubiquitination.
To provide further evidence that the higher Mr bands represent ubiquitinated α-syn, the SDS-soluble fraction from DLB brain was subjected to treatment with UCH-L1. A significant reduction in the level of higher Mr α-syn species was observed, consistent with deubiquitination of α-syn by UCH-L1 under these in vitro conditions (Figure 2E) ▶ .
A Subset of α-Syn Inclusions Are Regionally Ubiquitinated in a Mouse Model of α-Synucleinopathies
The M83 and M91 transgenic mouse lines that overexpress the A53T mutant human α-syn protein under the control of the murine PrP promoter were previously described. 19 These animals develop late-onset severe motor impairment associated with the formation of pathological inclusions containing fibrillar α-syn. 19 Inclusions in the spinal cord and pons of M83 transgenic mice were labeled by anti-α-syn antibody (Figure 3, A and D) ▶ as well as anti-ubiquitin antibody (Figure 3, B and E) ▶ in immunofluorescence microscopic analysis. By double-labeling, it was evident that a subset of α-syn inclusions in M83 mice is ubiquitin-immunoreactive, whereas many α-syn inclusions do not contain ubiquitin (Figure 3, C and F) ▶ .
Figure 3.

Ubiquitin immunostaining of α-syn inclusions in mice expressing A53T human α-syn. Double-label immunofluorescence of α-syn inclusions in the spinal cord (A–C) and pons (D–F) of M83 homozygous A53T human α-syn transgenic mice with rabbit anti-α-syn antibody SNL-4 (green, A and D) and mouse anti-ubiquitin antibody mAb 1510 (red, B and E). The overlays are shown in C and F. Arrows indicate α-syn inclusions that are not ubiquitin-positive. Scale bar, 80 μm.
α-Syn Is Ubiquitinated in Pathological Inclusions of A53T Human α-Syn Transgenic Mice
Sequential biochemical fractionation of mouse cortex and spinal cord was performed to determine whether α-syn in the inclusions of A53T human α-syn transgenic mice (lines M83 and M91) is ubiquitinated. This analysis was performed in parallel with nontransgenic mice (nTg) and M7 transgenic mice expressing wild-type human α-syn, which do not develop inclusions or display any phenotypic changes. 19 Higher Mr variants of α-syn accumulate in the SDS-soluble fraction of the spinal cord of A53T α-syn transgenic mice, but not in nTg or M7 transgenic (Figure 4A) ▶ . M83 transgenic mice predominantly accumulate inclusions in the spinal cord compared to cortex, 19 which correlates with the specific accumulation of monomeric and higher Mr α-syn species in the SDS-soluble fraction from the spinal cord of A53T α-syn transgenic mice. The abundance, however, of higher Mr species of α-syn varied in different transgenic mice (Figure 4B) ▶ , which exhibited end-stage motor impairment at the time that they were sacrificed. The amount of higher Mr α-syn-positive bands paralleled the amount of corresponding bands detected with the anti-ubiquitin antibody mAb 1510 (Figure 4B) ▶ .
Figure 4.

α-Syn is ubiquitinated in inclusions in A53T α-syn transgenic mice. A: Western blot analysis of the SDS-soluble fractions of cortex (C) and spinal cord (S) from 12-month-old nontransgenic (nTg) mice, homozygous transgenic mice expressing wild-type human α-syn (line M7), and homozygous transgenic mice expressing A53T human α-syn (line M83) using the anti-α-syn antibody LB509. Note the accumulation of α-syn and higher molecular mass species in the SDS-soluble fraction of M83 transgenic mice. B: Accumulation of higher molecular mass species of α-synuclein in the spinal cord of M83 and M91 transgenic mice coincides with presence of ubiquitin immunoreactive bands. Ten μl of each SDS-soluble fraction was loaded in separate lanes of a 15% polyacrylamide gel. Arrows indicate ubiquitinated forms of α-syn in the M91 mouse. The bracket indicates ubiquitinated proteins that are unlikely to be α-syn because they are not labeled by the anti-α-syn antibody LB509. The mobility of molecular mass markers (kd) is indicated on the left of each panel.
