

Abstract
Dysfunction of the autonomic nervous system is a recognized complication of diabetes. Neuroaxonal dystrophy (NAD), a distinctive axonopathy involving distal axons and synapses, represents the neuropathologic hallmark of diabetic sympathetic autonomic neuropathy in human and several insulinopenic experimental rodent models. Recent studies have suggested that loss of the neurotrophic effects of insulin and/or IGF-I on sympathetic neurons and not hyperglycemia per se, may underlie the development of sympathetic NAD. The streptozotocin (STZ)-diabetic and BB/W rat, the most commonly used experimental rodent models, develop marked hyperglycemia and concomitant deficiency in both circulating insulin and IGF-I. These animals reproducibly develop NAD in nerve terminals in the prevertebral sympathetic ganglia and the distal portions of noradrenergic ileal mesenteric nerves. The Zucker Diabetic Fatty (ZDF) rat, an animal model of type 2 diabetes, also develops severe hyperglycemia comparable to that in the STZ- and BB/W-diabetic rat models, although in the presence of hyperinsulinemia. In our study, ZDF rats maintained for 6 to 7 months in a severely diabetic state, as assessed by plasma glucose and glycated hemoglobin levels, maintained significant hyperinsulinemia and normal levels of plasma IGF-I at sacrifice. NAD did not develop in diabetic ZDF rat sympathetic ganglia and ileal mesenteric nerves as assessed by quantitative ultrastructural techniques, which is in dramatic contrast to neuropathologic findings in comparably hyperglycemic 6-month STZ-diabetic insulinopenic rats. These data combined with our previous results argue very strongly that hyperglycemia is not the critical and sufficient element in the pathogenesis of diabetes-induced NAD, rather that it is the loss of trophic support, most likely of IGF-I or insulin, that causes NAD.
Clinical presentations of diabetic neuropathy include symmetric sensory polyneuropathy, asymmetric mononeuropathies involving cranial and somatic nerves, and autonomic neuropathy. 1 Although symmetrical sensory polyneuropathy (resulting in classical “stocking-glove” limb anesthesia) is pathologically the most completely studied form, the clinical manifestations of diabetic autonomic neuropathy result in increased patient morbidity and are associated with substantially increased mortality. 2-4 Symptoms of diabetic autonomic neuropathy range widely from minor pupillary and sweating problems to significant disturbances in cardiovascular, alimentary and genitourinary function and involve both the sympathetic and parasympathetic nervous systems. Studies of autopsied diabetic patients 5 have established that the neuropathologic hallmark of diabetic sympathetic autonomic neuropathy is the reproducible development of markedly enlarged distal axons and nerve terminals (“neuroaxonal dystrophy”, NAD), thought to represent aberrant intraganglionic sprouting 5,6 in prevertebral sympathetic ganglia in the apparent absence of significant neuron loss. Degenerating, regenerating, and pathologically distinctive, markedly swollen dystrophic axons and nerve terminals also develop in prevertebral superior mesenteric ganglia (SMG) and noradrenergic ileal mesenteric nerves in the streptozotocin (STZ)-induced diabetic rat in the proven absence of neuron or axon loss. 7,8
The pathogenesis of diabetic autonomic neuropathy remains uncertain. Although hyperglycemia directly results in a variety of abnormal metabolic reactions in nerve (eg, disordered polyol and phosphoinositide metabolism, increase in glycated proteins and exaggerated oxidative stress), which may contribute to the development of neuropathy, other processes may also play a role. One such mechanism may involve the neurotrophic action of insulin or insulin-like growth factor-I (IGF-I), independent of their glycemic effects. We have previously demonstrated 8 that 6-month STZ-diabetic rats (ie, a duration of diabetes resulting in established NAD) treated for two additional months with systemic recombinant human IGF-I (rhIGF-I) resulted in nearly complete normalization of NAD in the SMG and ileal mesenteric nerves in the absence of an effect on the metabolic severity of diabetes. Although these results suggest a critical role for decreased IGF-I in the development of NAD, interpretation of these results was complicated by the use of pharmacological doses of rhIGF-I (1 mg/kg, s.c.) in that study.
To further test the hypothesis that more physiological levels of IGF-I or insulin can exert effects independent of their glycemic effects, we have taken advantage of the Zucker Diabetic Fatty (ZDF) rat model of type 2 diabetes. The male ZDF rat (ZDF/Gmi, fa/fa), derived from inbreeding of hyperglycemic Zucker obese rats, is characterized by a mutation in the leptin receptor with resultant high circulating leptin levels. Male ZDF rats exhibit marked hyperglycemia (400 to 500 mg/dl), developing between 7 to 10 weeks of age, which is maintained for at least 6 months. These animals demonstrate hyperlipidemia, insulin resistance, and hyperinsulinemia, reaching maximum circulating insulin values approximately eightfold those of lean controls at 7 weeks and decreasing thereafter with β-cell exhaustion. 9-11 Male ZDF rats gain weight in excess of lean controls for the first 6 months of life to a maximum of approximately 500 grams, decreasing somewhat thereafter. Prior to this study circulating levels of IGF-I in chronically diabetic ZDF rats have not been reported. To determine whether hyperglycemia alone is sufficient to produce sympathetic NAD or if decreased circulating levels of insulin or IGF-I play a critical role, the current study compares sympathetic autonomic neuropathy in the chronically diabetic ZDF rat model to that previously characterized in the STZ rat.
