

Abstract
Acute renal injury causes accumulation of free and esterified cholesterol (FC, CE) in proximal tubules, mediated, at least in part, by increased cholesterol synthesis. Normally, this would trigger compensatory mechanisms such as increased efflux and decreased influx to limit or reverse the cholesterol overload state. This study sought to determine the integrity of these compensatory pathways following acute renal damage. Rhabdomyolysis-induced acute renal failure was induced in mice by glycerol injection. Normal mice served as controls. After 18 hours, BUN levels and renal cortical FC/CE content were determined. Expression of ABCA-1 and SR-B1 (cholesterol efflux proteins) were assessed by Western blot. Renal cortical LDL receptor (LDL-R; a cholesterol importer) regulation was gauged by quantifying its mRNA. To obtain proximal tubule cell-specific data, the impact of oxidant (Fe) stress on cultured HK-2 cell LDL-R, SR-B1, and ABCA-1 proteins and their mRNAs (versus controls) was assessed. Glycerol evoked marked azotemia and striking FC/CE increments (44%, 384%, respectively). Paradoxically, renal cortical SR-B1 and ABCA-1 protein reductions and LDL-R mRNA increments resulted. Fe-induced injury suppressed HK-2 cell SR-B1, ABCA-1, and their mRNAs. LDL-R protein rose with the in vitro Fe challenge. Renal tubular cell injury causes dysregulation of SR-B1, ABCA-1, and LDL-R protein expression, changes which should contribute to a cholesterol overload state. Reductions in HK-2 cell SR-B1 and ABCA-1 mRNAs and increases in renal cortical LDL-R mRNA imply that this dysregulation reflects, at least in part, altered genomic/transcriptional events.
Recent studies have demonstrated that cholesterol accumulation within proximal tubular cells is an integral component of the renal stress response. 1 This is based on observations that all forms of acute renal injury tested to date (toxic, ischemic, thermal, obstructive, inflammatory) induce accumulation of free cholesterol (FC) and cholesteryl esters (CEs) within the renal cortex. 1-9 These changes occur within 18 to 24 hours following the onset of renal injury, and they persist for at least as long as the underlying renal damage. 7 Proximal tubules isolated from damaged renal cortex have striking cholesterol increases, 2,9 and cultured proximal tubule (HK-2) cells accumulate cholesterol following in vitro injury, 4 indicating that the observed renal cortical cholesterol elevations reflect, at least in part, proximal tubule cell events. The pathophysiologic consequences of these stress-induced cholesterol increments have not been fully defined. However, a large body of experimental data indicate that they protect the kidney from further injury, a phenomenon known as the acquired cytoresistant state. 2-4,9,10-12
The mechanism(s) which trigger and maintain post-injury tubular cell cholesterol accumulation have not been completely defined. To date, our laboratory has most fully evaluated this issue in the glycerol model of rhabdomyolysis-induced acute renal failure (ARF). Because this form of in vivo renal injury is, in large part, mediated via heme Fe-induced oxidative stress, 13-16 we have simulated it in cultured proximal tubular (HK-2) cells by the addition of a catalytic Fe (free-radical-generating) challenge. The results of these parallel in vivo and in vitro investigations indicate that increased tubular synthesis contributes to the cholesterol overload state, based on the following information: first, statin-induced inhibition of HMG CoA reductase (HMGCR) prevents cholesterol accumulation in Fe-challenged HK-2 cells; 4 and second, acute cell injury increases renal cortical HMGCR protein and its mRNA. 1,8
Cellular cholesterol levels are normally tightly regulated, maintaining near constant values. 17 Thus, increased synthesis, under otherwise normal circumstances, should trigger a compensatory increase in cholesterol efflux and a decrease in influx. Efflux occurs, at least in part, via the ATP binding cassette (ABC) A1 transporter, which shuttles cholesterol to apo-A1 at, and then away from, the plasma membrane. 18-22 FC efflux may also occur via scavenger receptor B1 (SR-B1 23-26 ; ie, an HDL receptor 27,28 ). Following esterification, the effluxed cholesterol is transported to, and then taken up by, a hepatic SR-B1-dependent pathway. Thus, by both extruding FC from extrahepatic cells, and by importing CEs into the liver, SR-B1 helps mediate reverse cholesterol transport (ie, trafficking extrahepatic cholesterol back to the liver 29 ). In contrast to ABCA-1 and SR-B1, the LDL receptor (LDL-R) facilitates cellular cholesterol uptake. 17 Given these considerations, it would be predicted that a normal compensatory response to increased renal tubular cell cholesterol synthesis would be increased ABCA-1, increased SR-B1, and decreased LDL-R expression.
The above suggests that increased synthesis alone is insufficient to cause post- injury cholesterol overload unless dysregulation of normal compensatory pathways co-exists. Indeed, a recent study from this laboratory provides some limited support for this hypothesis, since SR-B1 levels were found to be decreased following sepsis (or glycerol)- induced ARF. 9 Hence, the present study was undertaken to further explore the status of cellular cholesterol transporters and their mRNAs in a post-injury cholesterol overload state.