In Vitro Ubiquitination of Filamentous α-Syn Recapitulates the Pattern of α-Syn Ubiquitination Observed in Pathological Inclusions
To determine whether α-syn can serve as a substrate for ubiquitination, in vitro ubiquitination reactions were performed with monomeric and fibrillized recombinant human α-syn. α-syn was reacted with ubiquitin-conjugation fractions derived from a mammalian cell lysate and analyzed by Western blotting. Monomeric α-syn served as a substrate for ubiquitination under these in vitro conditions, generating a ladder of α-syn species (Figure 5) ▶ resembling the classically reported pattern of protein ubiquitination. 46 Polyubiquitinated forms of α-syn predominated when α-syn monomer was ubiquitinated, as indicated by the abundance of immunoreactive protein with molecular mass >75 kd. Filamentous α-syn was also ubiquitinated in vitro, but the pattern of higher Mr species was distinct from that generated from monomeric α-syn. Protein bands with apparent Mr of ∼24 kd and ∼32 kd, consistent with mono- and di-ubiquitinated α-syn, were the predominant species generated from ubiquitination of polymerized α-syn (Figure 5) ▶ . This pattern of α-syn ubiquitination resembled that seen in the SDS-soluble fraction of DLB brains (Figures 2 and 5) ▶ and A53T mutant mouse spinal cord (Figure 4) ▶ .
Figure 5.

In vitro ubiquitination of monomeric and fibrillar α-syn. Left: Unassembled or fibrillar α-syn (0.5 mg/ml) was subjected to in vitro ubiquitination as described in Materials and Methods. Equal volumes of unassembled α-syn, ubiquitination reaction without α-syn, ubiquitination reaction with unassembled α-syn, and ubiquitination reaction with fibrillar α-syn were loaded in separate lanes of 15% SDS-polyacrylamide gels that were transferred electrophoretically onto nitrocellulose and analyzed by Western blotting with LB509. Right: Western blot analysis (using LB509) of the SDS-soluble fraction of cingulate cortex from three DLB cases. Arrows indicate ubiquitinated forms of α-syn. The mobility of molecular mass markers (kd) is depicted to the left of the panel.
Discussion
Previous reports have documented that LBs, LNs, and GCIs are stained with antibodies to ubiquitin. 7,47-53 These studies, however, did not resolve whether the accumulation of ubiquitin in these inclusions was due to sequestration of free ubiquitin, which is a heat shock protein, or the ubiquitination of one or more proteins within the inclusions. Furthermore, the exact identities of ubiquitinated substrate(s) in the inclusions had not been resolved. These uncertainties have become important, because it has been proposed that ubiquitination of α-syn may be a prerequisite for the formation of inclusions. 30 This proposal was based on findings that patients with null mutations in the parkin gene 22 developed autosomal-recessive juvenile parkinsonism without the presence of α-syn pathological inclusions. 23,54 Parkin is associated with an E3 ubiquitin ligase activity, 25,27 and it was reported that a novel O-glycosylated isoform of α-syn (αSp22) is a substrate for ubiquitination. 30
The presence of a ladder of higher Mr species of α-syn in the SDS-soluble fraction of the cingulate cortex from DLB patients, and the ubiquitin immunolabeling of protein bands with a similar Mr suggests that α-syn in inclusions may be covalently conjugated to ubiquitin (Figure 2; A to C) ▶ . The anti-ubiquitin reactivity of these α-syn isoforms immunopurified from inclusions (Figure 2D) ▶ and their susceptibility to in vitro deubiquitination by UCH-L1 (Figure 2E) ▶ , demonstrate the existence of ubiquitinated α-syn in pathological inclusions of DLB brains. This notion is further supported by recent biochemical and protein sequencing results indicating that phosphorylated α-syn in inclusions of various α-synucleinopathies is ubiquitinated. 55 The finding here that UCH-L1 can deubiquitinate α-syn is consistent with the established hydrolytic activity of UCHs. 26,56,57 A recent study, however, has demonstrated that under certain conditions (such as μmol/L concentrations of UCH-L1 versus the nmol/L concentrations used here), UCH-L1 may exhibit an activity that elongates ubiquitin chains in protein-ubiquitin conjugates. 58
The protein bands with apparent Mr of ∼24 kd, ∼32 kd, and ∼40 kd observed in the SDS-soluble fractions of DLB brains correspond to the relatively abundant mono-, di-, and tri-ubiquitinated forms of α-syn, respectively, whereas the higher Mr bands that are less abundant may represent polyubiquitinated α-syn. It is presently unknown whether any of the 15 lysine residues within α-syn 59 are selectively targeted by the ubiquitination machinery or if ubiquitination of α-syn reflects the conjugation of monoubiquitin to various residues versus attachment of complex ubiquitin chains to a single lysine residue. The pattern, however, of closely spaced protein bands (see Figure 5 ▶ ) suggests that these isoforms are derived from the conjugation of ubiquitin chains to single lysine residues in α-syn.
