

Abstract
Platelet microthrombi are present in the diabetic retinal vasculature of humans and rodents; however, the mechanisms and consequences of their presence have not been defined. The current study demonstrates that platelet containing microthrombi accumulate in the retinal vasculature of the rat within 2 weeks of experimental diabetes, a timepoint at which leukocyte-mediated endothelial cell injury and death are known to occur. Platelet accumulation increased with the duration of diabetes, and crossover experiments revealed that maximal platelet accumulation required both diabetic platelets and a diabetic endothelium. Platelet accumulation also coincided with the expression of Fas and FasL in the diabetic retina. When endothelial cell apoptosis was inhibited with an anti-FasL neutralizing antibody, platelet accumulation was effectively suppressed. When platelets were depleted from the systemic circulation with an anti-platelet antibody, blood-retinal barrier breakdown worsened in the diabetic animals. These findings suggest that platelet accumulation in the diabetic retinal vasculature is secondary to endothelial cell death and serves, in part, to suppress blood-retinal barrier breakdown.
Platelet microthrombi are present in the retinas of diabetic rats 1 and humans, 2 and have been spatially associated with apoptotic endothelial cells in the diabetic retinal vasculature. 2 Platelets are best understood for their hemostatic properties, which are altered in diabetes. Platelet adhesiveness is increased in diabetic patients 3,4 and diabetic platelets are more sensitive to aggregating agents and produce more thromboxane A2. 5-7 The final common pathway of platelet aggregation requires the platelet glycoprotein receptor IIb-IIIa (GP IIb-IIIa or αIIbβ3). Based in part on these data, the Early Treatment Diabetic Retinopathy Study (ETDRS) administered low (anti-platelet) doses of aspirin to diabetic patients with non-proliferative retinopathy; however, no beneficial effects were observed. 8
In addition to their hemostatic role, platelets can also participate in inflammation. Within seconds of platelet activation, the platelet inflammatory mediator CD40 ligand (CD40L, CD154), a transmembrane protein structurally related to the cytokine TNF-α, is translocated to the cell surface. 9 CD40L binds to CD40 on endothelial cells, triggering the expression of ICAM-1, 10,11 VCAM-1, 11 IL-8, 12 and MCP-1, 12 among others. These proinflammatory molecules mediate the extravasation of leukocytes at sites of vascular injury. In addition, platelets contain pre-formed vascular endothelial growth factor (VEGF) 13 and platelet-derived growth factor (PDGF), 14 which are released on platelet activation. Thus, in addition to their hemostatic role, platelets can participate in inflammation and release growth factors that directly affect the vasculature.
Leukocytes adhere to the retinal vasculature very early in diabetes, a phenomenon that requires ICAM-1. 15 Recently, it was shown that adherent leukocytes can induce retinal endothelial cell apoptosis in the diabetic retinal vasculature via a Fas/FasL dependent mechanism. 16 Since injured and dying vascular endothelial cells are pro-coagulant in nature, 17 we hypothesized that this mechanism may underlie the accumulation of platelet microthrombi in the diabetic retinal vasculature. The current study explored the mechanisms underlying platelet accumulation in the diabetic retinal vasculature and characterized the functional consequences to the blood-retinal barrier.
Materials and Methods
Materials
The anti-rat MFL4 antibody against FasL (Armenian hamster IgG) and control antibody (Armenian hamster anti-Trinitrophenol (TNP) IgG) were purchased from PharMingen (San Diego, CA). Rabbit anti-rat Fas polyclonal antibody (FL-335, sc-7886) and rabbit anti-rat FasL polyclonal antibody (C-178, sc6237) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit antiserum against coagulation factor XIIIA subunit was purchased from Calbiochem (San Diego, CA). The antibodies were free of endotoxin, as determined by the manufacturer.