Materials and Methods
Glycerol-Induced Acute Renal Injury
Male CD-1 mice (25 to 35 g; Charles River Laboratories, Wilmington, MA) were used for all in vivo experiments. Rhabdomyolysis-associated ARF was induced by intramuscular glycerol injection (50%; 9 ml/kg, administered in equally divided doses into the upper hind limbs under light anesthesia with isoflurane). Following glycerol injection, the mice were allowed to recover from anesthesia and provided with free food and water access. At either 4 or 18 hours following glycerol injection, the mice were deeply anesthetized with pentobarbital (1 to 2 mg; i.p. injection). For those mice which had received glycerol injection 4 hours prior, the kidneys were resected through a midline abdominal incision and placed on ice. For mice injected with glycerol ∼18 hours previously, a ∼200-μl heparinized blood sample was withdrawn from the inferior vena cava for urea nitrogen (BUN) analysis. This was immediately followed by kidney resection. The renal cortices were dissected on ice with a razor blade and then used for either: 1) free cholesterol and cholesteryl ester quantification (chloroform:methanol extraction, followed by gas chromatographic analysis, as previously described in detail 4 ); or 2) Western blot analysis of either ABCA-1 or SR-B1, as described below. In each experiment, an equal number of sham-treated mice and glycerol-injected mice were simultaneously collected and analyzed together (n for each group presented in figure legends).
Renal Cortical LDL-R mRNA Levels Following Glycerol-Induced Renal Injury
LDL-R protein could not be quantified in renal cortical samples due to the lack of a suitable antibody for Western blotting. Hence, to gain insight into its expression, LDL-R mRNA was assessed. To this end, mice were injected with glycerol and kidneys were resected at either 3, 4, or 18 hours post-glycerol injection (n = 5, 8, and 8 mice respectively). Kidneys from 14 sham-treated mice were used as controls. Cortical samples were immediately placed in TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA) and total RNA was extracted as per the manufacturer’s instructions. The final RNA pellet was brought up in RNase-free water to an approximate concentration of 3 mg/ml. The samples were electrophoresed for 30 minutes through 1.2% agarose containing ethidium bromide (Sigma, St. Louis, MO) to ensure lack of degradation of the samples (preservation of 18S and 28S ribosomal RNA). The samples were then analyzed for LDL-R and GAPDH (as a housekeeping gene) mRNAs as described below. (Note: Results from the 3 and 4 hours post-glycerol injection groups did not significantly differ, and hence, they were combined for further statistical analysis.)
HK-2 Cell Experiments
Fe-Induced Oxidative Stress
HK-2 cells, an immortalized human proximal tubular cell line, 30 were used for all in vitro experiments. They were cultured in T-75 flasks with keratinocyte serum-free medium (K-SFM) and passaged by trypsinization every 5 to 6 days, as previously described. 30 After splitting and allowing an ∼8- to ∼16-hour recovery period, the cells were either maintained under continued control culture conditions (control cells), or they were subjected to either a 4-hour or an 18-hour oxidant challenge. This consisted of ferrous ammonium sulfate, complexed to equimolar hydroxyquinoline (HQ), a siderophore which allows Fe to gain ready intracellular access. 8 The 4-hour and 18-hour challenges used 10-μmol/L FeHQ and 7.5-μmol/L FeHQ concentrations, respectively. The cells were then recovered, first by centrifugation of any detached cells, and then by removing adherent cells with a cell scraper, as previously described. 8 This was followed by extraction of either protein or total RNA for use in Western blotting or multiplexing RT-PCR respectively, as described below. Note: these Fe challenges have previously been shown to cause <10% and ∼30% to 40% cell death (LDH release) at the 4- and 18-hour time points, respectively.
Effect of Statin Treatment on HK-2 cell SR-B1, ABCA-1, and LDL-R mRNAs
Eight T75 flasks of near confluent HK-2 cells were divided into two equal groups: 1) control incubation for 18 hours, and 2) incubation for 18 hours with 10 μmol/L of mevastatin, as previously described. 4 At the completion of the experiments, total RNA was extracted from each flask and subjected to multiplexing RT-PCR for quantification of SR-B1, ABCA-1, LDL-R, and GAPDH mRNAs.
Western Blot Analyses of Cortical and HK-2 Cell Protein Samples
Renal cortical tissue samples were probed by Western blot for SR-B1 and ABCA-1 as previously described. 9,31 In the case of SR-B1, 7 μg of protein extract were electrophoresed through a 12% Bis-Tris acrylamide Nupage gel (Invitrogen) and probed with rabbit anti-mouse SR-B1 antibody (catalog number NB-400–104; Novus Biologicals, Littleton, CO) according to manufacturer’s instructions. For ABCA-1 detection, a 30-μg protein extract sample was electrophoresed through a 4% to 12% gradient Bis-Tris acrylamide Nupage gel (Invitrogen). Rabbit anti-mouse ABCA-1 (catalog number NB-400–15: Novus Biologicals) was used as the primary antibody per manufacturer’s instructions. Secondary detection for both antibodies was performed with horseradish peroxidase-labeled donkey anti-rabbit IgG (catalog number NA 934; Amersham-Pharmacia, Piscataway, NJ) and enhanced chemiluminescence (ECL Kit; Amersham-Pharmacia). 9,31
HK-2 cell protein extracts were prepared for Western blotting as previously noted 31 and probed for SR-B1, ABCA-1, and LDL-R proteins. SR-B1 and ABCA-1 detection was performed as noted above, with 3.5 μg and 30 μg of protein being electrophoresed, respectively. In the case of LDL-R, 25 μg of protein extract were electrophoresed through a 4% to 12% gradient Bis-Tris acrylamide Nupage gel (Invitrogen). Mouse anti-LDL-R (catalog number LP02: Oncogene Research Products, Boston, MA) was used as the primary antibody according to the manufacturer’s instructions. Secondary detection was performed with horseradish peroxidase-labeled sheep anti-mouse IgG (catalog number NA 931; Amersham Pharmacia) and enhanced chemiluminescence. 31
Nonspecific secondary antibody staining in the above Western blots was excluded by the fact that the secondary antibody, in the absence of the primary antibody, did not identify the relevant protein band(s). Equal protein loading/transfer was verified by India ink staining. Relevant protein band analysis was performed by optical density scanning (ABCA-1, 220 kd; SR-B1, 82 kd; nonglycosylated LDL-R 120 kd; glycosylated LDL-R, 160 kd).