The data presented here support the hypotheses that ubiquitination of α-syn is not required for α-syn inclusion formation and that α-syn aggregation precedes ubiquitination. Immunohistochemical analysis of cingulate cortex from DLB patients demonstrates that most, but certainly not all, cortical LBs contain ubiquitin (Figure 1 ▶ , Table 1 ▶ ), in agreement with previous reports. 11,32,33 An analysis of LBs and LNs in various regions of PD and DLB brains demonstrated that although most of these pathological structures were immunopositive for both α-syn and ubiquitin, lesions stained for α-syn were more numerous than those that contained both ubiquitin and α-syn, and no structures were seen to be ubiquitin-positive but α-syn-negative. 11,33 It has also been previously reported that only a subset of pale bodies, which are α-syn-containing inclusions that have been suggested to be LB precursors, are immunoreactive for ubiquitin. 32,60,61 Furthermore, consistent with a previous study, 10 it is shown here that the number of GCIs in MSA stained with anti-α-syn antibodies exceeds that stained by anti-ubiquitin antibodies (Figure 1) ▶ . Quantitative analysis revealed that the percentage of ubiquitinated α-syn containing GCIs in the cerebellum varies widely from 19 to 96% across the MSA cases studied here (Table 1) ▶ .
In a transgenic mouse model of α-synucleinopathies, in which the expression of A53T human α-syn results in motor impairment associated with formation of α-syninclusions, 19 a subset of these inclusions are immunolabeled with anti-ubiquitin antibodies. The immunoreactivity of these inclusions is, at least in part, due to the ubiquitination of α-syn, as shown biochemically in the SDS-soluble fraction from spinal cords of diseased A53T α-syn transgenic mice (Figure 4) ▶ . The extent of ubiquitination varied between animals, and the incomplete ubiquitination of α-syn inclusions further suggests that inclusion formation does not require prior ubiquitination.
In vitro ubiquitination of polymerized synuclein recapitulated the pattern of α-syn ubiquitination seen in DLB brains as well as in the α-synucleinopathy mouse model. Mono- and di-ubiquitinated forms of α-syn predominated over polyubiquitinated forms of α-syn when filamentous α-syn was used as substrate. In contrast, the amount of polyubiquitinated α-syn species generated was much greater when using monomeric α-syn as a substrate in the in vitro reaction. This finding supports the possibility that ubiquitination of α-syn in the brains of α-synucleinopathy patients occurs after polymerization of α-synuclein, and this could occur if cells contain fibril-specific ubiquitin-ligase machinery that allows recognition of α-syn polymers for ubiquitination. Mono- and di-ubiquitinated forms could accumulate if the conformation of α-syn in polymerized fibrils after conjugation with one or two ubiquitin molecules to exposed lysine residues in α-syn sterically prevents additional ligation reactions from occurring. Such fibrils, in turn, may be inefficiently recognized for degradation by proteasomes because tetra-ubiquitin appears to be the shortest ubiquitin chain that binds well to proteasomes. 62
Although the data here suggest that ubiquitination likely occurs after the formation of α-syn inclusions, it remains possible that under certain pathophysiological conditions, ubiquitination of inclusions may contribute to the pathogenesis of α-synucleinopathies. For instance, the attempts by cells to remove inclusions via the ubiquitin-proteasomal pathway may result in overall diminished proteasomal activity, which could have toxic effects because of the accumulation of proteins that would normally be degraded. This, in addition to the persistence of α-syn inclusions, could have detrimental effects on cellular viability.