Experimental Diabetes
All experiments were performed in accordance with the Statement for the Use of Animals in Ophthalmology and Vision Research from the Association for Research in Vision and Ophthalmology and were approved by the Animal Care and Use Committee of the Massachusetts Eye and Ear Infirmary. Male Long-Evans rats weighing approximately 200 g received a single 60 mg/kg intraperitoneal injection of streptozotocin (Sigma, St. Louis, MO) in 10 mmol/L citrate buffer (pH 4.5) after an overnight fast. Control non-diabetic animals received citrate buffer alone. Animals with blood glucose levels greater than 250 mg/dl after 24 hours were considered diabetic. The animals were allowed free access to standard laboratory chow and water in an air-conditioned room with a 12-hour light-dark cycle until they were used for the experiments. Blood glucose levels and body weights at the time of experimentation are shown in Table 1 ▶ .
Table 1.
Blood Glucose Levels and Body Weights of Rats in Diabetes Experiments
| Parameters | Control | Diabetes | |||
|---|---|---|---|---|---|
| 0 weeks | 2 weeks | 1 week | 2 weeks | 4 weeks | |
| Blood glucose (mg/dl) | 120 ± 8 | 116 ± 9 | 341 ± 29 | 329 ± 25 | 364 ± 27 |
| Body weight (g) | 204 ± 5 | 319 ± 11 | 232 ± 9 | 251 ± 9 | 294 ± 11 |
Platelet Isolation
The method to evaluate platelet-endothelial interactions in the retinal vessels has been previously described elsewhere. 18-20 Blood samples from donor rats were harvested from the abdominal artery and collected in polypropylene tubes containing 2 ml of 38 mOsM citric acid, 75 mmol/L trisodium citrate, and 100 mOsM dextrose. The blood was centrifuged at 250 × g for 10 minutes. Platelet-rich plasma was gently transferred to a fresh tube and centrifuged at 2000 × g for 10 minutes. The platelet pellet was resuspended in Hanks’ balanced salt solution and incubated with 78 μmol/L of carboxyfluorescein diacetate succinimidyl ester (CFDASE; Molecular Probes, Eugene, OR) for 30 minutes at 37°C. CFDASE is a non-fluorescent precursor that diffuses into cells and forms a stable fluorochrome carboxyfluorescein succinimidyl ester (CFSE; peak absorbance, 492 nm; peak emission, 518 nm) after catalysis by esterase. After incubation, the platelet suspension was centrifuged again at 2000 × g for 10 minutes. The platelet pellet was resuspended in Hanks’ balanced salt solution at a concentration of 1 × 109 platelets/0.3 ml.
Platelet Infusion
Immediately before platelet injection, the rats were anesthetized with a 1:1 mixture of xylazine hydrochloride (4 mg/kg) and ketamine hydrochloride (10 mg/kg). Each rat had CFSE-labeled platelets (1 × 109/0.3 ml) infused into the tail vein. Twenty-four hours after platelet infusion, the rats were again anesthetized and the pupils were dilated with 1% tropicamide. A contact lens was placed on the eye and the fundus was observed with the scanning laser ophthalmoscope (SLO; Rodenstock Instruments, Munich, Germany) with 40°-field setting. The argon blue laser was used as the illumination source and the standard fluorescein angiography filter was in place. Trapped platelets were recognized as fluorescent dots in the retina. The images were recorded on S-VHS videotape at a rate of 30 frames/second. The number of trapped platelets was counted within a radius of 5-disk diameters from the center of the optic disk.
Western Blot Analysis
The retinas were homogenized in lysis buffer and centrifuged at 4°C for 10 minutes. The supernatant was collected and mixed with sample buffer. Each sample containing 100 μg of total protein was then boiled for 2 minutes, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and electroblotted to polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). After blocking nonspecific binding with 5% normal goat serum, the membranes were incubated with a rabbit anti-rat Fas polyclonal antibody (1:200) or a rabbit anti-rat FasL polyclonal antibody (1:200) at room temperature for 1 hour, followed by incubation with a horseradish peroxidase-conjugated goat antibody against rabbit immunoglobulin. The signals were visualized with the Opti-4CN substrate kit (Bio-Rad) according to the manufacturer’s protocol.