Renal Cortical Analysis for LDL-R mRNA
Cortical RNA samples, harvested as noted above, were subjected to reverse transcription with the first strand synthesis kit for RT-PCR (Ambion Inc., Austin, TX). The RT reaction was carried out in RNase-free tubes by first adding ∼1 μg of RNA to a volume of RNase-free water (giving a total volume of 10 μl). After adding 2 μl of oligo-dt primer and 4 μl of dNTP mix (0.25 mmol/L each), the reaction was incubated at 72°C for 3 minutes, and then rapidly quenched on ice. RT components included 2 μl of 10X buffer (final composition of 50 mmol/L Tris-HCl, pH 8.3 + 75 mmol/L KCl + 3 mmol/L MgCl2, 1 μl of recombinant RNase inhibitor, 10 unit/μL), and 1 μl of M-MLV reverse transcriptase (100 units/μg RNA), added as per the manufacturer’s instructions. The total reaction volume of 20 μl was mixed and incubated at 42°C for 1 hour. The reaction was stopped by heating at 94°C for 10 minutes and storing the products on ice until PCR.
PCR was performed using the first strand synthesis kit for RT-PCR with mouse LDL-R and GAPDH-specific primers (Integrated DNA Technologies, Coralville, IA). The specific primers were designed with 50% to 60% GC composition (see Table 1 ▶ ). The resultant calculated high melting temperature (Tm) (>75°C) allowed for a stringent annealing temperature in the PCR cycles. After RT, 47 μl of a PCR master mix, containing all PCR components and primers for both LDL-R and GAPDH (50 pmol of sense and antisense for each), were added to tubes containing 3 μl of cDNA. Deoxynucleotides were added to a final concentration of 0.125 mmol/L. Reaction buffer (10X) was diluted (1:10) to a final composition of 10 mmol/L Tris-HCl, pH 8.3, 50 mmol/L KCl, and 1.5 mmol/L MgCl2. After the samples were overlaid with mineral oil, the tubes were placed in a DNA thermal cycler. The thermal cycler was programmed as delineated in Table 1 ▶ (rows 1 and 2). PCR product analysis was conducted with 2% agarose gel electrophoresis and ethidium bromide staining. cDNA bands were visualized and quantified by densitometry using a Typhoon 8600 scanner (Amersham Pharmacia). LDL-R cDNA bands were expressed as a ratio to the simultaneously obtained GAPDH cDNA band (housekeeping gene).
Table 1.
Mouse and Human Primers Used for Assessment of ABCA-1, SR-B1, and LDL-R mRNAs in Mouse Renal Cortex or in HK-2 (Human) Proximal Tubular Cells
| Genes | Primer sequences | PCR conditions | Product size |
|---|---|---|---|
| Mouse | 5′-CAA TCG GAA AAC CAT TTT GG-3′ | 94°C, 60 s; 55°C, 60 s; | 198 bp |
| LDL-R | 5′-TGT GAC CTT GTG GAA CAG GA-3′ | 72°C, 60 s; 23 cycles | |
| Mouse | 5′-CTG CCA TTT GCA GTG GCA AAG TGG-3′ | 94°C, 60 s; 55°C, 60 s; | 439 bp |
| GAPDH | 5′-TTG TCA TGG ATG ACC TTG GCC AGG-3′ | 72°C, 60 s; 23 cycles | |
| Human | 5′-CAA TGT CTC ACC AAG CTC TG-3′ | 94°C, 60 s; 55°C, 60 s; | 260 bp |
| LDL-R | 5′-TCT GTC TCG AGG GGT AGC TG-3′ | 72°C, 60 s; 23 cycles | |
| Human | 5′-CTG TGG GTG AGA TCA TGT GG-3′ | 94°C, 60 s; 55°C, 60 s; | 215 bp |
| SR-B1 | 5′-GCC AGA AGT CAA CCT TGC TC-3′ | 72°C, 60 s; 23 cycles | |
| Human | 5′-CAA CTA CAA AGC CCT CTT TG-3′ | 94°C, 60 s; 55°C, 60 s; | 310 bp |
| ABCA-1 | 5′-CTT GGC TGT TCT CCA TGA AG-3′ | 72°C, 60 s; 30 cycles | |
| Human | 5′-GTC TTC ACC ACC ATG GAG AAG-3′ | 94°C, 60 s; 55°C, 60 s; | 490 bp |
| GAPDH | 5′-GCT TCA CCA CCT TCT TGA TGT CAT C-3′ | 72°C, 60 s; 23 cycles |
RT-PCR Analysis of HK-2 Cells for ABCA-1, SR-B1, and LDL-R mRNAs
Total RNA was harvested from control and Fe-treated HK-2 cells and electrophoresed through ethidium bromide to ensure RNA integrity. 8 RT was conducted as above. The samples were then subjected to multiplexing PCR using specific sense/antisense primers designed for human ABCA-1, SR-B1, LDL-R, and GAPDH (Table 1 ▶ , rows 3 to 6) using the conditions described. After PCR product analysis, ABCA-1, SR-B1, and LDL-R cDNA bands were expressed as a ratio to the simultaneously obtained GAPDH cDNA.