The existence of ubiquitinated α-syn in pathological inclusions and the ubiquitination of α-syn by ubiquitin conjugation fractions of mammalian cell lysates raise the question as to whether α-syn is degraded by proteasomes. Several recent reports examining the mode of degradation of α-syn in cell culture models have yielded conflicting results. Studies using HEK293 cells showed that proteasomes did not degrade α-syn, 63,64 but other studies in SH-SY5Y and COS-7 cells suggested that proteasomal inhibition leads to a increase in levels of nonubiquitinated synuclein, 58,65,66 thereby implicating the 20S proteasome in α-syn degradation. Although cell-specific differences or epitope tagging of α-syn may underlie the discrepancies among these studies, ubiquitination of untagged α-syn has yet to be reported in cultured cells.
Several observations presented here argue against the idea that ubiquitination of α-syn is required for inclusion formation: 1) a significant percentage of α-syn inclusions in human brains and a transgenic mouse model of α-synucleinopathies do not contain ubiquitin (Figures 1 and 3 ▶ , Table 1 ▶ ), 2) nonubiquitinated species of α-syn in SDS-soluble fractions from diseased brains are more abundant than ubiquitinated forms of α-syn (Figures 2 and 4) ▶ , and 3) the pattern of α-syn ubiquitination observed in diseased brains closely resembles the pattern of in vitro ubiquitination obtained when filamentous rather than monomeric α-syn is used as a substrate (Figure 5) ▶ .
The ubiquitination of α-syn may be a secondary event that occurs late in the process of inclusion formation. It is speculated that ubiquitination reflects attempts by affected cells to target misfolded proteins for proteasomal degradation but that α-syn fibrillization and inclusion formation are not ubiquitination-dependent processes. This is consistent with the view that proteasomal dysfunction may contribute to the pathobiology of α-synucleinopathies, 31 reducing the ability of cells to remove aberrant aggregates of α-syn and further enhancing cellular dysfunction. Further work, however, is required to test these hypotheses and improve our understanding of disease mechanisms in α-synucleinopathies.
Acknowledgments
We thank Dr. C. Pickart for kindly providing anti-ubiquitin antibody Conj8, the Michigan Alzheimer’s Disease Research Center for providing frozen brain tissue, Mrs. S. Hamilton for technical assistance, and the families of patients who made this research possible.
Footnotes
Address reprint requests to Dr. Virginia M.-Y. Lee, Ph.D., Center for Neurodegenerative Disease Research, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, 3600 Spruce St., 3rd floor Maloney Bldg., Philadelphia, PA 19104. E-mail: vmylee@mail.med.upenn.edu.
Supported by the National Institutes of Health [training grant T32 AG 00255 (Training in Age-Related Neurodegenerative Diseases) to D. M. S. and A. C. P.], a Pioneer Award from the Alzheimer Association, and the Canadian Institutes of Health Research (fellowship to B. I. G.).
V. M.-Y. L. is the John H. Ware III Chair of Alzheimer’s Research.
References
- 1.Spillantini MG, Goedert M: The α-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann NY Acad Sci 2000, 920:16-27 [DOI] [PubMed] [Google Scholar]
- 2.Duda JE, Lee VM-Y, Trojanowski JQ: Neuropathology of synuclein aggregates. J Neurosci Res 2000, 61:121-127 [DOI] [PubMed] [Google Scholar]
- 3.Goedert M: α-Synuclein and neurodegenerative diseases. Nat Rev Neurosci 2001, 2:492-501 [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL: Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276:2045-2047 [DOI] [PubMed] [Google Scholar]
- 5.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O: Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 1998, 18:106-108 [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M: α-Synuclein in Lewy bodies. Nature 1997, 388:839-840 [DOI] [PubMed] [Google Scholar]
- 7.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E: Abnormal accumulation of NACP/α-synuclein in neurodegenerative disorders. Am J Pathol 1998, 152:367-372 [PMC free article] [PubMed] [Google Scholar]
- 8.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H: NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett 1997, 239:45-48 [DOI] [PubMed] [Google Scholar]
- 9.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM-Y, Trojanowski JQ, Iwatsubo T: Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998, 152:879-884 [PMC free article] [PubMed] [Google Scholar]
- 10.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M: Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 1998, 251:205-208 [DOI] [PubMed] [Google Scholar]
- 11.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 1998, 95:6469-6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conway KA, Harper JD, Lansbury PT: Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med 1998, 4:1318-1320 [DOI] [PubMed] [Google Scholar]