Materials and Methods
Male ZDF rats (ZDF/Gmi, fa/fa) and their lean littermates (ZDF/Gmi, +/?) were obtained from Charles Rivers Genetic Models, Inc. (Indianapolis, IN) Animals were maintained on Purina 5008 (16.7 kcal% fat) diet, being followed at intervals by monitoring blood glucose and body weight. ZDF rats were sacrificed 6 to 7 months after the onset of diabetes. Male Sprague-Dawley rats (∼300 grams; Charles Rivers, Belmont, MA) received a single dose of STZ (65 mg/kg, i.v.; Upjohn, Kalamazoo, MI) and within a few days developed plasma glucose levels ≥350 mg/dl which were maintained until sacrifice 6 months later. Animals were housed and cared for in accordance with the guidelines of the National Institutes of Health and the Washington University Committee for the Humane Care of Laboratory Animals. The frequency of NAD in this group of STZ-induced diabetic rats formed part of a previously reported investigation 8 and is included in the current study for comparison to ZDF rats.
Acid/ethanol extracted rat serum IGF-I was measured in the Washington University Diabetes Research and Training Center (DRTC) RIA Core laboratory at the time of sacrifice of ZDF rats by double-antibody radioimmunoassay using antibody produced in rabbits against human IGF-I and human IGF-I calibrators. Rat insulin was measured in a double-antibody radioimmunoassay, using antibody produced in guinea pigs and rat insulin calibrators. HbA1c values were determined using the Bayer DCA 2000+ analyzer (ZDF experiment) or Glycogel B kit (STZ experiment; Pierce, Rockford, IL).
The superior mesenteric ganglia (SMG) and ileal mesenteric nerves were fixed by perfusion in 3% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.4, dissected, cleaned of extraneous tissue, and remained fixed overnight in the same buffer. Tissue samples were postfixed in phosphate-buffered 2% OsO4 containing 1.5% potassium ferricyanide, dehydrated in graded concentrations of alcohol and embedded in Epon with propylene oxide as an intermediary solvent. One-μm-thick plastic sections were examined by light microscopy after staining with toluidine blue. Ultrathin sections of individual SMG and ileal mesenteric nerve pedicles were cut onto formvar-coated slot grids, which permits visualization of entire ganglionic and nerve cross-sections, and stained with uranyl acetate and lead citrate and examined with a JEOL 1200 electron microscope. The frequency of NAD is expressed as a ratio of lesions/nucleated neuronal perikarya in the SMG, as a percentage of total mesenteric nerve axon number, and as lesions per ileal mesenteric nerve fascicle (two main paravascular fascicles, largely destined for alimentary targets, are contained in each ileal mesenteric neurovascular arcade) using methods previously described in detail. 8 Statistical comparison was performed using a two-sample t-test assuming unequal variances.
Results
ZDF rats were markedly hyperglycemic in comparison to their lean littermate controls (Table 1) ▶ , with levels of blood glucose and diabetic/control ratios of HbA1c somewhat increased or comparable, respectively, to STZ-diabetics. Body weight of ZDF rats at the time of sacrifice was not significantly different from that of lean controls (Table 1) ▶ , which reflects the known phenomenon of weight loss that occurs in ZDF rats after 6 months of age. 11
Table 1.
Metabolic Characteristics of Diabetes in ZDF and STZ-Diabetic Rats
| Group | n | Serum glucose (mg %) | HbA1c (%) | Final weight (g) |
|---|---|---|---|---|
| ZDF rat model | ||||
| Lean control | 7 | 98 ± 3 | 3.7 ± 0.2 | 448 ± 4 |
| Diabetic | 9 | 476 ± 41* | 9.6 ± 0.4* | 419 ± 14 |
| STZ rat model | ||||
| Control | 7 | 190 ± 9 | 4.6 ± 0.2 | 460 ± 22 |
| Diabetic | 6 | 431 ± 19* | 12.6 ± 0.6* | 378 ± 17† |
Values represent the means ± SEM of n rats.
Statistical comparison:
*P ≤ 0.0001;
†P ≤ 0.01 vs. control group.
Serum insulin levels were increased approximately threefold in ZDF rats at the time of sacrifice compared to lean controls (Controls, 0.44 ± 0.05 ng/ml, mean ± SEM, n = 7 rats; Diabetic, 1.13 ± 0.26, n = 9 rats, P = 0.03). IGF-I concentration in serum was unchanged in ZDF rats (1462 ± 88 ng/ml, n = 9) compared to lean controls (1320 ± 52, n = 7). Although IGF-I and insulin levels were not determined in the STZ-group which had formed part of a previous publication, 8 numerous studies 12-17 of the STZ-rat model have reported serum insulin values decrease >75% and serum IGF-I content decrease 50% to 86% in STZ-rats diabetic for variable durations.
