

Abstract
Despite intensive high-dose chemotherapy and autologous hematopoietic stem cell transplantation, disseminated neuroblastoma (NB) frequently proves to be chemosensitive but not chemocurable, and more often so in NB-presenting MYCN amplification. To assess the direct relationship between the MYCN oncogene and chemoresistance acquisition during NB metastatic dissemination, we have studied MYCN and MDR1 genes using the human IGR-N-91 ectopic xenograft metastatic model. This characterized experimental in vitro model includes human neuroblasts derived from a subcutaneous primary tumor xenograft, disseminated blood cells, myocardium, and bone marrow (BM) metastatic cells. All IGR-N-91-derived neuroblasts harbor a consistent MYCN genomic content but, unlike primary tumor xenograft, BM, and myocardium, human neuroblasts elicit a concomitant increase in MYCN and MDR1 transcripts levels, consistent with chemoresistance phenotype and active P-gp. In contrast, no variation of MRP1 transcript level was associated with the metastatic process in this model. Using an MDR1 promoter-CAT construct, we have shown that the MycN protein activates MDR1 transcription both in exogenous transient MYCN-transfected SK-N-SH cells and in endogenous BM metastatic neuroblasts with an increase in the MYCN transcript level. Band-shift experiments indicate that IGR-N-91 cells enriched with the MycN transcription factor do bind to two E-box motifs localized within the MDR1 promoter. Overall, our data indicate that MYCN overexpression increment contributes to the acquired drug resistance that occurs throughout the NB metastatic process.
Neuroblastoma (NB), the most common solid tumor of early childhood, originating in tissues of the sympathetic nervous system, presents extreme heterogeneity clinically, histologically, and genetically. From a clinical point of view, NBs are classified according to stage (localized stages 1, 2, and 3, and metastatic stage 4) and the child’s age at diagnosis (<1 year and >1 year). On presentation, stage-4 NBs systematically present involved bone marrow (BM). From a biological point of view, following cytogenetic and genetic research throughout the past 2 decades, NB tumors can be classified into three types depending on genomic and genetic alterations. 1-3 Among genotypic changes, MYCN oncogene activation through amplification is the hallmark of advanced disease and very severe prognosis. 4,5 According to both clinical and biological criteria, high-risk NBs include MYCN-amplified NB, whatever the stage, and stage 4 NBs in children older than 1 year old.
High-risk NB treatments include initial induction chemotherapy followed by high-dose chemotherapy supported by myeloablative treatment with autologous hematopoietic stem cell transplantation; 13-cis-retinoic acid is proposed in maintenance regimens. 6,7 However, despite such an intensive multimodal therapy, the majority of high-risk patients (50% of all NBs) present a poor prognosis (overall survival rate of 30%). Generally, treatment failure in patients with disseminated cancer, including NB, can be primarily because of metastases that are resistant to conventional therapies. In fact, drug-resistance acquisition by tumor cells is a multifactorial process that includes alterations of molecules involved in DNA repair and drug metabolism, activation of oncogenes (bcl-2, c-fos, ras, p53, MDM2), as well as modulation of detoxifying molecules such as multidrug resistance transporters (MDR1, MRP, LPR). 8,9
It has been demonstrated that apoptotic pathways contribute to the cytotoxic action of most chemotherapeutic agents. 10 In this regard, p53 is a critical factor and p53 mutation occurring in ∼60% of human cancers is an important mechanism of de novo chemoresistance in many of them. 11 In a high percentage of NB tumors so far surveyed at diagnosis, p53 is of wild type but inactive. Recently, it was shown that p53 is frequently mutated after NB cytotoxic chemotherapy. 12 Such an irreversible loss of p53 function confers high-level multidrug resistance in NB cell lines. 13 Alterations of various apoptotic proteins, such as caspase-8 or caspase-10, have also been related to malignancy and apoptotic resistance in MYCN-amplified NB. 14,15 Furthermore, in many solid, chemotherapy-treated tumors from various organs (colon, kidney, liver), the increased expression of two well-known genes, the MDR1 gene encoding the transmembrane transport protein P-glycoprotein (P-gp) 16 and the multidrug resistance-associated protein (MRP) gene, characterize multidrug resistance. 17 These proteins belong to the superfamily of ABC transporters that act as energy-dependent efflux pumps for numerous substrates with varying chemical structures, including anti-cancer drugs. 18
In our laboratory, an experimental NB metastatic model has been obtained in vitro from high-risk NB BM cells. This physiopathological experimental model, the IGR-N-91 human NB xenograft, includes human neuroblasts in vitro derived from a subcutaneous primary tumor xenograft (PTX), disseminated blood cells, myocardium (Myoc), and BM metastatic cells, as previously characterized. 19 Variations of gene transcript levels, in particular the co-activation of MYCN and MDR1 genes, were observed in metastatic neuroblasts from this model. MycN protein increase is clearly related to NB tumor angiogenesis and invasiveness. 20-22 Furthermore, MYCN amplification recorded at diagnosis does not vary during the tumor’s progression. 23 Nonetheless, a direct MycN implication in chemoresistance acquisition has not yet been established throughout the metastatic process of human NB in the absence of drug treatment. Therefore, we have examined MDR1 gene status in the IGR-N-91 metastatic model and its relation to MYCN expression. This study shows that the MycN transcription factor directly regulates MDR1 gene expression in human metastatic NB cell lines.
Materials and Methods
Cell Lines and Culture
A human NB cell line SK-N-SH was purchased from the European Collection of Cell Cultures (ECACC, Wiltshire, UK). An IGR-N-91 cell line was established in our laboratory from an involved BM collected from a stage 4 NB belonging to an 8-year-old boy. 19 Human IGR-N-91 NB cells were injected subcutaneously into nude mice (Figure 1) ▶ : a PTX cell line, blood cells, Myoc, and BM sublines were obtained from mechanically dissociated tumor samples and were grown as previously described. 19 Each cell line was cultured in Dulbecco’s modified Eagle’s medium supplemented with 2 mmol/L l-glutamine, 10 μg/ml gentamicin, and 10% fetal calf serum (BioMedia, Canada Inc. Drummondville, Canada), at 37°C in a 5% CO2 humidified atmosphere. The seeding density varied according to the type of experiment: for methyl tetrazolium sulfate (MTS) study (Promega, Madison, WI), cells (1 × 104) were seeded into 96-well tissue culture plates (Costar, France); for apoptotic studies, cells (1.5 × 106) were seeded into 25-cm2 tissue culture flasks in 5 ml of culture medium. cis-platin (CDDP) and etoposide (VP16) (Sigma, St. Louis, MO) were both dissolved in dimethyl sulfoxide, added the next day for 48 hours, and cells were harvested for protein and mRNA analyses. An equal volume of vehicle control (dimethyl sulfoxide) was used to treat control cells. Results are the mean ± SD of three independent experiments and duplicates were used in each individual experiment.
Figure 1.
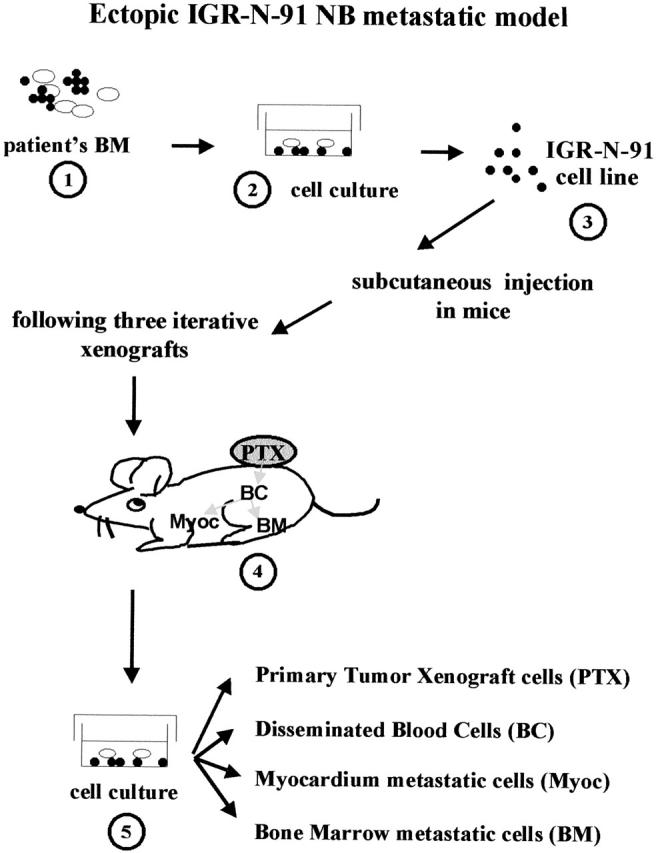
The IGR-N-91-derived human xenograft NB model. The IGR-N-91 cell line was established from an involved BM collected from a high-risk NB (stage 4-NB, 8-year-old boy). 19 Neuroblasts were injected subcutaneously into nude mice and a PTX was isolated. The PTX neuroblasts were then minced to be subcutaneously xenografted to other mice. This procedure was repeated three times. Blood-disseminated as well as metastatic neuroblasts from the Myoc and BM were then cultured on bovine corneal extracellular matrix. These malignant human neuroblasts were further subcultured in vitro to established cell lines: PTX and blood cells, Myoc, and BM metastatic sublines.