Calculations and Statistics
All values are presented as means ± 1 SEM. Statistical comparisons were performed by unpaired Student’s t-test. Significance was judged by a P value of <0.05. The number of replicates for each experiment are given in the figure legends.
Results
Glycerol-Induced ARF: Renal Cortical Cholesterol Assessments
Glycerol injection caused severe ARF, as denoted by a marked rise in BUN concentrations (185 ± 22 mg/dl; controls, 29 ± 2; P < 0.0001). The corresponding renal cortical cholesterol assessments demonstrated 45% and 384% increments in FC and CE levels, respectively (see Figure 1 ▶ ).
Figure 1.

Free cholesterol (FC) and cholesteryl ester (CE) levels in renal cortex from control (C) mice and from mice 18 hours post-induction of glycerol mediated acute renal failure (Gly). Striking FC and CE levels were apparent following glycerol treatment, compared to controls (n = 7 for each determination).
Renal Cortical SR-B1 and ABCA-1 Protein Expression 18 Hours Post-Glycerol Injection
As shown in the left panel of Figure 2 ▶ , at 18 hours post-glycerol injection there was a statistically significant decrease in SR-B1 expression, as observed by Western blot analysis (protein detection at ∼58 kd). Of note, these SR-B1 data were previously presented in written form 9 and are presented here for comparison with subsequent renal cortical/HK-2 cell data.
Figure 2.

SR-B1 and ABCA-1 levels in renal cortical samples obtained from control (Cont) mice and from mice 18 hours post-induction of glycerol (Gly)-induced renal failure. As shown in the left panel, there was a marked reduction in SR-B1 in the glycerol-treated mice (n = 6), compared to controls (n = 5). As shown in the right panel, reductions in ABCA-1 bands were also observed (n = 12 for both the control and glycerol-treated group). SR-B1 appeared as a single dense band at 82 kd. In contrast, there was much weaker expression of ABCA-1, assuming a characteristic triplet appearance at ∼220 kd. Each of the three bands were diminished in the glycerol group. (Note the different y axis scales for ABCA-1 and SR-B1, reflecting much lesser apparent expression for the former versus the latter protein.)
Western blot analysis for ABCA-1 demonstrated its characteristic 3-band appearance at ∼220 kd (Figure 2 ▶ , right). As previously noted, this isoform complex is only weakly expressed in nonsteroidogenic (eg, kidney) versus steroidogenic (eg, adrenal) tissues, 31 and as per Santa Cruz Scientific, the producer of the ABCA-1 primary antibody. Eighteen hours after the glycerol challenge, a significant reduction in all three ABCA-1 bands were apparent with the lower two being barely detectable. Note the different y axis scales for heavily and lightly expressed HK-2 cell SR-B1 and ABCA-1, respectively.
Renal Cortical LDL-R mRNA Expression Post in Vivo Injury
A stepwise increase in LDL-R mRNA developed following glycerol injection. These values increased from a baseline value of 0.7 ± 0.03 to 0.82 ± 0.05 at the 3- to 4-hour time point (P < 0.001 vs. controls), and to 1.0 ± 0.03 at the 18-hour time point (P < 0.00001 vs. controls; results expressed as a ratio to the simultaneously obtained GAPDH values).
HK-2 Cell SR-B1 and ABCA-1 Protein Expression in Response to an Fe Challenge
Assessments at 4 Hours Post-Fe Challenge
As shown in Figure 3 ▶ , left panel, a marked reduction in SR-B1 expression was noted in the 4-hour Fe-challenged HK-2 cells, compared to their simultaneously processed controls. A statistically significant reduction in ABCA-1 expression at 4 hours post-FeHQ treatment was also observed (Figure 3 ▶ , right).
Figure 3.

SR-B1 and ABCA-1 in HK-2 cell extracts obtained 4 hours following the addition of an Fe challenge. Paralleling the changes observed in renal cortex, significant reductions in each protein were observed in the Fe-challenged cells, compared to the controls (Cont) (n = 6 separate cell preparations for each of the 4 groups). As with the renal cortical samples, ABCA-1 expression was, in general, remarkably lower than SR-B1, reflecting the fact that low levels of ABCA-1 exist in non-steroidogenic tissues.
Assessments at 18 Hours Post-Fe Challenge
As shown in Figure 4 ▶ , the results obtained at 18 hours post-Fe treatment largely recapitulated the results seen at 4 hours, as discussed above. These consisted of statistically significant reductions in both SR-B1 (P < 0.001; left panel) and ABCA-1 (P < 0.03; right panel). Thus, when viewed together, all of the in vivo data (obtained at one time point) and all of the in vitro data (obtained at 2 time points) yielded completely congruent results: significantly reduced SR-B1 and ABCA-1 protein levels following Fe/oxidant-induced tubular injury.