Immunofluorescence
Twenty-four hours after the infusion of 1 × 109 CFSE-labeled platelets, the rats were anesthetized, the chest cavity was opened, and a 14-gauge perfusion cannula was introduced into the aorta. Achieving drainage from the right atrium, each rat was perfused with PBS (500 ml/kg b.wt.) to remove the non-adherent blood cells. After the perfusion, the eyes were enucleated and the retinas were carefully removed. Flat-mounted retinas were then fixed with 4% paraformaldehyde for 1 hour at 4°C. After fixation, they were washed three times (30 minutes each) with 0.01% Triton (Sigma) in PBS at 4°C. Nonspecific binding was blocked with 5% normal goat serum in PBS containing 0.05% Triton X for 24 hours at 4°C. The retinas were incubated overnight at 4°C with Factor XIIIA antiserum diluted 1:1000, followed by incubation with a Texas Red-conjugated goat antibody against rabbit immunoglobulin. The flat mounts were prepared using a mounting medium for fluorescence and imaged using a fluorescence microscope.
Propidium Iodide Labeling
Dead and injured endothelial cells were labeled in vivo using propidium iodide (PI; Molecular Probes). 16,21,22 Twenty-four hours after the infusion of 1 × 109 CFSE-labeled platelets, the rats were anesthetized and PI (1 mg/ml in PBS) was injected intravenously via the tail vein at a concentration of 5 μmol/kg. After 20 minutes, the chest cavity was opened and a 14-gauge perfusion cannula was introduced into the aorta. Achieving drainage from the right atrium, each rat was perfused with PBS (500 ml/kg b.wt.) to clear the remaining intravascular PI. After the perfusion, the eyes were enucleated and the retinas were carefully isolated and removed. Retinal flat mounts were examined by fluorescence microscopy as described above.
Measurement of Blood-Retinal Barrier Breakdown with Fluorescein Isothiocyanate (FITC)-Dextran
After deep anesthesia with xylazine hydrochloride and ketamine hydrochloride, fluorescein isothiocyanate (FITC)-conjugated dextran (4.4 kd, 50 mg/ml in PBS, 50 mg/kg b.wt.; Sigma) was injected intravenously. After 10 minutes, the chest cavity was opened and a 14-gauge perfusion cannula was introduced into the aorta. A blood sample was collected immediately before perfusion. Achieving drainage from the right atrium, each rat was perfused with PBS (500 ml/kg b.wt.) to clear the remaining intravascular dextran. The blood sample was centrifuged at 7000 rpm for 20 minutes at 4°C, and the supernatant was diluted at 1:1000. Immediately following perfusion, the retinas were carefully removed, weighed, and homogenized to extract the FITC-dextran in 0.4 ml of water. The extract was processed through a 30,000-molecular weight filter (Ultrafree-MC; Millipore, Bedford, MA) at 7000 rpm for 90 minutes at 4°C. The fluorescence in each 300-μl sample was measured (excitation, 485 nm; emission, 538 nm) using a spectrofluorometer (SPECTRAmax GEMINI XS; Molecular Devices, Sunnyvale, CA) with water as a blank. Corrections were made by subtracting the autofluorescence of retinal tissue from rats without FITC-dextran injection. The amount of FITC-dextran in each retina was calculated from a standard curve of FITC-dextran in water. For normalization, the retinal FITC-dextran amount was divided by the retinal weight and by the concentration of FITC-dextran in the plasma. Blood-retinal barrier breakdown was calculated using the following equation, with the results being expressed in μl × g−1 × h−1.