Light microscopic examination of the SMG of 6- to 7-month diabetic ZDF rats failed to show active neuronal degeneration or neuron loss, apoptosis, dystrophic neuritic swellings, cytoplasmic inclusions, chromatolysis, or significant perikaryal atrophy. Ultrastructural examination of the SMG in ZDF rats showed normal neurons surrounded by axonal and dendritic processes comprising the adjacent neuropil and a complement of normal nerve terminals (arrows, Figure 1,A and B ▶ ). Significant numbers of dystrophic swollen axons were not encountered in either 6- to 7-month diabetic ZDF rats or lean controls. This result is in marked contrast to the SMG of STZ rats diabetic for the same interval. Dystrophic axon terminals (arrow, Figure 1C ▶ ) often contain neurotransmitter granules (arrowheads, Figure 1D ▶ ) and, occasionally, synaptic densities (arrow, Figure 1D ▶ ). Quantitative ultrastructural determination of the NAD ratio (dystrophic axon number/nucleated neurons) in the SMG showed no increase in ZDF diabetic rats compared to lean controls (Figure 2) ▶ in contrast to a fivefold increase in 6-month STZ-diabetic rats compared to their age-matched controls.
Figure 1.
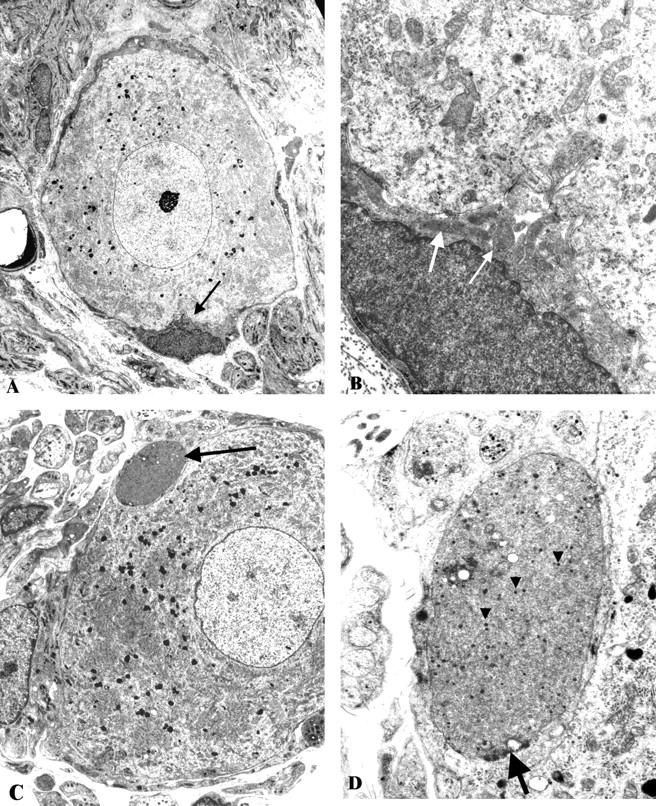
Neuroaxonal dystrophy in ZDF- and STZ-diabetic rat SMG. A and B: A typical principal sympathetic neuron from a 6- to 7-month ZDF-diabetic SMG is shown surrounded by axonal and dendritic processes composing the neuropil. Note the comparative size of normal nerve terminals (arrow, A), seen at higher magnification (arrows, B) compared to the dystrophic terminal shown at the same magnification in C,D. (Original magnifications: A, ×2000; B, ×12,000). C and D: The SMG of a 6-month STZ-diabetic rat is shown consisting of a normal-appearing principal sympathetic neuron and an intimately apposed swollen dystrophic nerve ending (arrow, C, shown at higher magnification in D). The dystrophic neurite contains dense neurotransmitter containing granules (arrowheads, D) and a possible synaptic density (arrow, D). (Original magnifications: C, ×2000; D, ×12,000).
Figure 2.

Quantitative comparison of sympathetic autonomic neuropathy in ZDF- and STZ-induced diabetic rats.