Flow Cytometry Analysis and P-gp Functionality
Cells were cultured in 25-cm2 flasks as described above. Cells were exposed to various concentrations of drugs for 48 hours and then washed with phosphate-buffered saline (PBS), trypsinized, collected by centrifugation, and fixed in 70% ethanol. After having been washed twice with PBS, the cells were incubated for 15 minutes at room temperature in PBS containing 100 μg/ml RNase A (Sigma, St. Louis, MO) and 10 μg/ml propidium iodide (Sigma) and cell-cycle distribution was determined by flow cytometric analysis of DNA content by FACScalibur (Becton Dickinson). For assessment of P-gp function, 5 × 105 cells were trypsinized and incubated at 37°C for 2 hours in a complete culture medium containing 0.5 μg/ml rhodamine-123 (Rho-123; Calbiochem, La Jolla, CA). For uptake studies, the reaction was stopped at 4°C and samples were analyzed on an XL/MCL flow cytometer (Beckman, Coulter, France). For efflux studies, cells were washed and further incubated for 30 minutes at 37°C, in a Rho-123-free medium before flow cytometric analysis. Results are presented as histograms of Rho-123 fluorescence and expressed as an uptake/efflux ratio of mean fluorescence intensity. Control flow cytometric materials were human epidermoid carcinoma cell lines, KB-3–1, and their multidrug adriamycin-resistant KB-A1 (a gift from Dr. M. Gottesman, National Cancer Institute, Bethesda, MD, with the kind assistance of Dr. J. Y. Charcosset, Toulouse, France).
Western Blot Analysis
Western blots were performed on cultured cells homogenized in RIPA buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholate (DOC), 0.1% sodium dodecyl sulfate) on ice for 15 minutes. Protein extracts (50 μg/lane) were loaded on a 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then developed with ECL enhancer (Amersham Pharmacia Biotech). Immunoblots were probed with the following antibodies diluted at 1/500 for MycN (Ab-1, Oncogene Research), 1/200 for Max (C124, Santa Cruz), 1/80 for c-myc (Ab-1, Oncogene Research), 1/40 for P-gp (Ab-1, Oncogene Research), 1/1000 for p53 (DO-7, DAKO), 1/25 for Bcl-2 (C124, DAKO). Controls were performed using β-actin antibody (1/2000, monoclonal antibody 1501; Chemicon International). Protein concentrations were determined by the Bradford method.
Isolation of RNA and Northern Blotting
Malignant neuroblasts were trypsinized at the end of the growth exponential phase. The resulting pellet was dipped and stored in liquid nitrogen. Nucleic acids were extracted from cell lines using a Qiagen RNA/DNA midi kit according to the conditions recommended by the supplier. The quality of gDNA and total RNA was then assessed by agarose gel electrophoresis. Northern blot hybridizations were performed as previously described, 19 with 10 μg of total RNA and using [32P]-labeled probes (Amersham Pharmacia, Uppsala, Sweden). Qualitative and quantitative controls of the RNA preparations were performed through ethidium bromide staining and rehybridization of the membrane with an actin probe. Blots were exposed for various periods of time to Amersham Hyper Films M.P. The gene transcript levels were determined by densitometric scanning of the autoradiograms, obtained at various times of exposure, using a Phosphorimager (Storm 840, Molecular Dynamics). The human MDR1 gene probe was the cDNA probe HDR5A, which encompasses a coding region of the gene (a gift from Dr. M. Gottesman, National Cancer Institute, Bethesda, MD). The human MYCN probe was pNb-1, which covers the second exon (a gift from Dr. M. Schwab, German Cancer Research Center, Heidelberg, Germany). The human MRP1 gene probe was a 1-kb EcoR1 cDNA fragment (a gift from Dr. Susan Cole, Kingston, Canada).
Real-Time Quantitative Polymerase Chain Reaction (PCR) Analysis
Nucleic acids were extracted from cell lines using a Qiagen RNA/DNA purification kit (Qiagen, Hilden Germany). The RNAs (1 μg) were subjected to quantitative real-time reverse transcriptase (RT)-PCR analyses. The quality of gDNA and total RNAs was then assessed by agarose gel electrophoresis. The MYCN copy number and level of expression using TaqMan 5′ nuclease fluorigenic real-time quantitative PCR assay were measured as previously described. 24 The technique used the ABI Prism 7700 sequence detection system (PE Biosystems). Ct values (defined as the fractional cycle number at which the fluorescence generated by cleavage of the probe crosses a fixed threshold) were determined. Calibration curves plotting Ct against reference DNA quantity or cDNA quantity were generated and the gene copy number or cDNA level for the test sample were determined by extrapolation. Control SK-N-SH cells with one MYCN gene copy per haploid genome were used. The MYCN gene copy number in the sample was normalized by a copy number of two internal control genes: GAPDH and albumin. PCR primers for the MYCN gene target were as follows: gMYCN forward primer 5′-GGC GTT CCT CCT CCA ACA C-3′, gMYCN reverse primer 5CGT TTG AGG ATC AGC TCG C-3′, and the TaqMan probe FAM 5′-ACA TTC ACC ATC ACT GTG CGT CCC AAG- 3′TAMRA. MYCN expression levels were quantified by using the following primers: cMYCN forward primer 5′-CAC CCT GAG CGA TTC AGA TGA-3′, cMYCN reverse primer 5′-CCG GGA CCC AGG GCT-3′ and the same TaqMan probe as for gDNA. The calibration curve was generated using cDNA from SK-N-SH as a relative reference of MYCN expression. MDR1 and MRP1 gene expression levels were quantified by using the following primers: cMDR1 forward primer: 5′-GTC CCA GGA GCC CAT CCT-3′, cMDR1 reverse primer 5′-TGT ATG TTG GCC TCC TTT GCT-3′, and the TaqMan probe FAM 5′-TGA CTG CAG CAT TGC TGA GAA CAT TGC-3′TAMRA; cMRP1 forward primer 5′-TGG TGC CCG TCA ATG CTG-3′, cMRP1 reverse primer 5′-CGA TTG TCT TTG CTC TTC ATG TG-3′, and the TaqMan probe FAM 5′-ATG GCG ATG AAG ACC AAG ACG TAT CAG GT-3′TAMRA. Samples were normalized using 18S mRNA expression with the following primers: 18S forward primer 5′-CGG CTA CCA CAT CCA AGG AA-3′, 18S reverse primer 5′-GCT GGA ATT ACC GCG GCT-3′, and the TaqMan probe FAM 5′-TGC TGG CAC CAG ACT TGC CCT C-3′TAMRA.
Transient Transfection Analysis
The plasmids used for transfection and CAT assays were pMDR1-CAT containing the MDR1 gene promoter (−4741, +286) just ahead of the CAT gene (kindly donated by Dr. K. Cowan, Omaha, NE), pMYCNhu containing the MYCN gene under a cytomegalovirus promoter (a gift from Dr. M. Schwab, Heidelberg, Germany), pSVECAT (American Type Culture Collection, Rockville, MD) containing the chloramphenicol acetyltransferase (CAT) gene under the control of the SV40 early region used as a positive control with a high promoter strength, pSb1 (American Type Culture Collection, Rockville, MD) with a low background promoterless CAT expression vector used as a negative control, and pCH110 (Pharmacia LKB), which contains the β-galactosidase gene under the control of the SV40 early promoter.