Figure 4.

SR-B1 and ABCA-1 in HK-2 cell extracts obtained 18 hours following the addition of an Fe challenge. The 18-hour results mimicked those seen at 4 hours, with significant reductions in SR-B1 and ABCA-1 (n = 6 per group).
HK-2 Cell LDL-R Protein Expression Post Fe-Induced Injury
After 4 hours of Fe treatment, a slight and insignificant increase in LDL-R protein expression was observed (FeHQ, 657 ± 34; controls 621 ± 60 density units; NS). However, as shown in Figure 5 ▶ , by 18 hours post-FeHQ treatment, a significant increase in HK-2 cell LDL-R protein was observed: a prominent band was observed at 120 kd (nonglycosylated form). A less prominent band, corresponding with the glycosylated LDL-R, was seen at 160 kd and appeared increased in the Fe-treated cells. These two bands were barely discernible in the control cells.
Figure 5.
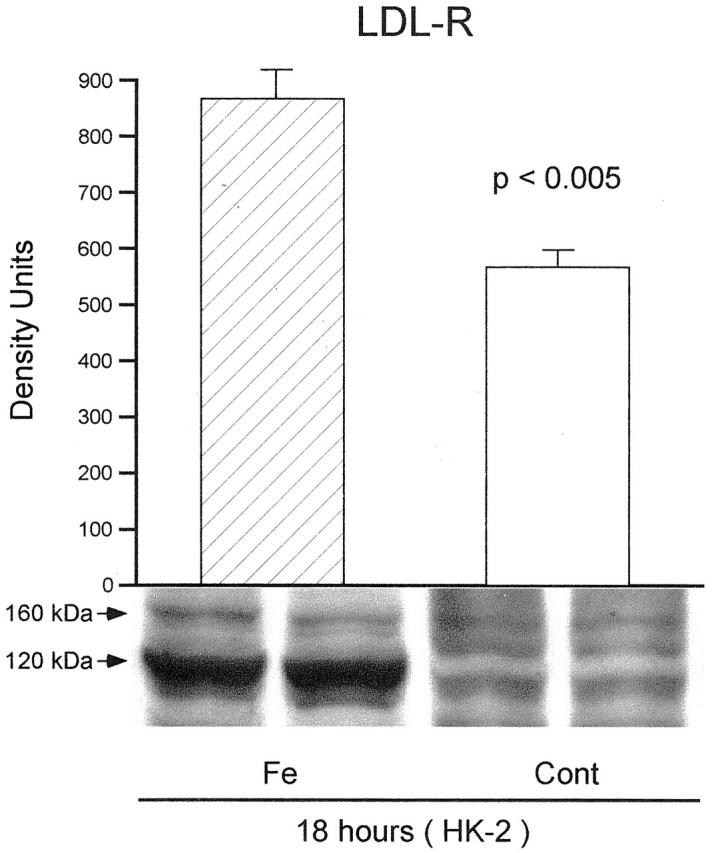
LDL-R protein expression in HK-2 cells 18 hours following the addition of an Fe challenge. LDL-R appears as a major (nonglycosylated) and a minor (glycosylated) band at 120 and 160 kd, respectively. There was greater expression of the dominant band in the Fe-treated cells versus the control group (P < 0.005; n = 5 per group).
HK-2 Cell SR-B1, ABCA-1, and LDL-R mRNAs Following FeHQ Injury
As shown in Figure 6 ▶ , left, ABCA-1 mRNA showed a progressive decline in FeHQ-challenged HK-2 cells, reaching values which were 40% of controls at the 18-hour time point. By 18 hours post Fe-induced injury, a significant decline in SR-B1 mRNA levels was also apparent. HK-2 cell LDL-R mRNA values also demonstrated time-dependent reductions following the induction of the Fe challenge. Of note, this finding was in contrast to the results observed in renal cortical samples, where LDL-R values showed stepwise increments, not decrements, following glycerol-induced injury.
Figure 6.

mRNA levels for ABCA-1, SR-B1, and LDL-R in HK-2 cells at either 4 or 18 hours post Fe-induced injury, as determined by multiplexing RT-PCR. The value for each mRNA (x) was factored by simultaneously obtained GAPDH values (x/GAPDH; y axis). Significant depressions in ABCA-1 and LDL-R mRNAs were apparent at both 4 and 18 hours post-Fe addition, compared to controls. In the case of SR-B1, a significant reduction was apparent in the Fe-challenged cells, but only at the 18-hour time point. (4-hour time points, n = 8 samples for each; 18-hour time points, n = 15 determinations for each).
Impact of Statin Therapy on HK-2 Cell ABCA-1, LDL-R, and SR-B1 mRNAs
As shown in Figure 7 ▶ , left, treating HK-2 cells with mevastatin for 18 hours caused an approximate 80% suppression in ABCA-1 mRNA levels. This is consistent with an appropriate physiological response whereby a statin-induced reduction in cellular cholesterol would be expected to induce a compensatory decrease in ABCA-1 mRNA. Statin therapy also suppressed SR-B1 mRNA (Figure 7 ▶ , middle panel). While the degree of SR-B1 mRNA suppression was quantitatively much less dramatic than it was for ABCA-1 mRNA, it, too, appears to be a physiologically appropriate response (a decrease in an FC efflux mechanism, compensating for a reduction in cholesterol levels). In contrast, statin treatment caused a 50% increase in LDL-R mRNA. Again, this is a physiologically appropriate response, given that a statin-induced block in intracellular cholesterol synthesis should theoretically lead to an increase in the LDL-R cholesterol uptake pathway. In sum, these results illustrate normal proximal tubular cell mRNA responses for a cholesterol reduction state. They also provide biological validation of the used multiplexing PCR technique for human cells.