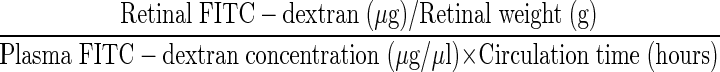 |
The formula is identical to that used in the Evans blue dye leakage assay. 23-27
Statistical Analysis
All values are presented as means ± SEM. The data were analyzed by Whitney-Mann-U-test with post hoc comparisons tested using Fisher’s protected least significant difference procedure. Differences were considered statistically significant at P < 0.05.
Results
Platelets Accumulate in the Diabetic Retinal Vasculature in a Time-Dependent Manner
When the retina was observed with the SLO 24 hours after CFSE-labeled platelet infusion, the platelets trapped in the retinal vasculature were observed as stable fluorescent dots (Figure 1, A and B) ▶ . The number of trapped platelets was elevated in non-diabetic rats, and 1-, 2-, and 4-week diabetic rats. Platelet accumulation was significantly elevated at 2 weeks of diabetes (P < 0.01) and continued to increase up to 4 weeks (Figure 1C) ▶ . Measurements beyond the 4-week time points were precluded by cataract.
Figure 1.

Platelet accumulation in the diabetic retinal circulation. Labeled platelets were visualized as white dots (arrows) in the retinas of non-diabetic rats (A) and 2-week diabetic rats (B). C: Time course of platelet adhesion in the retinas of diabetic rats. Values represent the mean ± SEM *P < 0.01 versus non-diabetic rats.
Platelets Are Contained within Microthrombi
To confirm that the platelets seen with the SLO are present within microthrombi, retinas from 2-week diabetic rats were isolated 24 hours after CFSE-labeled 2-week diabetic platelet injection, and were immunostained for coagulation factor XIIIA. Non-immune rabbit serum was used as a negative control. Factor XIIIA staining colocalized with the CFSE-positive platelets in the retinal vessels (Figure 2) ▶ . Staining was absent in the negative control (data not shown).
Figure 2.

Immunofluorescence for Factor XIIIA in the retinas of rats 24 hours after platelet infusion. Green fluorescence from the CFSE-labeled adhering platelets (arrowheads in A) and red fluorescence from the anti-Factor XIIIA antibody (arrows in B). Co-localization was confirmed when the images were superimposed (C).
Platelet Accumulation Requires Diabetic Platelets and a Diabetic Vasculature
When the platelets of 2-week diabetic rats were infused into non-diabetic rats, platelet accumulation was slightly increased, but the degree of accumulation did not differ from that observed when non-diabetic platelets were infused into non-diabetic rats (Figure 3) ▶ . Similarly, when the platelets of 2-week non-diabetic rats were infused into the diabetic rats, the degree of accumulation was not significantly increased above that observed when non-diabetic platelets were infused into non-diabetic rats.
Figure 3.

Effect of diabetic platelets and endothelial cells on platelet adhesion. N to N, platelets from non-diabetic rats infused into non-diabetic rats; D to D, platelets from 2-week diabetic rats infused into 2-week diabetic rats; N to D, platelets from non-diabetic rats infused into 2-week diabetic rats; D to N, platelets from 2-week diabetic rats infused into non-diabetic rats. Values represent the mean ± SEM *P < 0.01 versus N to N.
The Expression of Fas and FasL Are Increased in Early Diabetes
Two weeks following the onset of diabetes, the expression of Fas and FasL in diabetic retina was evaluated via Western blotting and compared to non-diabetic retina. Figure 4 ▶ demonstrates that Fas and FasL levels were increased in the retinas of 2-week diabetic rats.
Figure 4.

Western blotting for Fas (top) and FasL (bottom) showed that retinal Fas and FasL protein levels in 2-week diabetic rats (DM2w) were elevated compared to non-diabetic rats (non-DM). Similar results were obtained in three separate experiments.