Examination of the ileal mesenteric nerves of ZDF rats and littermate controls showed nerve fascicles (Figure 3A) ▶ containing large numbers of normal unmyelinated axons (arrows, Figure 3B ▶ ) without significant numbers of dystrophic axons. In contrast, STZ-induced diabetic rats showed large numbers of dystrophic axons in ileal paravascular mesenteric nerves (arrows, Figures 3C,D ▶ ) in comparison to their age-matched controls. Quantitative comparison of the frequency of NAD in ZDF and STZ-diabetic rats in ileal mesenteric nerves confirmed the failure of ZDF rats to develop NAD, which is in contrast to the 15-fold increase in the frequency of NAD in ileal paravascular mesenteric nerves of 6-month STZ-diabetic rats expressed as a percentage of total axon number (Figure 2) ▶ . Expression of numbers of dystrophic axons as number/fascicle (each mesenteric neurovascular arcade typically is composed of 2 paravascular nerve fascicles) showed no increase in ZDF rats compared to controls (ZDF, 0.27 ± 0.08 dystrophic axons/fascicle, mean ± SEM, n = 9 rats versus control, 0.28 ± 0.13, n = 7 rats). In STZ diabetics, expression of the frequency of NAD in this manner demonstrated marked increase in diabetic compared to controls (STZ-diabetic, 5.3 ± 1.3, n = 6 versus age-matched controls, 0.34 ± 0.1, n = 7, P ≤ 0.01). Neither ZDF nor STZ-diabetic rats showed significant axon loss in paravascular ileal mesenteric nerves (Figure 2) ▶ .
Figure 3.
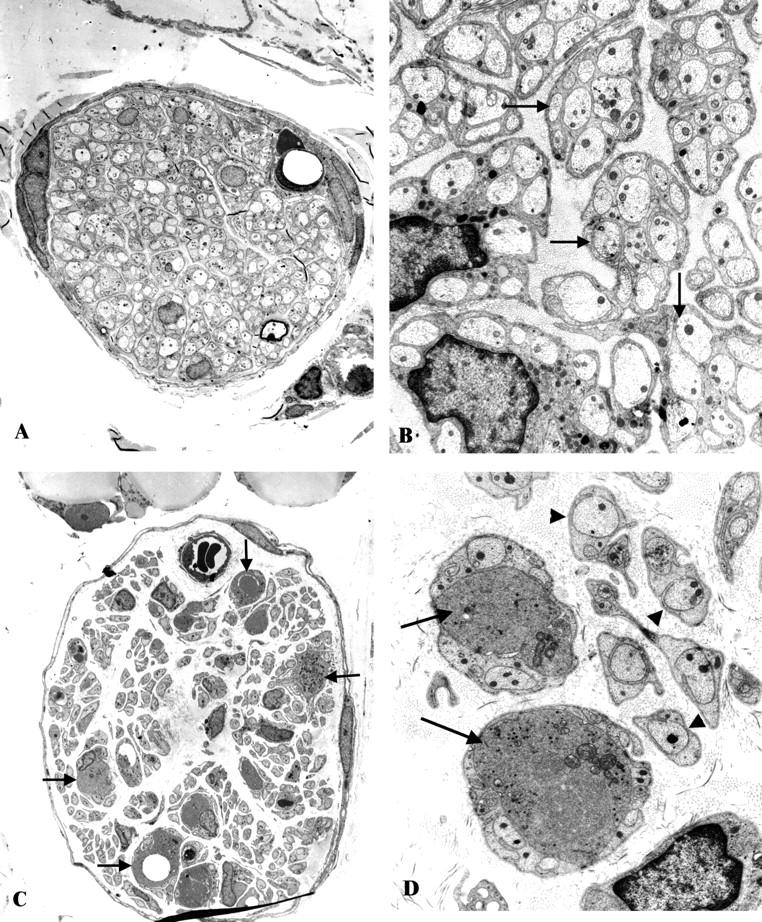
Neuroaxonal dystrophy in the ileal mesenteric nerves of ZDF- and STZ-diabetic rats. A and B: A typical mesenteric nerve fascicle in the ZDF rat (A) contains numerous normal appearing unmyelinated axons (arrows, B) without significant numbers of dystrophic axons. (Original magnification: A, ×1200; B, ×7500). C and D: A single mesenteric STZ-diabetic rat nerve fascicle (C) shows numerous unmyelinated axons (arrowheads, D) some of which demonstrate the ultrastructural appearance of neuroaxonal dystrophy (arrows, C and D). (Original magnification: C, ×1200; D, ×7500)
Discussion
The results of these experiments demonstrate that the type 2 diabetic ZDF rat model does not develop sympathetic NAD. ZDF rats maintain marked hyperglycemia in the presence of normal circulating levels of IGF-I and significantly increased circulating insulin levels. This is in striking contrast to STZ and BB/W rat models of type 1 diabetes in which NAD develops in a setting of comparable hyperglycemia and decreased circulating levels of insulin and IGF-I. 12-19 Similarly, NAD is a characteristic finding in the sympathetic ganglia of genetically diabetic Chinese hamsters 20 and STZ-treated and non-obese diabetic mice (Schmidt et al, unpublished data), which are also hyperglycemic, hypoinsulinemic and deficient in serum IGF-I. 21-23 These findings are consistent with the hypothesis that sympathetic NAD develops in response to loss of the neurotrophic effect of insulin and/or IGF-I, rather than hyperglycemia per se. Although a rat strain effect selectively conferring resistance to the development of NAD in ZDF rats could complicate this interpretation, all strains of insulinopenic rats (BB/W, 18 STZ-induced Lewis, 24,25 and STZ-induced Sprague-Dawley 8 ), mice (Schmidt et al, unpublished data), Chinese hamsters 20 and man examined to date have shown a diabetes-induced increase in NAD in prevertebral sympathetic ganglia. In addition, examination of ZDF rat somatic (sciatic) nerve has shown evidence of neuropathy similar to STZ-rats as characterized by reduced conduction velocity and morphological changes in myelinated axons. 9
These data combined with previous results, 8 are consistent with the argument that it is the loss of trophic support of insulin or IGF-I, rather than hyperglycemia, that causes NAD. However, the results in the ZDF rat do not permit us to absolutely distinguish between insulin, IGF-I or an as yet unidentified diabetes-induced substance as the critical determinant in the development of NAD. Nonetheless, a variety of observations suggest that IGF-I may be most relevant. We have found that pharmacological doses of rhIGF-I reverse established NAD within two months. 8 In that experiment a separate group of 6 month diabetic rats received a small daily dose of regular insulin (0.3 U/kg/day, s.c. to mimic the transient hypoglycemic effect of high doses of rhIGF-I) which did not affect levels of glycated hemoglobin nor did it influence the frequency of established NAD in the SMG and ileal mesenteric nerves. 8 The results in this experiment are consistent with the contention that IGF-I has a neurotrophic effect in this paradigm. The salutary effect of insulin at higher doses on NAD in the SMG and ileal mesenteric nerves in the STZ-rat in the absence of tight metabolic control 24,25 may reflect a direct insulin neurotrophic effect or an insulin-induced increase in hepatic synthesis of circulating IGF-I 26-28 which secondarily result in decreased sympathetic NAD.