Malignant human neuroblasts (2 × 106), plated 24 hours in a 100-mm plastic Petri dish, were transfected using a CaPO4 precipitation procedure after Gorman and colleagues 25 with a mixture of CAT (10 μg pMDR1) + pCH110 (5 μg) plasmids. The SK-N-SH cell line was transfected with 1 μg pMYCNhu + 5 μg pCH110. The cells were exposed for 24 hours to the CaPO4/DNA precipitate, shocked for 2 minutes with 10% buffered glycerol, then rinsed with culture medium and incubated further in fresh medium for 24 hours. The cells were then washed, scrapped off, and frozen and thawed four times. The cell extracts were processed for β-galactosidase and CAT assays, as reported elsewhere. 25,26 For CAT assays, cell extracts were incubated with [C14]-chloramphenicol and acetyl-CoA for 3 hours. The resulting material was submitted to silica thin-layer chromatography in chloroform methanol, 19/1 (v/v), to separate nonacetylated into monoacetylated and diacetylated chloramphenicol. Autoradiography of silicate plates and scrapping of spots allowed CAT activity to be measured, which was expressed as the ratio between acetylated (monoacetylated and diacetylated)/total [C14]-chloramphenicol. Each set of experiments was repeated at least twice. Acetyl CoA was purchased from Pharmacia LKB (Les Ulis, France) and silica gel plates from OSI France. [C14]-Chloramphenicol (50 μCi/μmol) was from Amersham.
Gel Shift Assays
Nuclei from different cell lines were prepared as described previously. 27 To perform the gel shift experiments, we chose oligonucleotides containing two putative E-box sequences, CACGTG at position −272 and −444 located within the proximal promoter region of the human MDR1 gene 28 as shown in Figure 5C ▶ . Binding reactions for band shift assays were performed in 15 μl of a reaction mixture containing 10 mmol/L Hepes buffer, pH 7.9, 30 mmol/L KCl, 9 mmol/L MgCl2, 9 mmol/L spermidine, 0.5 mmol/L dithiothreitol, 10% glycerol, 5 μg/ml Boehringer-Mannheim protease inhibitor cocktail, 1.5 μg of poly(dI-dC), and 1 μg of sonicated salmon sperm DNA. Ten μg of nuclear protein extract were preincubated in this mixture for 5 minutes at 4°C. One ng of kinase-labeled double-stranded oligonucleotide as a probe and competitor oligonucleotides was then added, and incubated for 15 minutes on ice. To prove the binding specificity, reactions were performed in the presence of 25-fold excess of unlabeled probe. DNA-protein complexes were loaded onto a low ionic strength 4% native polyacrylamide gel (1:29 acrylamide/bisacrylamide ratio) in 0.25× TBE (89 mmol/L Tris, 89 mmol/L borate, 2.0 mmol/L ethylenediaminetetraacetic acid), and electrophoresed at 12 V/cm. After electrophoresis, the gel was dried and exposed to an X-ray film at −70°C with an intensifying screen.
Figure 5.
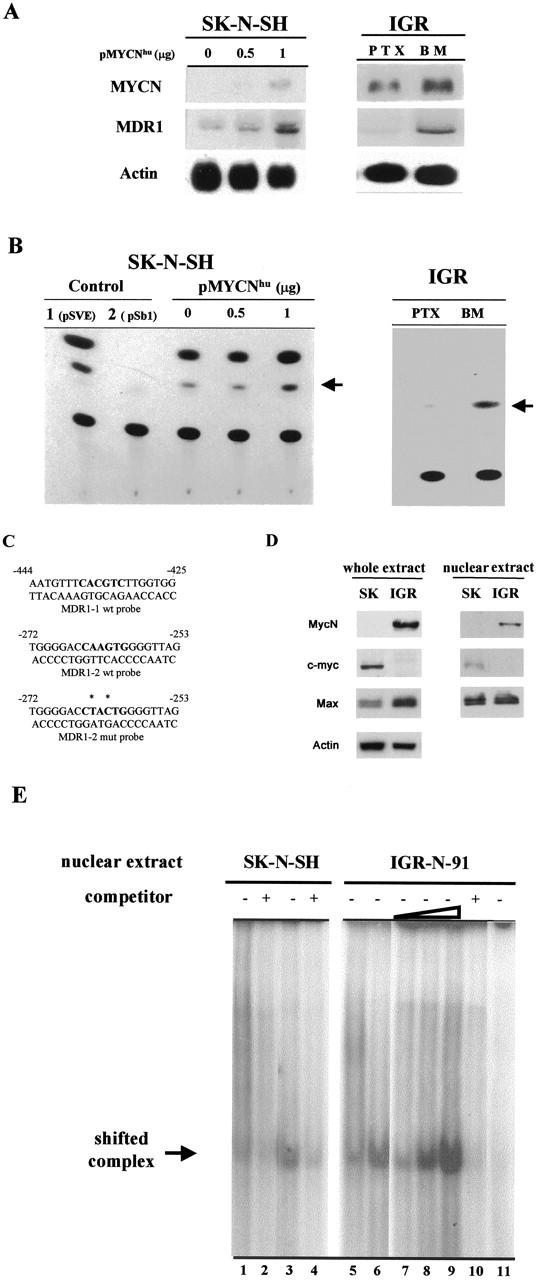
MycN-mediated induction of MDR1 promoter activity. A: IGR xenograft model (IGR): MYCN and MDR1 transcript levels for MYCN-transfected SK-N-SH neuroblasts and for PTX and BM neuroblasts. Northern blots of 10 μg total from parental SK-N-SH cells, from 0.5 and 1 μg of pMYCNhu-transfected SK-N-SH cells (left), and from PTX and BM neuroblasts (right), hybridized with 32P-labeled cDNA probes. Autoradiography of actin probe blotting demonstrated similar loadings. MYCN and MDR1 gene mRNA levels increase in parallel. B: CAT activity of the pMDR1-CAT construct transfected into the SK-N-SH and IGR cells. The SK-N-SH neuroblasts were co-transfected with 0.5 or 1 μg of pMYCNhu and 10 μg of pMDR1-CAT. The PTX and BM cells were transfected with 10 μg of pMDR1-CAT construct. Controls were performed in SK-N-SH cells transfected with 5 μg of pSVE-CAT construct or co-transfected with 10 μg of pSb1-CAT + 1 μg pMYCNhu constructs. PSVE and pSb1 constructs are used as positive (1) or negative (2) controlwith strong or weak promoter activity. The experiment was repeated at least twice and a representative transfection is shown. C: Synthetic oligonucleotides are used for gel shift assays. They correspond to the two E-boxes located within the proximal promoter region of the MDR1 gene, either wild-type (MDR1-1 wt and MDR1-2 wt) or mutated (MDR1-2mut) probes. The E-box binding sites are indicated in bold, and mutated bases with asterisks. D: Absence of c-myc protein expression in MYCN-amplified neuroblasts (IGR-N-91), and its presence in MYCN nonamplified neuroblasts (SK-N-SH), as demonstrated by immunoblots of total and nuclear extracts. Max expression is higher in whole extracts of IGR-N-91 cells. E: Electrophoretic mobility shift of oligonucleotide/nuclear protein complexes. Gel shifts were performed using two labeled double-stranded oligonucleotides, MDR1-1 wt and MDR1-2 wt incubated with nuclear extracts from SK-N-SH and IGR-N-91 neuroblasts, without (lanes 1, 3, 5–9, 11) or with (lanes 2, 4, 10) a 25-fold excess of respective competitor. A faint shifted complex is noted with the MDR1-1 probe, and SK-N-SH or IGR-N-91 nuclear extracts (lanes 1 and 5), whereas a stronger shifted complex is evidenced with the MDR1-2 probe (lanes 3, 6–9) with SK-N-SH or IGR-N-91 extracts. No complex is observed in the presence of the MDR1-2 mutated probe or IGR-N-91 nuclear extracts (lane 11).
Statistical Analysis
The results of the MTS, cell death (fluorescence-activated cell sorting analysis), and TaqMan assays were analyzed using an unpaired Student’s t-test.
Results
MYCN Status in NB Cell Lines
MYCN genomic content and expression in NB cell lines were determined by quantitative RT-PCR using the TaqMan procedure (Table 1) ▶ . The SK-N-SH (SK) cells are not MYCN-amplified (1 copy/haploid genome) in contrast with IGR-N-91 (IGR) cells, which are MYCN-amplified (350 copies). The various metastatic sublines derived from the IGR-N-91 NB xenograft model are also MYCN-amplified, with a gDNA copy number, which does not vary significantly within the xenograft model (300 copies). In this model, however, MYCN-transcript levels of Myoc and BM metastatic cells are higher than that of primary tumor (PTX) cells: 347 ± 10 mRNA expression/18S in PTX versus 404 ± 8 in Myoc (P ≤ 0.01) and 545 ± 14 in BM (P ≤ 0.001) (Figure 2A) ▶ . RT-PCR data were confirmed by Northern blotting (Figure 2B) ▶ and MYCN mRNA correlated with protein levels as shown in Figure 3A ▶ .