Figure 7.

mRNA levels for ABCA-1, SR-B1, and LDL-R in control HK-2 cells and in HK-2 cells following 18 hours of HMG CoA reductase inhibition with mevastatin treatment (statin). The value of each mRNA (x) was factored by the simultaneously obtained GAPDH values (x/GAPDH). Statin treatment caused a marked reduction in ABCA-1 mRNA and a modest but highly significant reduction in SR-B1 mRNA. In contrast, LDL-R mRNA values manifested a highly significant increase in response to statin treatment. Each of these statin-mediated responses were judged to be biologically appropriate for a statin-induced cholesterol reduction state (n = 4 determinations for each depicted group).
Discussion
As previously reported, 1,4,6,7 glycerol-induced ARF leads to dramatic increases in renal cortical FC and CE levels, a phenomenon again documented in the present study. Since this change has been associated with an increase in both HMGCR protein and its mRNA, 1,8 it has been assumed that increased cholesterol synthesis mediates, or at least contributes to, this cholesterol overload state. For reasons detailed above, increased synthesis should initiate compensatory mechanisms to increase cholesterol efflux/decrease influx to normalize cholesterol levels. However, the present study provides three compelling pieces of evidence to support the concept that dysregulation of these compensatory processes occurs in a post-rhabdomyolysis cholesterol overload state, as follows: first, the cholesterol efflux protein ABCA-1 is reduced, not increased, in renal cortex following glycerol-induced injury, as evidenced by Western blotting; second, SR-B1 protein was also decreased post-renal injury despite the marked cholesterol overload state. Of note, SR-B1 can either mediate cellular CE influx (eg, in liver) 32-34 or enhance FC efflux. 23-26 To ascertain whether the influx or efflux pathway dominates in renal tubules, HK-2 cells were subjected to statin-induced partial cholesterol depletion, and SR-B1 mRNA levels were assessed. A significant decrease resulted, supporting the concept that SR-B1 predominantly functions in tubular cells as a cholesterol efflux pathway (ie, a decrease in efflux would compensate for statin-mediated cholesterol decrements). The third piece of evidence indicating dysregulation of cell cholesterol homeostasis post-renal injury is that glycerol-induced ARF caused stepwise increments in renal cortical LDL-R mRNA. It was not possible for us to probe for LDL-R protein in mouse cortex because of the lack of a suitable antibody. However, that LDL-R mRNA progressively rose rather than fell as renal cortical cholesterol loading occurred illustrates inappropriate regulation of this cholesterol homeostatic pathway. That LDL-R mRNA levels rose in HK-2 cells in response to statin treatment underscores that LDL-R message inversely correlates with tubular cell cholesterol content.
While the above documented decrements in renal cortical ABCA-1/SR-B1 proteins and the LDL-R mRNA increments were all physiologically inappropriate for a cholesterol overload state, it is conceivable that these changes could have reflected nonproximal tubular cell events. Since the proximal tubule is the prime target of glycerol-induced rhabdomyolysis, further attempts were made to strengthen the above conclusions with experiments conducted on Fe-challenged cultured proximal tubular (HK-2) cells. In large part, parallel changes to those observed in renal cortex were observed; at both 4 and 18 hours after the Fe challenge, ABCA-1 and SR-B1 protein decrements were observed. Conversely, LDL-R protein was significantly elevated at the 18-hour time point, consistent with the observed increases in renal cortical LDL-R mRNA. To complement these protein results, SR-B1, ABCA-1, and LDL-R mRNA levels after HK-2 cell injury were also assessed. Both ABCA-1 and SR-B1 mRNAs were suppressed at 4 and 18 hours post cell injury, directly correlating with the reduced protein levels. Thus, the physiologically aberrant changes in SR-B1 and ABCA-1 protein expression presumably reflected, at least in part, genomic/transcriptional events. Conversely, LDL-R mRNA reductions were observed. The reason for this discrepancy in LDL-R message in HK-2 cells versus renal cortex (decreases versus increases, respectively) remains elusive at this time. However, that HK-2 cell LDL-R protein increased despite reductions in its mRNA suggests that post-transcriptional events may also be involved in injury-evoked alterations in LDL-R protein expression.