Human subjects with type 2 diabetes (non-insulin-dependent diabetes mellitus, NIDDM) do develop sympathetic NAD, 29 a result which would seem to be at odds with our current findings in the type 2 diabetes ZDF rat model. However, a study of large numbers of adult human diabetics with NIDDM and insulin-dependent diabetes showed a 40% to 50% decrease in serum IGF-I in both groups. 30 Most studies have shown a decrease in circulating IGF-I levels in human diabetes, 30-36 although no change 37 or even an increase 38 has occasionally been reported. Moreover, other studies have reported that serum IGF-I levels are preferentially decreased in patients with sensory and autonomic neuropathic symptoms compared to either non-neuropathic diabetics or controls. 39 Levels of serum IGF-I also decline (≥30 to 40%) with aging in diabetic and control human subjects 5,30 suggesting a possible shared mechanism which may underlie the observed development of sympathetic ganglionic NAD (identical in distribution, ultrastructure, and immunohistochemistry to that which develops in diabetics) in aged rats, mice, Chinese hamsters, and humans. 5,40
A role for insulin and IGF-I deficiency has been proposed previously in the pathogenesis of diabetic sensory and autonomic neuropathies. 41,42 Insulin and IGF-I are known to bind to specific receptors on sensory and sympathetic neuronal perikarya, axons, and nerve terminals, 43 and support their survival in culture. 44,45 In vivo studies immunolocalize IGF-I to the cytoplasm of lumbar sympathetic neurons, 46 although it is not known if it is locally synthesized in sympathetic ganglia, perhaps by satellite cells, synthesized within endorgans and retrogradely transported within sympathetic axons or is derived from circulating IGF-I facilitated by the lack of a sympathetic ganglionic blood-nerve barrier. Decreased levels of mRNA transcripts for IGF-I and its receptor as well as diminished 125I-IGF-I binding to a crude membrane preparation have been reported in the SCG of STZ-treated rats diabetic for 3 months, 47 although this study did not determine whether message or 125I-IGF-I binding was localized to neurons, intraganglionic glia or both. Although little is known of satellite or Schwann cell IGF-I production in sympathetic ganglia, it is known that baseline IGF-I gene expression is reduced in Schwann cells of the sciatic nerves of STZ rats 48 and is poorly induced in response to nerve injury, 49 which may underlie the identified defect in somatic axonal regeneration in diabetic rats, 17,50 a defect ameliorated by exogenously administered IGF-I. 48 STZ-diabetic rats also show a 50% to 86% decrease in levels of circulating IGF-I and >75% loss of circulating insulin 12-17 as well as increased levels of IGFBP-1, one of several IGF-I binding proteins, which may sequester IGF-I, reducing its activity, or, alternatively, control its local availability. 41 Reduced levels of circulating IGFBP-3 are thought to result in faster elimination of rhIGF-I in STZ-diabetic rats. 51 Therefore, although there is evidence of a role for IGF-I in the pathogenesis of diabetic neuropathy, it is not clear if IGF-I is a circulating factor or provided by a local autocrine or paracrine process.
Evidence has also accumulated supporting the participation of abnormal collateral sprouting, frustrated axonal regeneration, and loss of synaptic plasticity (“synaptic dysplasia”) in the pathogenesis of neuroaxonal dystrophy. 6 Significantly, IGF-I is thought to have a role in normal synaptic development as well as nerve terminal plasticity, collateral axonal sprouting, and axonal regeneration. 41,52 A role for IGF-I in routine synaptic turnover and its deficiency in diabetes could underlie synaptic dysplasia and the development of NAD and represent a possible future therapeutic target.