Table 1.
MYCN Amplification and Expression Status in Neuroblastoma Cell Lines
| Cell line | gDNA copy number/haploid genome | mRNA expression/18S |
|---|---|---|
| SK-N-SH | 1 | 1 |
| IGR-N-91 | 350 | 450 |
| PTX | 300 | 350 |
| BC | 300 | 200 |
| Myoc | 300 | 400 |
| BM | 300 | 550 |
PTX, primary tumor xenograft; BC, blood cells; Myoc, myocardium; BM, bone marrow.
The MYCN copy number and mRNA level expression were measured by real-time quantitative PCR using the TaqMan 5′ nuclease fluorigenic assay. The SK-N-SH cell line is not MYCN-amplified (1 copy) while the IGR-N-91 cell line and the different NB cell lines of the IGR xenograft model harbor 350 copies and 300 copies, respectively.
Figure 2.
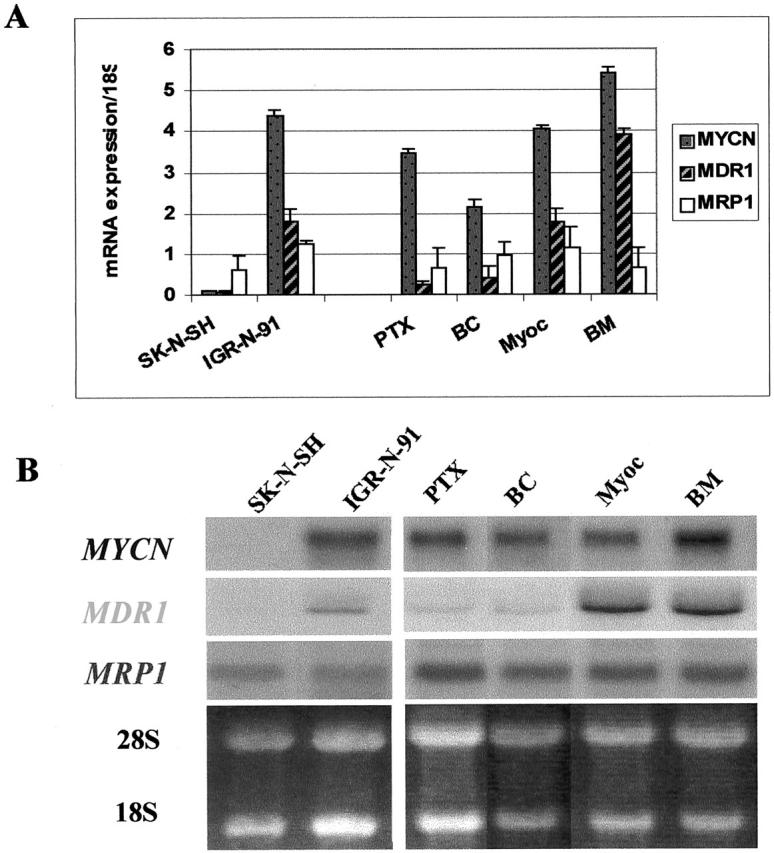
Analysis of MYCN and multi-drug resistance gene expression by RTQ-PCR (A) and Northern blotting (B) in NB cell lines. MYCN (1/100 mRNA expression/18S), MDR1 and MRP1 mRNA levels are shown. The increase in the MYCN gene mRNA level is correlated with the increase in the MDR1 gene mRNA level in the IGR-N-91 xenograft model for Myoc and BM metastatic neuroblasts. No variations in MRP1 gene expression in cell lines is determined by RTQ-PCR or Northern blotting. Data are the mean ± SD of three independent experiments.
Figure 3.
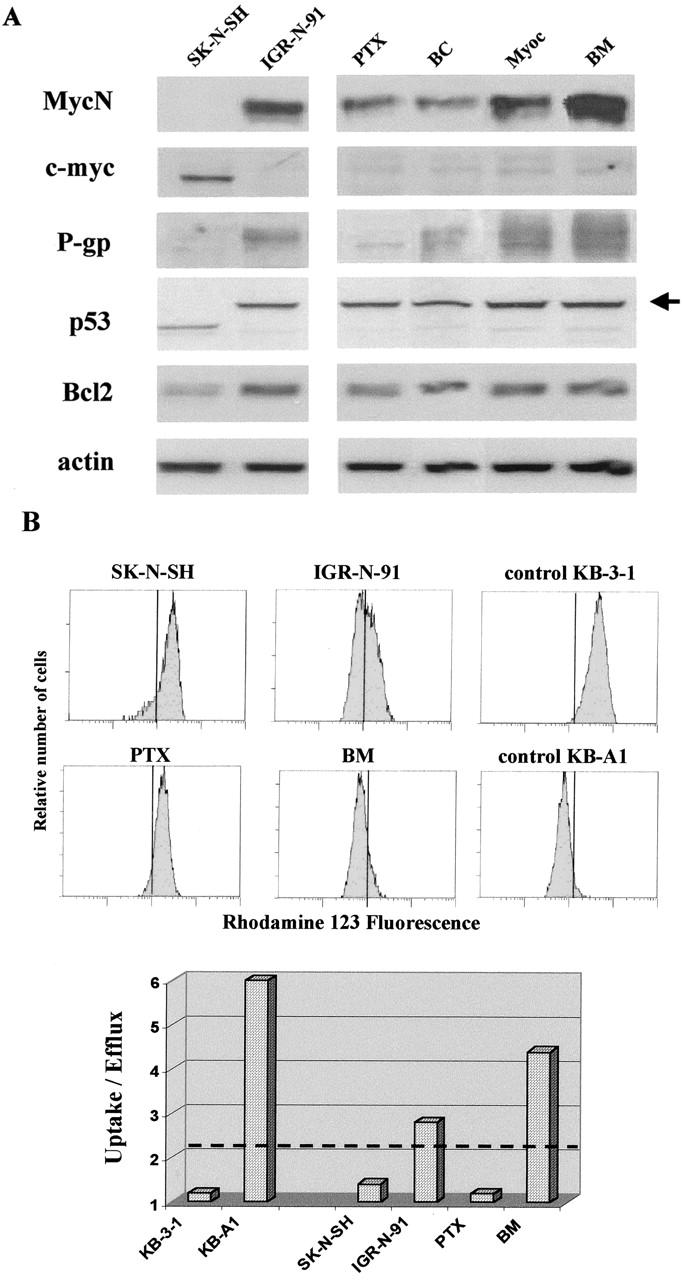
A: Western blots of MycN, c-myc, P-gp, P53, and Bcl2 proteins in NB cell lines. MycN protein expressions are well correlated with mRNA gene expressions in NB cell lines. A higher level of MycN protein expression is noted in BM and Myoc neuroblasts. Note the absence of c-myc protein expression in MYCN-overexpressing neuroblasts and of MycN protein in c-myc expressing neuroblasts. Wild-type p53 is present in SK-N-SH neuroblasts in contrast to IGR-N-91, PTX, and metastatic sublines, which all present similar heavier shifted-p53 protein (arrow). B: Flow cytometric analysis of Rho-123 uptake in NB cell lines. P-gp expression is determined by the uptake/efflux ratio of rhodamine-123. The cutoff value of this ratio was arbitrarily fixed at 2. A representative histogram of mean fluorescence intensity is shown for the different cell lines. Controls are KB3–1 and KB-A1 cell lines.