An Anti-FasL Antibody Suppresses Platelet Accumulation
Previous work has demonstrated that the Fas/FasL pathway plays an important role in the apoptosis of retinal endothelial cells in experimental diabetes. The administration of an anti-FasL antibody suppressed the diabetes-induced apoptosis of retinal endothelial cells. 16 To determine whether platelet accumulation is secondary to retinal cell apoptosis, 2-week diabetic rats received intraperitoneal injections of either an anti-FasL antibody (1 mg/kg) or an isotype control antibody (1 mg/kg). Labeled platelets from untreated 2-week diabetic rats were infused 2 days later. As shown in Figure 5 ▶ , the anti-FasL antibody significantly suppressed platelet accumulation in the retina (P < 0.01).
Figure 5.
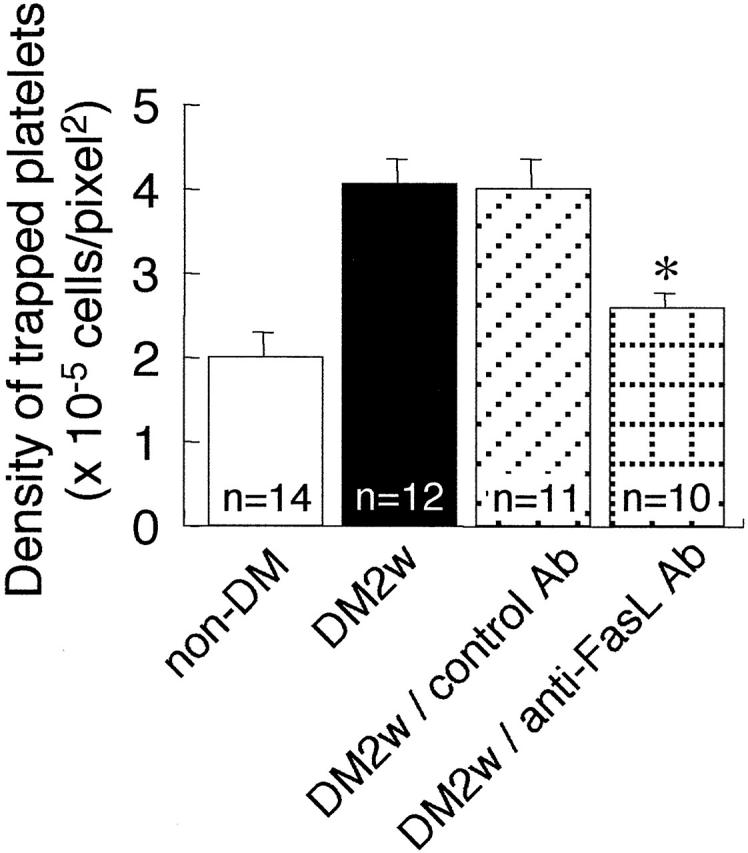
Effect of an anti-FasL neutralizing antibody on platelet trapping. non-DM, platelets from non-diabetic rats were infused into non-diabetic rats; DM2w, platelets from 2-week diabetic rats were infused into 2-week diabetic rats; DM2w/control Ab, platelets from 2-week diabetic rats were infused into 2-week diabetic rats treated with the control antibody; DM2w/anti-FasL Ab, platelets from 2-week diabetic rats were infused into 2-week diabetic rats treated with the anti-FasL antibody. Values represent the mean ± SEM *P < 0.01 versus DM2w.
Platelets Colocalize to Areas of Retinal Endothelial Injury
To further confirm the role of vascular injury in platelet accumulation, the PI labeling technique was used. Injured and/or dead endothelial cells in the retinal vasculature of 2-week diabetic rats were labeled with PI 24 hours after CFSE-labeled 2-week diabetic platelets were injected. PI-positive endothelial cells could be seen colocalizing with the platelets in the retinal vasculature (Figure 6) ▶ .
Figure 6.

Dying and/or injured retinal endothelial cells were labeled in vivo with propidium iodide (PI) 24 hours after platelet infusion into 2-week diabetic rats. Co-localization of green fluorescence from the CFSE-labeled trapped platelets (arrowheads in A) and red fluorescence from the PI-labeled endothelial cells (arrows in B) was confirmed when the images were superimposed (C).