- 13.Giasson BI, Uryu K, Trojanowski JQ, Lee VM-Y: Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem 1999, 274:7619-7622 [DOI] [PubMed] [Google Scholar]
- 14.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M: Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J Biol Chem 1999, 274:9843-9846 [DOI] [PubMed] [Google Scholar]
- 15.Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van der PH, Probst A, Kremmer E, Kretzschmar HA, Haass C: Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci 2000, 20:6365-6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G: Neuropathology in mice expressing human α-synuclein. J Neurosci 2000, 20:6021-6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feany MB, Bender WW: A Drosophila model of Parkinson’s disease. Nature 2000, 404:394-398 [DOI] [PubMed] [Google Scholar]
- 18.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L: Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287:1265-1269 [DOI] [PubMed] [Google Scholar]
- 19.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y: Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002, 34:521-533 [DOI] [PubMed] [Google Scholar]
- 20.Auluck PK, Chan HY, Trojanowski JQ, Lee VM-Y, Bonini NM: Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295:865-868 [DOI] [PubMed] [Google Scholar]
- 21.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH: The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395:451-452 [DOI] [PubMed] [Google Scholar]
- 22.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392:605-608 [DOI] [PubMed] [Google Scholar]
- 23.Giasson BI, Lee VM-Y: Parkin and the molecular pathways of Parkinson’s disease. Neuron 2001, 31:885-888 [DOI] [PubMed] [Google Scholar]
- 24.Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem 1998, 67:425-479 [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM: Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 2000, 97:13354-13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imai Y, Soda M, Takahashi R: Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 2000, 275:35661-35664 [DOI] [PubMed] [Google Scholar]
- 27.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000, 25:302-305 [DOI] [PubMed] [Google Scholar]
- 28.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM: Parkin ubiquitinates the α-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 2001, 7:1144-1150 [DOI] [PubMed] [Google Scholar]
- 29.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R: An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 2001, 105:891-902 [DOI] [PubMed] [Google Scholar]
- 30.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ: Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 2001, 293:263-269 [DOI] [PubMed] [Google Scholar]
- 31.McNaught KS, Jenner P: Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett 2001, 297:191-194 [DOI] [PubMed] [Google Scholar]
- 32.Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT: Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J Neuropathol Exp Neurol 1998, 57:334-337 [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT: α-Synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol (Berl) 2000, 99:352-357 [DOI] [PubMed] [Google Scholar]
- 34.Rideout HJ, Larsen KE, Sulzer D, Stefanis L: Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem 2001, 78:899-908 [DOI] [PubMed] [Google Scholar]
- 35.McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jennert P, Olanow CW: Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem 2002, 81:301-306 [DOI] [PubMed] [Google Scholar]
- 36.Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM-Y: A panel of epitope-specific antibodies detects protein domains distributed throughout human α-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res 2000, 59:528-533 [DOI] [PubMed] [Google Scholar]
- 37.Duda JE, Giasson BI, Mabon ME, Lee VM-Y, Trojanowski JQ: Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 2002, 52:205-210 [DOI] [PubMed] [Google Scholar]
- 38.Haas AL, Bright PM: The immunochemical detection and quantitation of intracellular ubiquitin-protein conjugates. J Biol Chem 1985, 260:12464-12473 [PubMed] [Google Scholar]
- 39.Schmidt ML, Murray J, Lee VM-Y, Hill WD, Wertkin A, Trojanowski JQ: Epitope map of neurofilament protein domains in cortical and peripheral nervous system Lewy bodies. Am J Pathol 1991, 139:53-65 [PMC free article] [PubMed] [Google Scholar]
- 40.Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VM-Y, Trojanowski JQ: Immunohistochemical and biochemical studies demonstrate a distinct profile of α-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol 2000, 59:830-841 [DOI] [PubMed] [Google Scholar]
- 41.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, De Vos RA, Wilcock GK, Jellinger KA, Perry RH: Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996, 47:1113-1124 [DOI] [PubMed] [Google Scholar]