Drug-Resistance Phenotype in MYCN-Expressing Neuroblasts
Responses of SK-N-SH, parental IGR-N-91, and IGR-N-91 xenograft model cells to CDDP and VP16 were then studied. Indeed these drugs are usually administered for induction regimens in high-risk NB treatment. Cytotoxicity and apoptosis were measured by MTS assay (Figure 4A) ▶ and cell-cycle analysis (Figure 4B) ▶ . After 48 hours of incubation, a greater sensitivity to VP16 and CDDP was observed in SK-N-SH over IGR-N-91 cells on the one hand, and in PTX over BM metastatic neuroblasts (Figure 4B) ▶ on the other hand. The same comparison was examined for apoptosis as assessed quantitatively by sub-G1 cell cycle analysis and qualitatively by PARP cleavage. The sub-G1 cell population treated for 48 hours with 10 μmol/L of VP16 or 10 μmol/L of CDDP was found in greater proportion in SK-N-SH cells than in IGR-N-91 cells. This difference was found consistently for both drugs; 38 ± 1.3% versus 25.8 ± 1.1% (P ≤ 0.001) for VP16, and 40 ± 2.6% versus 15 ± 1.8% (P ≤ 0.001) for CDDP (Figure 4B) ▶ . There was a significantly higher sub-G1 population in PTX cells (33.7 ± 1.9%) than in BM metastatic neuroblasts (22.5 ± 3.9%, P ≤ 0.05) with 10 μmol/L of VP16, but not with 10 μmol/L of CDDP treatment (18.5 ± 4.9% versus 18.0 ± 1.1%, nonsignificant). Nevertheless, PARP cleavage, an early signal of apoptosis in NB cells, 29 confirmed that all cell lines undergo apoptosis, whether treated with 10 μmol/L of CDDP or VP16 for 48 hours (data not shown). From these data, we can conclude that neuroblasts, ie, parental IGR-N-91 and metastatic sublines, are more drug-resistant than SK-N-SH and PTX cells.
Figure 4.
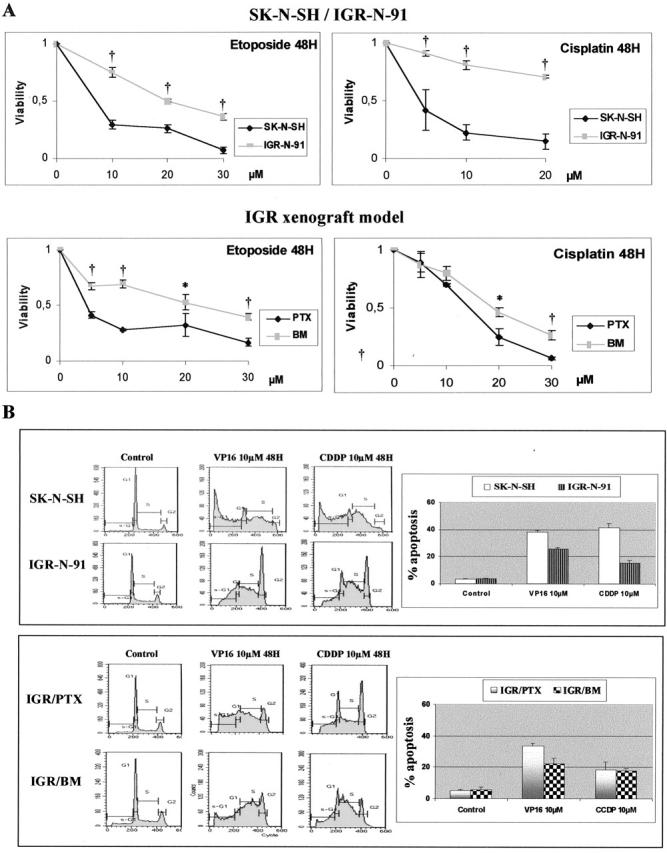
Response of NB cell lines to VP16 or CDDP cytotoxicity. A: Viability of NB cells treated throughout a concentration range of 0 to 30 μmol/L VP16 or CDDP for 48 hours was determined by MTS analysis. The arbitrary value of 1 is referred to dimethyl sulfoxide-treated cells (control) [data are the mean of three separate experiments (error bars, ± SD)]. A greater anti-cancer drug resistance is noted for IGR-N-91 cells and BM metastatic neuroblasts. The difference in viability between SK-N-SH and IGR-N-91 and between PTX and BM is significant and noted (*) if P ≤ 0.05 or (†) if P ≤ 0.001. B: Cycle analysis was performed in SK-N-SH, IGR-N-91, PTX, and BM cell lines 48 hours after a 10-μmol/L treatment with either VP16, CDDP, or vehicle control (dimethyl sulfoxide). Apoptosis was derived quantitatively by measuring the percentage of sub-G1 population (inset).
Concomitant Increase in MYCN and MDR1 Gene Expression in IGR-N-91 Metastatic Neuroblasts
To determine whether drug resistance noted at the cellular levels in NB cell lines correlates with higher multidrug resistance gene expression, we looked at MDR1 gene expression using RT-QPCR and Northern blotting (Figure 2, A and B) ▶ . The phenotypically more drug-sensitive IGR-PTX cells presented a lower MDR1 gene expression (0.25 ± 0.07 mRNA expression/18S) than the less drug-sensitive Myoc neuroblasts (1.75 ± 0.35, P ≤ 0.01) or BM neuroblasts (3.9 ± 0.14, P ≤ 0.001). The decrease in MDR1 transcript levels paralleled the decrease in MYCN expression (Figure 2A) ▶ . Data obtained from Northern blot analysis corroborate the TaqMan data (Figure 2A) ▶ : a strong intensity band was detected for MDR1 expression in both Myoc and BM cell lines. In contrast, no variation in MRP1 gene expression was shown by TaqMan or Northern blot assays in any of the cell lines (Figure 2, A and B) ▶ .
Western blot analysis performed on NB cell lines showed that the c-myc protein was only expressed in MYCN nonamplified SK-N-SH cells, and not in the MYCN-amplified IGR-N-91 xenograft model, and conversely for MycN protein expression (Figure 3A) ▶ . Significantly, P-gp protein expression parallels MDR1 gene expression in our range of NB cell lines.
As the apoptotic process in cancer cells was shown to be tightly regulated by the protein levels of three major protagonists (Myc, p53, and Bcl-2), p53 and Bcl-2 protein levels were simultaneously assessed in our NB cell lines (Figure 3A) ▶ . p53 mutations are rare in NBs at diagnosis but have been evidenced after cytotoxic therapy. 12 A wild-type p53 is noted in the MYCN nonamplified SK-N-SH cell line, as previously described. 30 In contrast, the IGR-N-91 cell line and the PTX and metastatic sublines were observed to present similar abnormal p53 transcripts as detected by Western blot showing a MW-shifted heavier p53 protein (Figure 3A ▶ , arrow). When the IGR-N-91 cell line was examined for p53 mutation in our laboratory, an insertional mutation was revealed (Goldschneider et al, submitted). Similar p53 abnormality was observed in the various IGR xenograft model cell lines. In addition to p53, the Bcl-2 family members also play a major role in regulating the intrinsic apoptotic pathway. In this study, a lower protein level of Bcl-2 expression is noted in SK-N-SH as compared to the IGR-N-91 model cell lines (Figure 3A) ▶ . However, no variation in Bcl-2 protein level was noted between the different IGR cell lines with various MDR1 gene transcript levels. Therefore no significant Bcl-2 and p53 protein variation parallels the MDR1 gene transcript level variations noted in the IGR-N-91 xenograft model.
P-gp Functionality in Metastatic IGR Neuroblasts
The P-gp functionality was tested in NB cell lines by analyzing the Rho-123 uptake obtained after 2 hours (see fluorescence histograms in Figure 3B ▶ ). Compared to SK-N-SH and IGR/PTX cells, Rho-123 accumulation was lower in IGR/BM cells indicating that these cells, which were derived from metastases in nude mice, overexpressed a functional P-gp. This data was confirmed by measuring fluorescence intensity after a 30-minute incubation in Rho-123-free medium. A significant decrease in fluorescence intensity was measured in adriamycin-resistant KB-A1 cells (used as a control for the P-gp-positive cell line) and IGR/BM neuroblasts yielding a 6.0 and 4.4 ratio of uptake/efflux values, respectively, far higher than the 2.0 limit level, with a 1.2 ratio for the P-gp-negative KB-3-1 cell line. For IGR-N-91 cells, a heterogeneous population was measured in terms of Rho-123 uptake as well as efflux (ratio uptake/efflux of 2.8), suggesting a mix of P-gp-positive and -negative cells. When combined, these data illustrate a significant P-gp expression and functionality in IGR/BM but not IGR/PTX cells.
MDR1 Gene Promoter Activation by MYCN in Human Malignant Neuroblasts
To investigate the effect of MycN on MDR1 gene expression regulation, we performed two complementary experiments. First, we studied the effect of exogenous MycN on the SK-N-SH cell line (1 copy/haploid genome) using pMYCN-transfection. Secondly, we examined the effect of endogenous MYCN by comparing PTX neuroblasts with a low constitutive MYCN expression to BM neuroblasts with a high constitutive MYCN expression level in the IGR-N-91 xenograft model. The pMDR1-CAT construct (see Materials and Methods) was co-transfected with pMYCN in SK-N-SH cells and transfected alone in IGR/PTX or IGR/BM cells.