Platelet Depletion Worsens Diabetic Blood-Retinal Barrier Breakdown
An anti-platelet serum (1 ml/kg) was administered via intraperitoneal injection and efficiently reduced the circulating platelet count within 3 hours (Figure 7) ▶ . Significant thrombocytopenia was present for at least 24 hours. Blood-retinal barrier was evaluated with using 4400 MW FITC-dextran in non-diabetic and 2-week diabetic rats 24 hours after the administration of the anti-platelet serum or rabbit control serum. In the non-diabetic rats, the anti-platelet serum has no effect on blood-retinal barrier breakdown (Figure 8) ▶ . In contrast, thrombocytopenia significantly increased the blood-retinal barrier breakdown in the diabetic rats.
Figure 7.

Time course of platelet counts in the peripheral blood of normal rats following the intraperitoneal administration of the anti-platelet serum (APS). Values represent the mean ± SEM (n = 6 at each time point).
Figure 8.

The effect of platelet depletion on blood-retinal barrier breakdown. non-DM, non-diabetic rats; non-DM + APS, non-diabetic rats treated with intraperitoneal anti-platelet serum; DM2w, 2-week diabetic rats; DM2w + APS, 2-week diabetic rats treated with the anti-platelet serum. Values represent the mean ± SEM *P, < 0.01 versus non-DM. †P, < 0.01 versus DM2w.
Discussion
The current study used a relatively new SLO methodology to track platelet dynamics and found that platelets accumulated in the retinal circulation within two weeks of experimental diabetes. Some of these platelets were localized within microthrombi and colocalized to damaged endothelial cells. Maximum platelet accumulation required both diabetic platelets and a diabetic vasculature. Fas and FasL expression coincided with the previously described onset of retinal endothelial cell death. 16,21 Moreover, the inhibition of endothelial cell apoptosis with an anti-FasL antibody prevented the accumulation of platelets. When the endothelial cell death was allowed to proceed in the absence of platelets, diabetic blood-retinal barrier breakdown worsened.
It is known that adhesiveness of diabetic platelets is increased. 3,4 Platelets from diabetic patients are more sensitive to aggregating agents and produce more thromboxane A2. 5-7 We speculate that these alterations likely contributed to the increased platelet adhesion observed in the current study. It is also known that adherent leukocytes begin to injure the diabetic retinal endothelium after only 1 week of experimental diabetes. 16,21 With the onset of endothelial cell injury and death, the retinal vasculature becomes more pro-coagulant in nature. We speculate that these two features, namely endothelial cell injury and activated diabetic platelets, likely underlie the maximal platelet accumulation observed with diabetic platelets and a diabetic endothelium.
The onset of platelet accumulation correlated with the increased expression of Fas and FasL; the latter two molecules were recently shown to be operative in the leukocyte-mediated apoptotic death of the diabetic retinal vasculature. 16 The same study demonstrated that Fas/FasL-mediated endothelial apoptosis was causal, in part, of the breakdown of the blood-retinal barrier in early diabetes. In this context, it is not surprising that the inhibition of FasL would lead to the suppression of platelet accumulation. Boeri et al 2 demonstrated an association of microthrombi with apoptotic vascular cells in the retinas of diabetic patients. The current data suggest that these apoptotic vascular cells are causal for the accumulation of platelets.
Some groups have reported that the apoptosis of endothelial cells cannot be detected in trypsin-digests of the retinal vasculature until after 6 to 8 months of experimental diabetes using the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) technique. 28,29 This observation stands in contrast to the finding of TUNEL- and M30-positive endothelial cells in paraffin-embedded retinal sections from 2-week diabetic rats. 16 In support of an earlier onset of death, De La Cruz and associates 30 demonstrated that the retinal vascular area decreases significantly after only 15 days of experimental diabetes in rats. Barber and associates 31 have also observed neural cell death within 1 month of experimental diabetes. In light of the methodological variations, we suspect that the time course differences listed above may be method-dependent.