- 42.Gilman S, Low PA, Quinn N, Albanese A, Ben Shlomo Y, Fowler CJ, Kaufmann H, Klockgether T, Lang AE, Lantos PL, Litvan I, Mathias CJ, Oliver E, Robertson D, Schatz I, Wenning GK: Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999, 163:94-98 [DOI] [PubMed] [Google Scholar]
- 43.Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM-Y, Trojanowski JQ: Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol 2000, 157:1439-1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM-Y, Trojanowski JQ: Tau and α-synuclein pathology in amygdala of parkinsonism-dementia complex patients of Guam. Am J Pathol 2002, 160:1725-1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giasson BI, Murray IV, Trojanowski JQ, Lee VM-Y: A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem 2001, 276:2380-2386 [DOI] [PubMed] [Google Scholar]
- 46.Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA: Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci USA 1980, 77:1783-1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ: Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol 1988, 155:9-15 [DOI] [PubMed] [Google Scholar]
- 48.Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y: Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol (Berl) 1988, 75:345-353 [DOI] [PubMed] [Google Scholar]
- 49.Papp MI, Kahn JE, Lantos PL: Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989, 94:79-100 [DOI] [PubMed] [Google Scholar]
- 50.Kato S, Nakamura H: Cytoplasmic argyrophilic inclusions in neurons of pontine nuclei in patients with olivopontocerebellar atrophy: immunohistochemical and ultrastructural studies. Acta Neuropathol (Berl) 1990, 79:584-594 [DOI] [PubMed] [Google Scholar]
- 51.Kato S, Nakamura H, Hirano A, Ito H, Llena JF, Yen SH: Argyrophilic ubiquitinated cytoplasmic inclusions of Leu-7-positive glial cells in olivopontocerebellar atrophy (multiple system atrophy). Acta Neuropathol (Berl) 1991, 82:488-493 [DOI] [PubMed] [Google Scholar]
- 52.Arima K, Murayama S, Mukoyama M, Inose T: Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 1. Neuronal cytoplasmic inclusions. Acta Neuropathol (Berl) 1992, 83:453-460 [DOI] [PubMed] [Google Scholar]
- 53.Murayama S, Arima K, Nakazato Y, Satoh J, Oda M, Inose T: Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 2. Oligodendroglial cytoplasmic inclusions. Acta Neuropathol (Berl) 1992, 84:32-38 [DOI] [PubMed] [Google Scholar]
- 54.Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H: An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord 2000, 15:884-888 [DOI] [PubMed] [Google Scholar]
- 55.Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM-Y, Trojanowski JQ, Mann D, Iwatsubo T: Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J Biol Chem 2002, 277:49071-49076 [DOI] [PubMed] [Google Scholar]
- 56.Pickart CM, Rose IA: Ubiquitin carboxyl-terminal hydrolase acts on ubiquitin carboxyl-terminal amides. J Biol Chem 1985, 260:7903-7910 [PubMed] [Google Scholar]
- 57.Wilkinson KD: Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin Cell Dev Biol 2000, 11:141-148 [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT, Jr: The UCH-L1 gene encodes two opposing enzymatic activities that affect α-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111:209-218 [DOI] [PubMed] [Google Scholar]
- 59.Jakes R, Spillantini MG, Goedert M: Identification of two distinct synucleins from human brain. FEBS Lett 1994, 345:27-32 [DOI] [PubMed] [Google Scholar]
- 60.Leigh PN, Probst A, Dale GE, Power DP, Brion JP, Dodson A, Anderton BH: New aspects of the pathology of neurodegenerative disorders as revealed by ubiquitin antibodies. Acta Neuropathol (Berl) 1989, 79:61-72 [DOI] [PubMed] [Google Scholar]
- 61.Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN: Relationships between Lewy bodies and pale bodies in Parkinson’s disease. Acta Neuropathol (Berl) 1992, 83:525-529 [DOI] [PubMed] [Google Scholar]
- 62.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM: Recognition of the polyubiquitin proteolytic signal. EMBO J 2000, 19:94-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ancolio K, Alves DC, Ueda K, Checler F: α-Synuclein and the Parkinson’s disease-related mutant Ala53Thr-α-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett 2000, 285:79-82 [DOI] [PubMed] [Google Scholar]
- 64.Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM-Y, Ischiropoulos H: Induction of α-synuclein aggregation by intracellular nitrative insult. J Neurosci 2001, 21:8053-8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM: Degradation of α-synuclein by proteasome. J Biol Chem 1999, 274:33855-33858 [DOI] [PubMed] [Google Scholar]
- 66.Tofaris GK, Layfield R, Spillantini MG: α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 2001, 509:22-26 [DOI] [PubMed] [Google Scholar]