A co-expression of MYCN and MDR1 was observed in MYCN-transfected SK-N-SH cells, as assessed by Northern blotting. Indeed, a strong MDR1 transcript signal was noted in 1-μg pMYCN-transfected cells (Figure 5A) ▶ . The MYCN-transfection efficiency was ∼10%, as determined through a subsequent synthesis of the oncoprotein, assessed by neuroblast-positive nuclear MycN Ab1 monoclonal antibody immunocytochemical staining (data not shown). Co-transfection of pMYCN and pMDR1-CAT containing the MDR1 gene proximal promoter (−4711, +286) into SK-N-SH cells induced a significant increase in CAT activity. CAT activity showed a twofold and fivefold increase after 0.5- and 1-μg pMYCN transfection, respectively (Figure 5B) ▶ . To determine whether the activation of the MDR1 gene promoter by MycN oncoprotein could lead to significant increases of MDR1 mRNA level and subsequent P-gp synthesis, we performed Northern blotting and immunocytochemical studies with P-gp C494 monoclonal antibody. A significant overexpression of the MDR1 gene was observed in MYCN-transfected SK-N-SH neuroblasts in a dose-dependent manner, ie, twofold and sixfold increase for 0.5 and 1 μg of pMYCN-transfected cells, respectively. A P-gp immunocytochemical study revealed a percentage of stained cells similar to that previously observed with MycN (data not shown). When combined, data indicate that exogenous MYCN-transfected SK-N-SH induces a marked increase of both MDR1 gene mRNA and P-gp through activation of the MDR1 gene promoter. Basal MDR1 gene promoter activity was analyzed in the IGR-N-91 xenograft model, including PTX and BM metastatic neuroblasts presenting a co-overexpression of MYCN and MDR1 genes (Figure 5A) ▶ . As shown in Figure 5B ▶ , the activation of the promoter is slightly detectable in PTX cells but significantly enhanced in BM cells. In these cells, the gradual activation of the MDR1 promoter parallels the gradual increase of both MYCN and MDR1 gene transcript levels. Hence, CAT activity in both the pMDR1 construct transfected into SK-N-SH, and PTX and BM neuroblasts increases in parallel to higher MYCN and MDR1 gene expression levels. We can conclude that high amounts of endogenous MycN oncoprotein are related to an increase in MDR1 gene mRNA level through promoter activation.
Interaction between MycN and E-Box within the MDR1 Promoter
Two putative Myc-binding sites (namely CACGTG, ie, E-box) were localized within the proximal promoter of the MDR1 gene, at −272 and −444 downstream from the transcription start site (Figure 5C) ▶ . To analyze DNA-protein complexes interacting with E-box, two synthetic oligonucleotides forming a double-stranded E-box binding site were annealed and used for electrophoretic mobility shift assays (Figure 5, C and E) ▶ . In addition, a third oligomeric-binding site was synthesized containing mutations in the E-box consensus sequence, as indicated in Figure 5C ▶ . These probes were, respectively, named MDR1-1 wt, MDR1-2 wt, and MDR1-2 mut.
MycN, c-myc, and Max protein expressions were controlled both in total and nuclear extracts of SK-N-SH (SK) and IGR-N-91 (IGR) neuroblasts (Figure 5D) ▶ . As previously mentioned, SK-cells expressed c-myc but not MycN, and conversely for IGR-cells. Both cell lines expressed Max. Similar results were noted in total and nuclear cell extracts.
Gel shift analysis showed that the wild-type E-box oligomers (MDR1-1 and -2 probes) formed a single band shift that is present both in SK-N-SH and IGR-N-91 cells (Figure 5E ▶ ; lanes 1, 3, 5, and 6). However, a shifted band of slight intensity was noted with the MDR1-1 probe and SK extracts (Figure 5E ▶ , lane 1) or IGR extracts (Figure 5E ▶ , lane 5). A more intense band appeared when the MDR1-2 probe was used for both SK-N-SH or IGR-N-91 nuclear extracts (Figure 5E ▶ , lanes 3 and 6). Incubation of the MDR1-2 probe with increasing levels of IGR nuclear extracts (5, 10 and 15 μg) revealed the presence of a shifted band of higher intensity (Figure 5E ▶ , lanes 7 to 9). Incubation of the nuclear extracts and MDR probes in the presence of an unlabeled competitor oligonucleotide resulted in a significant reduction of the respective shifted bands (Figure 5E ▶ ; lanes 2, 4, and 10). No band shift was observed with the MDR1-2 mutated probe Myc-binding element, indicating the specificity of the binding (Figure 5E ▶ , lane 11). Many attempts, performed to evidence supershifts with MycN and Max antibodies, were unsuccessful (data not shown). Thus, our data strongly suggest that MycN and c-myc proteins interact with the E-box motifs of the MDR1 gene promoter.
Discussion
High-risk NBs are very frequently associated with a poor clinical outcome. Initially sensitive to the first cycles of intensive chemotherapy, high-risk NBs tend to become chemo-incurable as the disease progresses, resulting in a dismal prognosis. 6,7 In these tumors, the extreme biological heterogeneity characterizing NB disease favors malignant neuroblasts with angiogenic, invasive, and metastatic properties. Moreover, the organ environment and the genetic selection of drug-resistant tumor cells may influence the response of metastases to chemotherapy. MYCN is the key oncogene that clearly contributes to the malignant phenotype of NB and is very likely to promote its metastatic process.
To investigate the role of MycN in drug resistance acquired during NB metastatic process, this study used the previously described human IGR-N-91 ectopic xenograft metastatic model. 19 To this end we controlled MYCN status in terms of DNA amplification and expression levels. Indeed this model mimics the clinical situation with blood-circulating malignant neuroblasts and BM as the major site for NB metastases. As such, the IGR-N-91 xenograft model may be considered the first physiopathological metastatic NB model so far described. Our results indicate that no variation in MYCN amplification was observed between these different NB cell lines (300 copies), whereas a significant and stable increase in MYCN gene expression occurs in Myoc and BM neuroblasts as compared to PTX neuroblasts. Quantitative RT-PCR data have been corroborated by Northern blot analysis: they show a significant MDR1 gene expression increase in Myoc and BM metastatic neuroblasts. Although these neuroblasts were maintained in vitro for many passages in the absence of drug treatment, this MDR1 gene expression increase is correlated with their chemoresistance phenotype, as evidenced by cell viability and P-gp functionality tests. Stable MYCN and MDR1 gene expressions in these cells therefore suggest a genetic selection leading to metastatic and chemoresistant cells throughout the progression of the NB tumor.
As assessed by the Rho-123 uptake, P-gp protein appears to be functional in BM neuroblasts but not in PTX neuroblasts; this is consistent with the VP16-resistant phenotype in BM cells, as measured by the MTS assay as well as the cytofluorimetric sub-G1 profile. Indeed VP16, but not CDDP, is recognized by P-gp as a substrate for the efflux function. In a previous study, a same resistance was observed in metastatic neuroblasts incubated with another P-glycoprotein substrate, adriamycin. 19 It is well accepted that P-gp acts as an ATP-dependent efflux pump and reduces the intracellular accumulation of many lipophilic drugs of various chemical structures. 17 Indeed, for many years, the model for drug resistance conferred by MDR1/P-gp focused on the cell’s xenobiotic efflux mechanism. A recent overview, however, highlights new biological regulatory functions for P-gp, such as cell differentiation, proliferation, and survival. 31 Therefore, in addition to drug transport function, P-gp may confer cell resistance by regulating some caspase-dependent apoptotic pathways. 32 Moreover, recent studies substantiated that soluble factors (cytokines, hormones, growth factors) or cell interaction with extracellular matrix (ECM) present in the tumor’s microenvironment are determining factors in cancer cell survival and the emergence of drug resistance. 33,34 In particular, the BM microenvironment is likely to promote tumor cell survival by conferring protection from cytotoxic drugs. 33 In that respect and strikingly, it must be reported that BM progenitors constitutively express the MDR1 gene and P-gp, conferring protection to the stem cell population against drug cytotoxicity. 35 Whether the BM microenvironment favors the proliferation of malignant neuroblasts that overexpress the MDR1 gene remains to be demonstrated.