Platelets are known to preserve the vascular integrity of various organs. Hemostasis in injured and compromised microvessels may prevent more serious hemorrhage and edema. Platelet-derived beneficial effects may also result from the release of VEGF 13 and PDGF, 14 growth factors which can promote the maintenance and healing of the vasculature. 32,33 We speculate that the low (anti-platelet) doses of aspirin used in the ETDRS may have suppressed these beneficial effects.
To summarize, these data are the first to demonstrate an underlying cause for the accumulation of platelet microthrombi in the diabetic retinal circulation. They also identify a beneficial effect arising from the accumulations of platelets, at least in early retinopathy, the suppression of blood-retinal barrier breakdown.
Footnotes
Address reprint requests to Anthony P. Adamis, Eyetech Research Center, 42 Cummings Park, Woburn, MA 01801. E-mail: tony.adamis@eyetk.com.
Supported by the Roberta W. Siegel Fund, the National Institutes of Health (grants EY12611 and EY11627), the Juvenile Diabetes Foundation, the Falk Foundation, the Iaccoca Foundation, and Eyetech Pharmaceuticals.
References
- 1.Ishibashi T, Tanaka K, Taniguchi Y: Platelet aggregation and coagulation in the pathogenesis of diabetic retinopathy in rats. Diabetes 1981, 30:601-606 [DOI] [PubMed] [Google Scholar]
- 2.Boeri D, Maiello M, Lorenzi M: Increased prevalence of microthromboses in retinal capillaries of diabetic individuals. Diabetes 2001, 50:1432-1439 [DOI] [PubMed] [Google Scholar]
- 3.Shaw S, Pegrum GD, Wolff S, Ashton WL: Platelet adhesiveness in diabetes mellitus. J Clin Pathol 1967, 20:845-847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bridges J, Dalby A, Millar J, Weaver J: An effect of d-glucose on platelet stickiness. Lancet 1965, 1:75-77 [DOI] [PubMed] [Google Scholar]
- 5.Colwell JA, Winocour PD, Halushka PV: Do platelets have anything to do with diabetic microvascular disease? Diabetes 1983, 32(Suppl 2):14-19 [DOI] [PubMed] [Google Scholar]
- 6.Bensoussan D, Levy Toledano S, Passa P, Caen J, Caniver J: Platelet hyperaggregation and increased plasma level of von Willebrand factor in diabetics with retinopathy. Diabetologia 1975, 11:307-312 [DOI] [PubMed] [Google Scholar]
- 7.Heath H, Brigden WD, Canever JV, Pollock J, Hunter PR, Kelsey J, Bloom A: Platelet adhesiveness and aggregation in relation to diabetic retinopathy. Diabetologia 1971, 7:308-315 [DOI] [PubMed] [Google Scholar]
- 8.: Early Treatment Diabetic Retinopathy Study Research Group: Effects of aspirin treatment on diabetic retinopathy: ETDRS report number 8. Ophthalmology 1991, 98:757-765 [PubMed] [Google Scholar]
- 9.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA: Cd40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391:591-594 [DOI] [PubMed] [Google Scholar]
- 10.Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, Aruffo A: Expression of functional cd40 by vascular endothelial cells. J Exp Med 1995, 182:33-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karmann K, Hughes CC, Schechner J, Fanslow WC, Pober JS: Cd40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci USA 1995, 92:4342-4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monaco C, Andreakos E, Young S, Feldmann M, Paleolog E: T cell-mediated signaling to vascular endothelium: induction of cytokines, chemokines, and tissue factor. J Leukoc Biol 2002, 71:659-668 [PubMed] [Google Scholar]