The failure of the ZDF rat model of type 2 diabetes to develop the hallmark neuropathologic lesions of neuroaxonal dystrophy in the SMG and ileal mesenteric nerves in the presence of sustained hyperglycemia is complete and dramatic. The ZDF model may allow investigators a different approach to interfere with various protective pathways to induce pathology without interference from established pathology, ie, to determine whether biochemical changes anticipate or produce NAD or if NAD induces biochemical alterations in ganglionic biochemistry, which is difficult to answer in the STZ-rat model. Specifically, it may be possible to interfere selectively with circulating levels of IGF-I in ZDF rats to begin to focus on its contribution to the pathogenesis and eventual therapy of diabetic autonomic neuropathy.
Acknowledgments
We thank Dr. Mike Landt and the DRTC RIA Core staff for measurement of IGF-I and insulin levels, and Drs. Kevin Roth and Eugene M. Johnson, Jr. for helpful discussions. Streptozotocin was a gift of the Upjohn Company.
Footnotes
Address reprint requests to Robert E. Schmidt, M.D., Ph.D., Department of Pathology and Immunology, Box 8118, Washington University School of Medicine, 660 South Euclid Avenue, Saint Louis, MO 63110. Email: reschmidt@pathology.wustl.edu.
Supported by grant R37 DK19645 from the National Institutes of Health, and DRTC RIA Core grant 5 P60 DK20579.
References
- 1.Vinik AI, Holland MT, LeBeau JM, Liuzzi FJ, Stansberry KB, Colen LB: Diabetic neuropathies. Diabetes Care 1992, 15:1926-1975 [DOI] [PubMed] [Google Scholar]
- 2.Ewing DJ, Campbell IW, Clarke BF: The natural history of diabetic autonomic neuropathy. Quart J Med 1980, 49:95-108 [PubMed] [Google Scholar]
- 3.Rundles RW: Diabetic neuropathy: general review with report of 125 cases. Medicine (Baltimore) 1945, 24:111-160 [Google Scholar]
- 4.Hosking DJ, Bennett T, Hampton JR: Diabetic autonomic neuropathy. Diabetes 1978, 27:1043-1055 [DOI] [PubMed] [Google Scholar]
- 5.Schmidt RE: Neuropathology and pathogenesis of diabetic autonomic neuropathy. Neurobiology of Diabetic Neuropathy. Edited by DR Tomlinson. Int Rev Neurobiol 2002, 50:267-292 [DOI] [PubMed] [Google Scholar]
- 6.Schmidt RE: Synaptic dysplasia in sympathetic autonomic ganglia. J Neurocytol 1996, 25:777-791 [DOI] [PubMed] [Google Scholar]
- 7.Schmidt RE: Neuronal preservation in the sympathetic ganglia of rats with chronic streptozotocin-induced diabetes. Brain Research 2001, 921:256-259 [DOI] [PubMed] [Google Scholar]
- 8.Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Miller MS: Insulin-like growth factor I reverses experimental diabetic autonomic neuropathy. Am J Pathol 1999, 155:1651-1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson RG, Neel MA, Little LA, Kincaid JC, Eichberg J: Neuropathic complications in the Zucker diabetic fatty rat. Shafrir E eds. Frontiers in Diabetic Research: Lessons from Animal Diabetes III. 1990:pp 456-458 Smith-Gordon, London
- 10.Corsetti JP, Sparks JD, Peterson RG, Smith RL, Sparks CE: Effect of dietary fat on the development of non-insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats. Atherosclerosis 2000, 148:231-241 [DOI] [PubMed] [Google Scholar]
- 11.Peterson RG: The Zucker diabetic fatty (ZDF) rat. Sima AAF Shafrir E eds. Animal Models of Diabetes-A Primer. 2001:pp 109-128 Harwood Academic Publishers, Amsterdam
- 12.Baxter RC, Brown AS, Turtle JR: Decrease in serum receptor reactive somatomedin in diabetes. Horm Metab Res 1979, 11:216-220 [DOI] [PubMed] [Google Scholar]
- 13.Norby FL, Wold LE, Duan J, Hintz KK, Ren J: IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol 2002, 283:E658-E666 [DOI] [PubMed] [Google Scholar]
- 14.Maes M, Ketelslegers J-M, Underwood E: Low plasma somatomedin-C in streptozotocin-induced diabetes mellitus: correlation with changes in somatogenic and lactogenic liver binding sites. Diabetes 1983, 32:1060-1069 [DOI] [PubMed] [Google Scholar]
- 15.Binz K, Zapf J, Froesch ER: The role of insulin-like growth factor I in growth of diabetic rats. Acta Endocrinol 1989, 121:628-632 [DOI] [PubMed] [Google Scholar]