To further investigate the correlation between MYCN and MDR1 expressions in this IGR-N-91 model (PTX versus BM and Myoc malignant neuroblasts), transcription studies and gel shift assays were performed. Data show that both exogenous MycN, transiently expressed in SK-N-SH cells, and endogenous MycN, primarily expressed in the IGR-N-91 neuroblasts, activate the MDR1 gene promoter. An inverse expression of MycN and c-myc is observed in SK-N-SH and IGR-N-91 cells, with both nuclear extracts expressing their common Max protein partner. As previously described, 36 the Myc-Max heterodimers induce transcription activation by recognizing the E-box-related sequence CACGTG, identified as the highest affinity target for myc binding, as well as a group of other E-box or non-E box-related sequences with lower affinity. 37 Indeed, additional helix-loop-helix proteins as the ubiquitous transcription factors USF, TFE3, TFEB, or cell type-specific proteins (TFEC, Mi), can bind and activate the typical Myc-Max sequences, CA(C/T)GTG. 38-40 Because two E-box-related motifs (CACGTC and CAAGTG) can be identified in the MDR1 gene promoter, 28 we present here a marked complex between the IGR-N-91 nuclear extracts and the E-box sequence (CAAGTG), adjacent to the MDR1 promoter ATG start codon. When combined, these observations support the idea that MycN is probably the key oncoprotein leading to MDR1 gene promoter activation through an interaction with the E-box motif.
Two multidrug resistance genes were found overexpressed in NB primary tumors, MDR1/P-gp 41,42 and MRP1. 43 With respect to the MDR1/P-gp expression prognostic value, Haber‘s team indicated that very high MDR1 gene expression was associated with poor outcome in MYCN gene single-copy NB tumors. 44 The authors suggest that the MDR1 gene plays a clinical role in specific subgroups of primary untreated NB. Regarding MRP1, gene expression was first correlated with poor prognosis 43 and secondly with MYCN expression. 45,46 A direct relationship between MRP1 and MYCN expressions was then established, 47 and studies concerning MRP promoter regulation are underway using the E-box in human NB (Manohar CF, personal communication). In the IGR xenograft model, no significant increase in MRP1 gene expression was observed, as assessed by RTQ-PCR or Northern blotting. We can therefore conclude that in the IGR xenograft model, MDR1/P-gp, but not MRP1, may play an important role in the acquisition of chemoresistance phenotype during the metastatic process in the absence of drug treatment. Whether other factors, such as cell adhesion, 34 also contribute to chemoresistance phenotype remains to be confirmed. Overall, the lack of correlation noted between MYCN and MDR1 gene expressions in primary NB may be explained by the phenotype of nonmetastatic malignant neuroblasts and possibly also by the heterogeneity of tumor tissues.
NB malignant phenotype is related not only to MYCN alteration but also to many other genetic abnormalities, including activation of other oncogenes and inactivation of tumor suppressor genes. Although p53 de novo mutations are rare in neuroblastic tumors and NB cell lines, 48,49 they are frequently found after cytotoxic treatment. 12 In this respect, acquisition of p53 mutations in NB after chemotherapy is likely to promote tumorigenesis through chemo- and radio-resistance mechanisms. 12 Such a gain in oncogenic function may constitute a critical step in the acquisition of therapeutic resistance and in particular chemoresistance. 50 Interestingly, it has been reported that wild-type p53 represses MDR1 or MRP1 in colon cancer. 51-53 This was recently corroborated in cell cultures by the fact that a loss of p53 repression leads specifically to MDR1 but not MRP1 up-regulation, 54 following a mechanism that involves an exclusive interaction between a p53 mutant (not wt-p53) and an Ets binding site in the MDR1 promoter.
In the IGR model, an identical p53 insertional mutation leading to high MW p53 is detected in the parental IGR-N-91 cell line, PTX, as well as in IGR-metastatic neuroblasts (Goldschneider et al, submitted). Likewise, a significant but stable Bcl-2 expression is noted in every IGR-N-91 xenograft model cell line. Consequently, as genomic amplification of MYCN is unchanged and unrelated to metastatic dissemination, it is suggested that the increment of MYCN transcript and MycN protein levels described in this study may be directly involved in the emergence of the metastatic process seen in our human IGR-N-91 xenograft model.
Future Directions
MYCN gene amplification is widely used for evaluating NB prognosis and designing therapeutic regimens for patients. 55 The current challenge is to find new predictive markers to evaluate the metastatic potential of NB primary tumors. Our study highlights the importance of two critical determining factors for high-risk metastatic NB, ie, MYCN alteration and acquired chemoresistance. The latest breakthrough in cDNA microarray technology will allow for the development of dynamic transcriptomic scanning using these models as well as tumors matched to BMs from patients under conventional chemotherapy. This technology will enable the identification of gene clusters involved in the pathways mediating NB dissemination and responsiveness to chemotherapy. As MYCN gene expression is increased in MYCN nonamplified tumors, it appears crucial to find new genetic markers. In this respect, the microarray analysis of PTX and BM neuroblasts in our IGR-N-91 xenograft model should be informative, allowing for the establishment of new criteria to predict the evolution of high-risk NB tumors.
Acknowledgments
We thank Dr. M. J. Terrier-Lacombe (Department of Pathology, Institut Gustave Roussy) for histological examination; Y. Lecluse (Service commun de cytométrie, Institut Fédératif de Recherche 54) for expert FACS analysis; edited by Englishbooster S.A.
Footnotes
Address reprint requests to Gilda Raguénez, Ph.D., Interactions Moleculaires et Cancer, Unité Mixte de Recherche 8126, Institut Gustave-Roussy, 39, rue Camille Desmoulins, 94805 Villejuif Cedex, France. E-mail: raguenez@igr.fr.
Supported by La Ligue Contre le Cancer, Comité de Montbéliard, Comité du Cher, and Bonus Qualité Recherche, Paris XI University.
Present address of E. F.: Institut IPSEN-Beaufour, Les Ulis, France.
References
- 1.Brodeur GM, Castleberry RP: Neuroblastoma: effect of genetic factors on prognosis and treatment. Cancer 1992, 70:1685-1694 [DOI] [PubMed] [Google Scholar]
- 2.Brodeur G: Molecular basis for heterogeneity in human neuroblastoma. Eur J Cancer 1995, 31A:505-509 [DOI] [PubMed] [Google Scholar]
- 3.Maris J, Matthay K: Molecular biology of neuroblastoma. J Clin Oncol 1999, 17:2264-2279 [DOI] [PubMed] [Google Scholar]
- 4.Schwab M, Ellison J, Busch M, Rosenau W, Varmus HE, Bishop JM: Enhanced expression of the human gene N-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastomas. Proc Natl Acad Sci USA 1984, 81:4940-4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwab M, Varmus HE, Bishop JM: Human N-myc gene contributes to neoplastic transformation of mammalian cell in culture. Nature 1985, 316:160-162 [DOI] [PubMed] [Google Scholar]
- 6.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP: Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation and 13-cis-retinoic acid. N Engl J Med 1999, 341:1165-1173 [DOI] [PubMed] [Google Scholar]
- 7.Valteau-Couanet D, Benhamou E, Vassal G, Stambouli F, Lapierre V, Couanet I, Lumbroso J, Hartmann O: Consolidation with a busulfan-containing regimen followed by stem cell transplantation in infants with poor prognosis stage 4 neuroblastoma. Bone Marrow Transplant 2000, 25:937-942 [DOI] [PubMed] [Google Scholar]
- 8.Johnstone RW, Ruefli AA, Lowe SW: Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002, 108:153-164 [DOI] [PubMed] [Google Scholar]
- 9.El-Deiry WS: Role of oncogenes in resistance and killing by cancer therapeutic agents. Curr Opin Oncol 1997, 9:79-87 [DOI] [PubMed] [Google Scholar]
- 10.Makin G, Hickman JA: Apoptosis and cancer chemotherapy. Cell Tissue Res 2000, 301:143-152 [DOI] [PubMed] [Google Scholar]
- 11.Levine A: p53, the cellular gatekeeper for growth and division. Cell 1997, 88:323-331 [DOI] [PubMed] [Google Scholar]
- 12.Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J: Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res 2001, 61:8-13 [PubMed] [Google Scholar]