- 13.Mohle R, Green D, Moore MA, Nachman RL, Rafii S: Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci USA 1997, 94:663-668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linder BL, Chernoff A, Kaplan KL, Goodman DS: Release of platelet-derived growth factor from human platelets by arachidonic acid. Proc Natl Acad Sci USA 1979, 76:4107-4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP: Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA 1999, 96:10836-10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joussen AM, Poulaki V, Mitsiades N, Cai WY, Suzuma I, Pak J, Ju ST, Rook SL, Esser P, Mitsiades CS, Kirchhof B, Adamis AP, Aiello LP: Suppression of fas-fasl-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. EMBO J 2003, 17:76-78 [DOI] [PubMed] [Google Scholar]
- 17.Bombeli T, Karsan A, Tait JF, Harlan JM: Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89:2429-2442 [PubMed] [Google Scholar]
- 18.Tsujikawa A, Kiryu J, Nonaka A, Yamashiro K, Nishiwaki H, Tojo SJ, Ogura Y, Honda Y: In vivo evaluation of platelet-endothelial interactions in retinal microcirculation of rats. Invest Ophthalmol Vis Sci 1999, 40:2918-2924 [PubMed] [Google Scholar]
- 19.Nishijima K, Kiryu J, Tsujikawa A, Honjo M, Nonaka A, Yamashiro K, Tanihara H, Tojo SJ, Ogura Y, Honda Y: In vivo evaluation of platelet-endothelial interactions after transient retinal ischemia. Invest Ophthalmol Vis Sci 2001, 42:2102-2109 [PubMed] [Google Scholar]
- 20.Tsujikawa A, Kiryu J, Yamashiro K, Nonaka A, Nishijima K, Honda Y, Ogura Y: Interactions between blood cells and retinal endothelium in endotoxic sepsis. Hypertension 2000, 36:250-258 [DOI] [PubMed] [Google Scholar]
- 21.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP: Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 2001, 158:147-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joussen AM, Poulaki V, Tsujikawa A, Qin W, Qaum T, Xu Q, Moromizato Y, Bursell SE, Wiegand SJ, Rudge J, Ioffe E, Yancopoulos GD, Adamis AP: Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol 2002, 160:1683-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Q, Qaum T, Adamis AP: Sensitive blood-retinal barrier breakdown quantitation using evans blue. Invest Ophthalmol Vis Sci 2001, 42:789-794 [PubMed] [Google Scholar]
- 24.Qaum T, Xu Q, Joussen AM, Clemens MW, Qin W, Miyamoto K, Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP: Vegf-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci 2001, 42:2408-2413 [PubMed] [Google Scholar]
- 25.Joussen AM, Poulaki V, Qin W, Kirchhof B, Mitsiades N, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP: Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol 2002, 160:501-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulaki V, Qin W, Joussen AM, Hurlbut P, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP: Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor-1α and vegf. J Clin Invest 2002, 109:805-815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP: Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via tnf-alpha suppression. EMBO J 2002, 16:438-440 [DOI] [PubMed] [Google Scholar]
- 28.Mizutani M, Kern TS, Lorenzi M: Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest 1996, 97:2883-2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kern TS, Tang J, Mizutani M, Kowluru RA, Nagaraj RH, Romeo G, Podesta F, Lorenzi M: Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci 2000, 41:3972-3978 [PubMed] [Google Scholar]
- 30.De la Cruz JP, Moreno A, Munoz M, Garcia Campos JM, Sanchez de la Cuesta F: Effect of aspirin plus dipyridamole on the retinal vascular pattern in experimental diabetes mellitus. J Pharmacol Exp Ther 1997, 280:454-459 [PubMed] [Google Scholar]
- 31.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW: Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 1998, 102:783-791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 33.Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M: Pdgf-bb modulates endothelial proliferation and angiogenesis in vitro via pdgf β-receptors. J Cell Biol 1994, 125:917-928 [DOI] [PMC free article] [PubMed] [Google Scholar]