- 16.Busiguina S, Fernandez AM, Barrios V, Clark R, Tolbert DL, Berciano J, Torres-Aleman I: Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiol Disease 2000, 7:657-665 [DOI] [PubMed] [Google Scholar]
- 17.Ekstrom PAR, Kanje M, Skottner A: Nerve regeneration and serum levels of insulin-like growth factor-I in rats with streptozotocin-induced deficiency. Brain Res 1989, 496:141-147 [DOI] [PubMed] [Google Scholar]
- 18.Yagihashi S, Sima AAF: The distribution of structural changes in sympathetic nerves of the BB rat. Am J Pathol 1985, 121:138-147 [PMC free article] [PubMed] [Google Scholar]
- 19.Pierson CR, Zhang W, Murakawa Y, Sima AAF: Early gene responses of trophic factors in nerve regeneration differ in experimental type 1 and type 2 diabetic polyneuropathies. J Neuropathol Exp Neurol 2002, 61:857-871 [DOI] [PubMed] [Google Scholar]
- 20.Schmidt RE, Plurad DA, Plurad SB, Cogswell BE, Diani AR, Roth KA: Ultrastructural and immunohistochemical characterization of autonomic neuropathy in genetically diabetic Chinese hamsters. Lab Invest 1989, 61:77-92 [PubMed] [Google Scholar]
- 21.Segev Y, Landau D, Rasch R, Flyvbjerg A, Phillip M: Growth hormone receptor antagonism prevents early renal changes in nonobese diabetic mice. J Am Soc Nephrol 1999, 10:2374-2381 [DOI] [PubMed] [Google Scholar]
- 22.Flyvbjerg A, Bennett WF, Rasch R, Kopchick JJ, Scarlett JA: Inhibitory effect of a growth hormone receptor antagonist (G120K-PEG) on renal enlargement, glomerular hypertrophy, and urinary albumin excretion in experimental diabetes in mice. Diabetes 1999, 48:377-382 [DOI] [PubMed] [Google Scholar]
- 23.Landau D, Segev Y, Eshet R, Flyvbjerg A, Phillip M: Changes in the growth hormone-IGF-I axis in non-obese diabetic mice. Int J Exp Diab Res 2000, 1:9-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt RE, Plurad SB, Olack BJ, Scharp DW: The effect of pancreatic islet transplantation and insulin therapy on neuroaxonal dystrophy in sympathetic autonomic ganglia of chronic streptozocin-diabetic rats. Brain Res 1989, 497:393-398 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt RE, Plurad SB, Olack BJ, Scharp DW: The effect of pancreatic islet transplantation and insulin therapy on experimental diabetic autonomic neuropathy. Diabetes 1983, 32:532-540 [DOI] [PubMed] [Google Scholar]
- 26.Pao CI, Farmer PK, Begovic S, Goldstein S, Wu GJ, Phillips LS: Expression of hepatic insulin-like growth factor-I and insulin-like growth factor-binding protein-1 genes is transcriptionally regulated in streptozotocin-diabetic rats. Mol Endocrinol 1992, 6:969-977 [DOI] [PubMed] [Google Scholar]
- 27.Scott CD, Baxter RC: Production of insulin-like growth factor I and its binding protein in rat hepatocytes cultured from diabetic and insulin-treated diabetic rats. Endocrinol 1986, 119:2346-2352 [DOI] [PubMed] [Google Scholar]
- 28.Crosby SR, Tsigos C, Anderton CD, Gordon C, Young RJ, White A: Elevated plasma insulin-like growth factor binding protein-1 levels in Type 1 (insulin-dependent) diabetic patients with peripheral neuropathy. Diabetologia 1992, 35:868-872 [DOI] [PubMed] [Google Scholar]
- 29.Schmidt RE, Plurad SB, Parvin CA, Roth KA: The effect of diabetes and aging on human sympathetic autonomic ganglia. Am J Pathol 1993, 143:143-153 [PMC free article] [PubMed] [Google Scholar]
- 30.Tan K, Baxter RC: Serum insulin-like growth factor I levels in adult diabetic patients: the effect of age. J Clin Endocrinol Metab 1986, 63:651-655 [DOI] [PubMed] [Google Scholar]
- 31.Strasser-Vogel B, Blum WF, Past R, Kessler U, Hoeflich A, Meiler B, Kiess W: Insulin-like growth factor I and II and IGF binding proteins 1, 2, 3 in children and adults with diabetes mellitus: correlation with metabolic control and height attainment. J Clin Endocrinol Metab 1995, 80:1207-1213 [DOI] [PubMed] [Google Scholar]
- 32.Amiel SA, Sherwin RS, Hintz RL, Gertner JM, Press CM, Tamborlane WV: Effect of diabetes and its control on insulin-like growth factors in the young subject with type I diabetes. Diabetes 1984, 33:1175-1179 [DOI] [PubMed] [Google Scholar]
- 33.Taylor AM, Dunger DB, Grant DB, Preece MA: Somatomedin-C/IGF-I measured by radioimmunoassay and somatomedin bioactivity in adolescents with insulin-dependent diabetes compared with puberty matched controls. Diabetes Res 1988, 9:177-181 [PubMed] [Google Scholar]