- 13.Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, Triche TJ, Reynolds CP: Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res 2001, 61:6185-6193 [PubMed] [Google Scholar]
- 14.Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look T, Lahti J, Kidd VJ: Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med 2000, 6:529-535 [DOI] [PubMed] [Google Scholar]
- 15.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM: Resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res 2001, 61:1314-1319 [PubMed] [Google Scholar]
- 16.Germann UA, Pastan I, Gottesman MM: P-glycoproteins: mediators of multidrug resistance. Semin Cell Biol 1993, 4:63-76 [DOI] [PubMed] [Google Scholar]
- 17.Cole SPC, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist CE, Stewart AJ, Kurz EU, Duncan AM, Deeley RG: Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258:1650-1654 [DOI] [PubMed] [Google Scholar]
- 18.Tan B, Piwnica-Worms D, Ratner L: Multidrug resistance transporters and modulation. Curr Opin Biol 2000, 12:450-458 [DOI] [PubMed] [Google Scholar]
- 19.Ferrandis E, Da Silva J, Riou G, Bénard J: Coactivation of the MDR1 and MYCN in human neuroblastoma during the metastatic process in the nude mouse. Cancer Res 1994, 54:2256-2261 [PubMed] [Google Scholar]
- 20.Schweigerer L, Breit S, Wenzel A, Tsunamoto K, Ludwig R, Schwab M: Augmented MYCN expression advances the malignant phenotype of human neuroblastoma cells: evidence for induction of autocrine growth factor activity. Cancer Res 1990, 50:4411-4416 [PubMed] [Google Scholar]
- 21.Breit S, Ashman K, Wilting J, Rossler J, Hatzi E, Fotsis T, Schweigerer L: The N-myc oncogene in human neuroblastoma cells: down-regulation of an angiogenesis inhibitor identified as activin A. Cancer Res 2000, 60:4596-4601 [PubMed] [Google Scholar]
- 22.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM: Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 1997, 16:2985-2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodeur GM, Hayes FA, Green AA, Casper JT, Wasson J, Wallach S, Seeger RC: Consistent N-myc copy number in simultaneous or consecutive neuroblastoma samples from sixty individual patients. Cancer Res 1987, 47:4248-4253 [PubMed] [Google Scholar]
- 24.Valent A, Bénard J, Clausse B, Barrois M, Valteau-Couanet D, Terrier-Lacombe MJ, Spengler B, Bernheim A: In vivo elimination of acentric double minutes containing amplified MYCN from neuroblastoma tumor cells through the formation of micronuclei. Am J Pathol 2001, 58:1579-1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorman CM, Moffat LF, Howard BH: Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol 1982, 2:1044-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall CV, Jacob PE, Ringold GM, Lee F: Expression and regulation of Escherichia coli lac Z gene fusions in mammalian cells. J Mol Appl Genet 1983, 2:101-104 [PubMed] [Google Scholar]
- 27.Crisanti P, Raguénez G, Blancher C, Néron B, Mamoune A, Omri B: Cloning and characterization of a novel transcription factor involved in cellular proliferation arrest: PATF. Oncogene 2001, 20:5476-5483 [DOI] [PubMed] [Google Scholar]
- 28.Ueda K, Pastan I, Gottesman MM: Isolation and sequence of the MDR1 gene promoter region. J Biol Chem 1987, 262:17432-17436 [PubMed] [Google Scholar]
- 29.Bursztajn S, Feng JJ, Berman SA, Nanda A: Poly (ADP-ribose) polymerase induction is an early signal of apoptosis in human neuroblastoma. Mol Brain Res 2000, 76:363-376 [DOI] [PubMed] [Google Scholar]
- 30.Moll UT, LaQuaglia M, Bénard J, Riou G: Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci USA 1995, 92:4407-4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnstone RW, Ruefli AA, Smyth MJ: Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem Sci 2000, 25:1-6 [DOI] [PubMed] [Google Scholar]
- 32.Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW: The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci USA 1998, 95:7024-7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shain KH, Landowski TH, Dalton WS: The tumor environment as a determinant of cancer cell survival: a possible mechanism for de novo drug resistance. Curr Opin Oncol 2000, 12:557-563 [DOI] [PubMed] [Google Scholar]
- 34.Shain KH, Dalton WS: Cell adhesion is a key determinant in de novo multidrug resistance (MDR): new targets for the prevention acquired MDR. Mol Cancer Ther 2001, 1:69-78 [PubMed] [Google Scholar]
- 35.Bunting KD: ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells 2002, 20:11-20 [DOI] [PubMed] [Google Scholar]
- 36.Wenzel A, Schwab M: The mycN/max protein complex in neuroblastoma. Short Review. Eur J Cancer 1995, 31A:516-519 [DOI] [PubMed] [Google Scholar]
- 37.Grandori C, Eisenman RN: Myc target genes. Trends Biochem Sci 1997, 22:177-181 [DOI] [PubMed] [Google Scholar]
- 38.Blackwell TK, Huang J, La A, Kretznze L, Alt FW, Eisenman RN, Weintraub D: Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol 1993, 13:5216-5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao GQ, Zhao Q, Zhou X, Mattei MG, de Crombrugghe B: TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol Cell Biol 1993, 13:4505-4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodgkinson CA, Moore KJ, Nakayama A, Steingrimsson E, Copeland NG, Jenk NA, Arnheiter H: Mutations at the mouse microphtalmia locus are associated in a gene encoding a novel basic-helix-loop-helix zipper protein. Cell 1993, 74:395-404 [DOI] [PubMed] [Google Scholar]
- 41.Bourhis J, Bénard J, Hartmann O, Boccon-Gibod L, Lemerle J, Riou G: Correlation of MDR1 gene expression with chemotherapy in neuroblastomas. J Nat Cancer Inst 1989, 81:1401-1405 [DOI] [PubMed] [Google Scholar]
- 42.Chan HS, Haddad G, Thorner PS, DeBoer G, Lin YP, Ondrusek N, Yeger H, Li V: P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N Engl J Med 1991, 325:1608-1614 [DOI] [PubMed] [Google Scholar]
- 43.Norris MD, Bordow SB, Marshall GM, Haber PS, Cohn SL, Haber M: Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N Engl J Med 1996, 334:231-238 [DOI] [PubMed] [Google Scholar]
- 44.Haber M, Bordow SB, Gilbert J, Madafiglio J, Kavallaris M, Marshall GM, Mechetner EB, Fruehauf JP, Tee L, Cohn SL, Salwen H, Schmidt ML, Norris MD: Altered expression of the MYCN oncogene modulates MRP gene expression and response to cytotoxic drugs in neuroblastoma cells. Oncogene 1999, 18:2777-2782 [DOI] [PubMed] [Google Scholar]
- 45.Haber M, Bordow SB, Haber PS, Marshall GM, Stewart BW, Norris MD: The prognostic value of MDR1 gene expression in primary untreated neuroblastoma. Eur J Cancer 1997, 33:2932-2036 [DOI] [PubMed] [Google Scholar]
- 46.Haber M, Kavallaris M: Multidrug resistance genes in neuroblastoma. Brodeur GM Sawada T Tsuchida Y Voûte PA eds. Neuroblastoma. 2000:pp 207-215 Elsevier Amsterdam, The Netherlands
- 47.Norris MD, Bordow SB, Haber PS, Marshall GM, Kavallaris M, Madafiglio J, Cohn SL, Salwen H, Schmidt ML, Hipfnen DR, Cole SP, Deeley RG, Haber M: Evidence that the MYCN oncogene regulates MRP gene expression in neuroblastoma. Eur J Cancer 1997, 33:1911-1916 [DOI] [PubMed] [Google Scholar]
- 48.Vogan K, Bernstein M, Leclerc JM, Brisson L, Brossard J, Brodeur GM, Pelletier J, Gros P: Absence of p53 gene mutations in primary neuroblastomas. Cancer Res 1993, 1:5269-5273 [PubMed] [Google Scholar]
- 49.Hosoi G, Hara J, Okamura T, Osugi Y, Ishihara S, Fukuzawa M, Okada A, Okada S, Tawa A: Low frequency of the p53 gene mutations in neuroblastoma. Cancer 1994, 73:3087-3093 [DOI] [PubMed] [Google Scholar]
- 50.Sigal A, Rotter V: Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res 2000, 60:6788-6793 [PubMed] [Google Scholar]
- 51.Thottassery JV, Zambetti GP, Arimori K, Schuetz EG, Schuetz JD: p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc Natl Acad Sci USA 1997, 94:11037-11042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Q, Beck WT: Transcriptional suppression of multidrug resistance-associated protein (MRP) gene expression by wild-type p53. Cancer Res 1998, 74:63-68 [PubMed] [Google Scholar]
- 53.Sullivan GF, Yang JM, Vassil A, Yang J, Bash-Babula J, Hait WN: Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J Clin Invest , 105:1261-1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD: Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 2001, 276:39359-39367 [DOI] [PubMed] [Google Scholar]
- 55.Savelyeva L, Schwab M: Amplification of oncogenes revisited: from expression profiling to clinical application. Cancer Lett 2001, 167:115-123 [DOI] [PubMed] [Google Scholar]