Given the panoply of changes in post-injury cholesterol homeostasis (increased HMGCR, 1,8 decreased ABCA-1, decreased SR-B1, increased LDL-R), and given that each of these changes favors cell cholesterol accumulation, it would be extremely difficult to quantify the functional contribution of each to cholesterol accumulation. This is particularly true since any attempt to experimentally manipulate one pathway would secondarily impact each of the others. However, what does seem clear, at least from a teleological perspective, is that injured cells appear to be totally committed (ie, activating essentially all available pathways) to increasing, and then maintaining, the cholesterol overload state. The genomic/post genomic stimuli which might initiate and then maintain these changes once cholesterol loading has occurred remain unknown at this time. However, given that cholesterol accumulation protects tubular as well as extrarenal 35,36 cells from injury, it is tempting to speculate that some highly conserved evolutionary cell “survival” pathway ultimately underlies the changes which have been observed. Noteworthy in this regard are recent observations that endotoxin-induced stress, imposed on a macrophage cell line, causes a down-regulation of SR-B1 and ABCA-1 expression. 37 Thus, these results both parallel our observations in injured tubular cells, and underscore the observation that alterations in cholesterol homeostasis may, indeed, have broad-based pathophysiologic relevance in regard to cellular adaptations to stress. Finally, it should be noted that changes in cholesterol homeostasis and HMGCR activity can potentially impact protein prenylation and related signaling events. 38 While previous studies from this laboratory have suggested that Fe-mediated oxidative stress does not appear to alter these pathways, 8 these findings do not exclude their potential participation in alternative forms of tissue stress. 38
Acknowledgments
We thank Benita Brenner, and Hemangini Shah, University of New Mexico Health Sciences Center, Albuquerque, NM for assistance with LDL receptor mRNA analyses performed on mouse renal cortical samples.
Footnotes
Address reprint requests to Richard A. Zager, M.D., Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue N, Room D2–190, Seattle, WA 98109. E-mail: dzager@fhcrc.org.
Supported by grants from the National Institutes of Health (RO1s: DK-38432 and DK 54200).
References
- 1.Zager RA, Johnson A: Renal cortical cholesterol accumulation is an integral component of the systemic stress response. Kidney Int 2001, 60:2299-2310 [DOI] [PubMed] [Google Scholar]
- 2.Zager RA, Burkhart KM, Johnson AC, Sacks BM: Increased proximal tubular cholesterol content: implications for cell injury and “acquired cytoresistance”. Kidney Int 1999, 56:1788-1797 [DOI] [PubMed] [Google Scholar]
- 3.Zager RA: Plasma membrane cholesterol: a critical determinant of cellular energetics and tubular resistance to attack. Kidney Int 2000, 58:193-205 [DOI] [PubMed] [Google Scholar]
- 4.Zager RA, Kalhorn TF: Changes in free and esterified cholesterol: hallmarks of acute renal tubular injury and acquired cytoresistance. Am J Pathol 2000, 157:1007-1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zager RA, Johnson A, Anderson K, Wright S: Cholesterol ester accumulation: an immediate consequence of acute in vivo ischemic renal injury. Kidney Int 2001, 59:1750-1761 [DOI] [PubMed] [Google Scholar]
- 6.Zager RA, Johnson A, Hanson S, dela Rosa V: Altered cholesterol localization and caveolin expression during the evolution of acute renal failure. Kidney Int 2002, 61:1674-1683 [DOI] [PubMed] [Google Scholar]
- 7.Zager RA, Andoh T, Bennett WM: Renal cholesterol accumulation: a durable response after acute and subacute renal insults. Am J Pathol 2001, 159:743-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zager RA, Shah VO, Shah HV, Zager PG, Johnson AC, Hanson S: The mevalonate pathway during acute tubular injury: selected determinants and consequences. Am J Pathol 2002, 161:681-692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zager RA, Johnson ACM, Hanson SY: Sepsis syndrome stimulates proximal tubule cholesterol synthesis and suppresses the SR-B1 cholesterol transporter. Kidney Int 2003, 63:123-133 [DOI] [PubMed] [Google Scholar]
- 10.Honda N, Hishida A, Ikuma K, Yonemura K: Acquired resistance to acute renal failure. Kidney Int 1987, 31:1233-1238 [DOI] [PubMed] [Google Scholar]
- 11.Nath KA, Balla G, Verrcelloti J, Balla H, Jacob H, Levitt M, Rosenberg M: Induction of heme oxygenase is a rapid protective response in rhabdomyolysis in the rat. J Clin Invest 1992, 90:267-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung N, Croatt AJ, Haggard JJ, Grande JP, Nath KA: Acute cholestatic liver disease protects against glycerol-induced acute renal failure in the rat. Kidney Int 2001, 60:1047-1057 [DOI] [PubMed] [Google Scholar]
- 13.Paller MS: Hemoglobin and myoglobin-induced acute renal failure: role of iron in nephrotoxicity. Am J Physiol 1998, 255:F539-F544 [DOI] [PubMed] [Google Scholar]
- 14.Shah SV, Walker PD: Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am J Physiol 1988, 255:F438-F443 [DOI] [PubMed] [Google Scholar]
- 15.Zager RA: Combined mannitol and deferoxamine therapy for myohemoglobinuric renal injury and oxidant tubular stress. J Clin Invest 1992, 90:711-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baliga R, Zhang Z, Baliga M, Shah SV: Evidence for cytochrome p450 as a source of catalytic iron in myoglobinuric acute renal failure. Kidney Int 1996, 49:362-369 [DOI] [PubMed] [Google Scholar]