- 34.Janssen JA, Jacobs ML, Derkx FH, Weber RF, van der Lely AJ, Lamberts SW: Free and total insulin-like growth factor I (IGF-I), IGF-binding protein-1 (IGFBP-1), and IGFBP-3 and their relationships to the presence of diabetic retinopathy and glomerular hyperfiltration in insulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1997, 82:2809-2815 [DOI] [PubMed] [Google Scholar]
- 35.Goke B, Fehmann HC: Insulin and insulin-like growth factor-I: their role as risk factors in the development of diabetic cardiovascular disease. Diabetes Res Clin Pract 1996, 30(Suppl 1):93-106 [DOI] [PubMed] [Google Scholar]
- 36.Clemmons DR: Role of peptide growth factors in development of macrovascular complications of diabetes. Diabetes Care 1991, 14:153-156 [DOI] [PubMed] [Google Scholar]
- 37.Zapf J, Morell B, Walter J, Laron Z, Froesch ER: Serum levels of insulin-like growth factor and its carrier protein in various metabolic disorders. Acta Endocrinol 1980, 95:505-517 [DOI] [PubMed] [Google Scholar]
- 38.Merimee TJ, Zapf J, Froesch ER: Insulin-like growth factors: studies in diabetics with and without retinopathy. N Engl J Med 1983, 309:527-530 [DOI] [PubMed] [Google Scholar]
- 39.Migdalis IN, Kalogeropoulou K, Kalantzis L, Nounopoulos C, Bouloukos A, Samartzis M: Insulin-like growth factor-I and IGF-I receptors in diabetic patients with neuropathy. Diab Med 1995, 12:823-827 [DOI] [PubMed] [Google Scholar]
- 40.Schmidt RE: The neuropathology of the aging autonomic nervous system. Duckett S eds. The Pathology of the Aging Human Nervous System. 2001:pp 431-448 Oxford University Press, New York
- 41.Ishii DN: Implication of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res Rev 1995, 20:47-67 [DOI] [PubMed] [Google Scholar]
- 42.Wuarin L, Guertin DM, Ishii DN: Early reduction in insulin-like growth factor gene expression in diabetic nerve. Exp Neurol 1994, 130:106-114 [DOI] [PubMed] [Google Scholar]
- 43.Karagiannis SN, King RHM, Thomas PK: Colocalization of insulin and IGF-I receptors in cultured rat sensory and sympathetic ganglion cells. J Anat 1997, 191:431-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernyhough P, Willars GB, Lindsay RM, Tomlinson DR: Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurons. Brain Res 1993, 607:117-124 [DOI] [PubMed] [Google Scholar]
- 45.Recio-Pinto E, Rechler MM, Ishii DN: Effects of insulin, insulin-like growth factor-II, and nerve growth factor in neurite formation and survival in cultured sympathetic and sensory neurons. J Neurosci 1986, 6:1211-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansson H-A, Rozell B, Skottner A: Rapid axoplasmic transport of insulin-like growth factor-I in the sciatic nerve of adult rats. Cell Tiss Res 1987, 247:241-247 [DOI] [PubMed] [Google Scholar]
- 47.Bitar MS, Pilchher CWT, Khan I, Waldbillig RJ: Diabetes-induced suppression of IGF-I and its receptor mRNA levels in rat superior cervical ganglia. Diab Res Clin Pract 1997, 38:73-80 [DOI] [PubMed] [Google Scholar]
- 48.Zhuang H-X, Snyder CK, Pu S-F, Ishii DN: Insulin-like growth factors reverse or arrest diabetic neuropathy: effects on hyperalgesia and impaired nerve regeneration in rats. Exp Neurol 1996, 140:198-205 [DOI] [PubMed] [Google Scholar]
- 49.Sima AAF, Merry AC, Levitan I: Increased regeneration in ARI-treated diabetic nerve is associated with upregulation of IGF-I and NGF receptors. Exp Clin Endocrinol Diabetes 1997, 105:60-62 [Google Scholar]
- 50.Longo FM, Powell HC, Lebeau J, Guerrero MR, Heckman H, Myers RR: Delayed nerve regeneration in streptozotocin diabetic rats. Muscle Nerve 1986, 9:385-393 [DOI] [PubMed] [Google Scholar]
- 51.Higaki K, Matsumoto Y, Fujimoto R, Kurosaki Y, Kimura T: Pharmacokinetics of recombinant human insulin-like growth factor-1 in diabetic rats. Drug Metab Dispos 1997, 25:1324-1327 [PubMed] [Google Scholar]
- 52.Cheng H-L, Randolph A, Yee D, Delafontaine P, Tennekoon G, Feldman EL: Characterization of insulin-like growth factor-I and its receptor and binding proteins in transected nerves and cultured Schwann cells. J Neurochem 1996, 66:525-536 [DOI] [PubMed] [Google Scholar]