- 17.Johnson WJ, Phillips MC, Rothblatt GH: Lipoproteins and cellular cholesterol homeostasis. Bittman R eds. Subcellular Biochemistry: Cholesterol: Its Functions and Metabolism in Biology and Medicine. 1997:pp 235-276 Plenum Press, New York [DOI] [PubMed]
- 18.Wang N, Silver DL, Costet P, Tall AR: Specific binding of ApoA-1, enhanced cholesterol efflux, and altered plasma membrane morphology in cell expressing ABC1. J Biol Chem 2002, 275:33053-33058 [DOI] [PubMed] [Google Scholar]
- 19.Schmitz G, Langmann T: Structure, function, and regulation of the ABC1 gene product. Curr Opin Lipidol 2001, 12:129-140 [DOI] [PubMed] [Google Scholar]
- 20.Fielding CJ, Fielding PE: Cellular cholesterol efflux. Biochim Biophys Acta 2001, 1533:175-189 [DOI] [PubMed] [Google Scholar]
- 21.Bortnick AE, Rothblat GH, Stoudt G, Hoppe KL, Royer LJ, McNeish J, Francone OL: The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J Biol Chem 2000, 275:28634-28640 [DOI] [PubMed] [Google Scholar]
- 22.Sparrow CP, Baffic J, Lam MH, Lund EG, Adams AD, Fu X, Hayes N, Jones AB, Macnaul KL, Ondeyka J, Singh S, Wang J, Zhou G, Moller DE, Wright SD, Menke JG: A potent synthetic LXR agonist is more effective than cholesterol loading at inducing ABCA-1 mRNA and stimulating cholesterol efflux. J Biol Chem 2002, 277:10021-10027 [DOI] [PubMed] [Google Scholar]
- 23.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH: Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem 1998, 273:5599-5606 [DOI] [PubMed] [Google Scholar]
- 24.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR: Scavenger receptor B1 promotes high-density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem 1997, 272:20982-20985 [DOI] [PubMed] [Google Scholar]
- 25.Kellner-Weibel G, de la Llera-Moya M, Connelly MA, Stoudt G, Christian AE, Haynes MP, Williams DL, Rothblat GH: Expression of scavenger receptor B1 in COS-7 cells alters cholesterol content and distribution. Biochemistry 2000, 39:221-229 [DOI] [PubMed] [Google Scholar]
- 26.de la Llera-Moya M, Rothblat GH, Connelly MA, Kellner-Weibel G, Sakr SW, Phillips MC, Williams DL: Scavenger receptor B1 (SR-B1) mediates free cholesterol flux independently of HDL tethering to the cell surface. J Lipid Research 1999, 40:575-580 [PubMed] [Google Scholar]
- 27.Acton S, Rigotti A, Landschulz KT, Xy S, Hobbs HH, Krieger M: Identification of scavenger receptor SR-B1 as a high-density lipoprotein receptor. Science 1996, 271:518-520 [DOI] [PubMed] [Google Scholar]
- 28.Landschultz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH: Regulation of scavenger receptor, class B, type 1, a high density lipoprotein receptor, in liver and steroidogneic tissues of the rat. J Clin Invest 1996, 98:984-985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krause BR, Auerbach BJ: Reverse cholesterol transport and future pharmacological approaches to the treatment of atherosclerosis. Curr Opin Invest Drugs 2001, 2:375-381 [PubMed] [Google Scholar]
- 30.Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA, Torok-Storb B: HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 1994, 45:48-57 [DOI] [PubMed] [Google Scholar]
- 31.Johnson ACM, Yabu J, Hanson S, Shah VO, Zager RA: Experimental glomerulopathy alters renal cortical cholesterol, SR-B1, ABCA-1, and HMG CoA reductase expression. Am J Pathol 2003, 162:273-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fluiter K, van Berkel TJ: Scavenger receptor B1 substrates inhibit the selective uptake of high-density lipoprotein cholesteryl esters by rat parenchymal cells. Biochem J 1997, 326:4515-4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imachi H, Murao K, Sato M, Hosokawa H, Ishida T, Takahara J: CD36 LIMPII analagous-1, a human homolog of the rodent scavenger receptor B1, provides the cholesterol ester for steroidogenesis in adrenocortical cells. Metabolism 1996, 48:627-630 [DOI] [PubMed] [Google Scholar]
- 34.Matveev S, van der Westhuyzen DR, Smart EJ: Co-expression of receptor-B1 and caveolin-1 is associated with enhanced selective cholesteryl ester uptake pathway in mice with deletion of the low density lipoprotein receptor function. J Lipid Res 1999, 40:1647-1654 [PubMed] [Google Scholar]
- 35.Stirewalt DL, Appelbaum FR, Willman CL, Zager RA, Banker DE: Mevastatin can increase toxicity in primary AMLs exposed to standard therapeutic agents, but statin efficacy is not simply associated with RAS hotspot mutations or overexpression. Leukemia Res 2002, 1572:1-13 [DOI] [PubMed] [Google Scholar]
- 36.Li HY, Appelbaum FR, Willman CL, Zager RA, Banker DE: Cholesterol modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003, 101:3628-3634 [DOI] [PubMed] [Google Scholar]
- 37.Baranova I, Vishnyakova T, Bocharov A, Chen Z, Remaley AT, Stonik J, Eggerman TL, Patterson AP: Lipopolysaccharide down-regulates both scavenger receptor B1 and ATP binding cassette transporter A1 in RAW cells. Infect Immun 2002, 70:2995-3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danesh FR, Sadeghi MM, Amro N, Phillips C, Zeng L, Sahai A, Kanwar YS: 3-hydroxymethylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase / p21 signaling pathway: implications for diabetic nephropathy. Proc Natl Acad Sci USA 2002, 99:8301-8305 [DOI] [PMC free article] [PubMed] [Google Scholar]